

ABSTRACT
Influenza A virus requires ongoing cellular transcription to carry out the cap-snatching process. Chromatin remodelers modify chromatin structure to produce an active or inactive conformation, which enables or prevents the recruitment of transcriptional complexes to specific genes; viral transcription thus depends on chromatin dynamics. Influenza virus polymerase associates with chromatin components of the infected cell, such as RNA polymerase II (RNAP II) or the CHD6 chromatin remodeler. Here we show that another CHD family member, CHD1 protein, also interacts with the influenza virus polymerase complex. CHD1 recognizes the H3K4me3 (histone 3 with a trimethyl group in lysine 4) histone modification, a hallmark of active chromatin. Downregulation of CHD1 causes a reduction in viral polymerase activity, viral RNA transcription, and the production of infectious particles. Despite the dependence of influenza virus on cellular transcription, RNAP II is degraded when viral transcription is complete, and recombinant viruses unable to degrade RNAP II show decreased pathogenicity in the murine model. We describe the CHD1–RNAP II association, as well as the parallel degradation of both proteins during infection with viruses showing full or reduced induction of degradation. The H3K4me3 histone mark also decreased during influenza virus infection, whereas a histone mark of inactive chromatin, H3K27me3, remained unchanged. Our results indicate that CHD1 is a positive regulator of influenza virus multiplication and suggest a role for chromatin remodeling in the control of the influenza virus life cycle.
IMPORTANCE Although influenza virus is not integrated into the genome of the infected cell, it needs continuous cellular transcription to synthesize viral mRNA. This mechanism implies functional association with host genome expression and thus depends on chromatin dynamics. Influenza virus polymerase associates with transcription-related factors, such as RNA polymerase II, and with chromatin remodelers, such as CHD6. We identified the association of viral polymerase with another chromatin remodeler, the CHD1 protein, which positively modulated viral polymerase activity, viral RNA transcription, and virus multiplication. Once viral transcription is complete, RNAP II is degraded in infected cells, probably as a virus-induced mechanism to reduce the antiviral response. CHD1 associated with RNAP II and paralleled its degradation during infection with viruses that induce full or reduced degradation. These findings suggest that RNAP II degradation and CHD1 degradation cooperate to reduce the antiviral response.
INTRODUCTION
Influenza A virus contains eight single-stranded RNA segments of negative polarity (viral RNA [vRNA]) that form viral ribonucleoproteins (vRNP) by association with a trimeric polymerase complex that consists of the PA, PB1, and PB2 subunits and the nucleoprotein (NP). These vRNP are the functional units for RNA transcription and replication, which are restricted to the nucleus of the infected cell (1). For viral RNA replication, the vRNAs are copied to form full-length positive-stranded RNAs (cRNA), which serve as templates for vRNA synthesis (2). During transcription, capped and polyadenylated viral mRNAs are synthesized by the viral polymerase through an initiation mechanism that uses as primers short-capped oligonucleotides scavenged from newly synthesized RNA polymerase II (RNAP II) transcripts by a viral endonuclease activity that resides in the PA subunit (3, 4). This transcription strategy involves functional coupling between viral and cellular transcription for the cap-snatching process. The viral polymerase is reported to interact with host cell transcription-related factors (5–9), among which is the largest subunit of the RNAP II itself (10).
Although influenza virus does not integrate into the infected-cell genome, its transcription mechanism involves absolute dependence on chromatin-based functions and thus on chromatin dynamics. vRNP are tightly bound to the nuclear matrix or to chromatin components (11–15), and viral RNA transcription and replication are proposed to take place in DNase-insensitive nuclear fractions that include chromatin and/or the cellular matrix (16). Specific interactions take place between chromatin remodelers and influenza virus proteins, including the association of CHD3 with the nonstructural protein NS2 (17). CHD6 interacts with the PA polymerase subunit and with the viral polymerase complex (8, 18), which relocates to inactive chromatin late in infection (18) and negatively modulates influenza virus multiplication (18).
CHD3 and CHD6 belong to the CHD (chromodomain-helicase DNA-binding) family of chromatin remodelers, which have two N-terminal chromodomains that interact with methylated histone tails (19, 20). This interaction contributes to the dynamics of chromatin structure, affecting transcription factor binding to modulate transcription initiation and elongation steps (21–23). Despite the coupling between viral and cellular transcription, reports showed that in a process triggered by the viral polymerase, influenza virus infection induces RNAP II degradation once viral transcription is complete and synthesis of cellular mRNA is no longer needed (24–26). We recently characterized the involvement pf the PB2 and PA subunits in RNAP II degradation, as well as the contribution of specific subunit residues to this process, which correlates with pathogenicity in mice (27). We also showed that the CHD6 chromatin remodeler is specifically degraded after influenza virus infection in cultured cells and in a mouse model (28).
CHD1 is the best-characterized member of the CHD chromatin remodeler family. It binds to histone 3 with a trimethyl group in lysine 4 (H3K4me3), a hallmark of active chromatin, both in vitro (22) and in vivo, near the beginning of active genes (29); it also associates with transcription complexes, such as Mediator, FACT, Paf1, or RUNX1/AML1 (29–32). The combined action of CHD1 binding to transcriptional complexes and CHD1 binding to H3K4me3 allows CHD1 recruitment to its responsive genes. Since influenza virus polymerase interacts with the RNAP II complex and is highly dependent on its activity, we explored the role of CHD1 in the influenza virus life cycle. We show that CHD1 interacts with influenza virus polymerase and positively modulates viral RNA transcription and virus multiplication. Like that of RNAP II, CHD1 degradation is initiated at midinfection, once viral RNA transcription is complete, suggesting a coordinated role for CHD1 and RNAP II in the influenza virus life cycle.
MATERIALS AND METHODS
Biological materials.
Human embryonic kidney 293T (HEK293T) cells, human respiratory cells (A549), and Madin-Darby canine kidney (MDCK) cells were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal calf serum. Plasmids pCMVPA, pCMVPB1, pCMVPB2, and pCMVNP have been described previously (33). Plasmid pHH-NS CAT expresses, under the control of an RNA polymerase I promoter, the chloramphenicol acetyltransferase (CAT) gene in an antisense orientation flanked by the untranslated region (UTR) sequences of the NS segment. This plasmid was constructed by insertion of the nonstructural (NS)-CAT fragment from pT7NSCAT-RT (34) into the pHH plasmid.
Virus infection.
Cells were infected with influenza viruses at a low or high multiplicity of infection (MOI) or with vesicular stomatitis virus (VSV) or adenovirus (AdV) at an MOI of 5 or 7.5 PFU/cell, respectively. At various times postinfection, cells were collected in phosphate-buffered saline (PBS) with a protease inhibitor cocktail (cOmplete; Roche), and virus titers were determined by plaque assay.
Lentiviral particle production and cell transduction.
Lentiviral particles were produced in HEK293T cells by cotransfection of plasmids psPAX2 and pMD2.G with each of the pLKO-based short hairpin RNA (shRNA) vectors, as described previously (35, 36). Supernatants were collected 40 to 48 h posttransfection, filtered through a 0.45-μm filter, and used to transduce A549 or HEK293T cells. Since the lentiviral vectors confer puromycin resistance, the minimum amount of supernatant necessary to confer 100% resistance to puromycin (5 μg/ml) was used. Silencing was tested by Western blotting, generally at 3 days postransduction. The viability of transduced cells was determined by the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (37).
Confocal immunofluorescence microscopy.
Cultured A549 cells were either left uninfected or infected with the A/Victoria/3/75 (VIC) strain at a multiplicity of infection of 5 PFU/cell. At 4 h postinfection (hpi), cells were fixed with 4% paraformaldehyde (for 20 min at room temperature; Sigma) and were stored in PBS. For immunofluorescence, cells were permeabilized in PBS with 0.5% Triton X-100 (5 min) and were incubated with the following primary antibodies diluted in PBS–0.1% (wt/vol) bovine serum albumin (BSA): rabbit anti-CHD1 (D8C2; dilution, 1:200; Cell Signaling), rabbit anti-H3K4me3 (C42D8; dilution, 1:200; Cell Signaling), a monoclonal anti-PB2 antibody (dilution, 1:100) (38), and a monoclonal anti-RNAP II antibody (8WG16; dilution, 1:500; Covance). Confocal microscopy was performed with a Leica TCS SP5 laser scanning system. Images (1,024 by 1,024 pixels; 8-bit gray scale depth) were acquired sequentially every 0.2 to 0.3 μm using LAS AF (version 2.2.1) software (Leica) and were analyzed with LAS AF and MetaMorph Premier (version 7.5.2) image analysis software (Molecular Devices). For colocalization analysis, single confocal sections and the colocalization mask that produces binary images showing only overlapping pixels (white spots) were used.
Western blotting.
Cells infected at various MOIs were collected at various times postinfection in Laemmli sample buffer. Western blotting was performed as described previously (24). The following rabbit polyclonal antibodies were used: anti-CHD1 (D8C2; dilution, 1:1,000; Cell Signaling), anti-CHD3 (A301-219A; dilution, 1:500; Bethyl), anti-CHD6 (A301-221A; dilution, 1:100; Bethyl), and anti-CHD9 (ABIN1007493; dilution, 1:500; Antibodies Online). A monoclonal anti-CHD5 antibody (D-10; dilution, 1:500; Santa Cruz) was also used. For RNA polymerase II, we used monoclonal antibody 8WG16 (dilution, 1:500; Covance), which recognizes predominantly unphosphorylated Ser2 of the C-terminal domain (CTD) (39). For influenza virus proteins, monoclonal anti-PA (antibodies 2 and 9; dilution, 1:250) and anti-PB2 (antibody 22; dilution, 1:100) antibodies (40), rabbit polyclonal anti-PB1 (dilution, 1:1,000) (41) and anti-NP (dilution, 1:5,000) (42) antibodies, and a mouse monoclonal anti-β-actin antibody (dilution, 1:1,000; Sigma) were used. For histone recognition, the following polyclonal antibodies were used: anti-H3K4me3 (dilution, 1:1,000; Cell Signaling), anti-H3K27me3 (dilution, 1:500; Active Motif), and anti-H3 (D1H2; dilution, 1:1,000; Cell Signaling).
CAT assays.
HEK293T cells were infected with control or specific lentiviruses for CHD1 silencing and were cotransfected with the pCMVPA, pCMVPB2, pCMVPB1, pCMVNP, or pHH-NS CAT plasmid and a plasmid expressing green fluorescent protein (GFP) under the control of the RNAP II promoter (24 h). Cell extracts were collected, and CAT accumulation was assayed by enzyme-linked immunosorbent assays (ELISA) (GE Healthcare) using purified CAT enzyme as the standard.
Immunoprecipitation.
Immunoprecipitation studies were performed as described previously (43). Briefly, 107 A549 cells were either mock infected or infected with influenza virus at 3 PFU/cell. At 4 hpi, cells were collected and were lysed in a buffer containing 150 mM NaCl, 5 mM EDTA, 1.5 mM MgCl2, 50 mM Tris-HCl (pH 8.5) and 0.5% IGEPAL, with cOmplete protease inhibitors. The lysate was centrifuged (10,000 × g, 10 min, 4°C), and the supernatant was used for immunoprecipitation studies with 1 μg of anti-CHD antibodies, anti-RNAP II (8WG16), or an unrelated antibody. Immune complexes were washed 10 times with the lysis buffer, and the immunoprecipitated proteins were analyzed by Western blotting.
RNA analysis.
For RNA extraction, cell pellets were resuspended in 1 ml TRIzol reagent (Invitrogen), and RNA was purified as recommended by the manufacturer. RNA was digested with RNase-free DNase (1 U/mg, 1 h, 37°C), extracted with phenol-chloroform-isoamyl alcohol, and precipitated with ethanol.
Primer extension analyses were performed as described previously (44). We used synthetic oligonucleotides complementary to mRNA or vRNA specific for the VIC strain sequence located ∼50 to 150 nucleotides downstream of the 5′ end. Primers were 5′ end labeled using [γ-32P]ATP and T4 polynucleotide kinase and were annealed to the specific RNA molecules in the RNA samples. Reverse transcriptase (RT), deoxyribonucleoside triphosphates, and appropriate buffer components were added to the primer-mRNA hybrids to catalyze primer elongation. The resulting radiolabeled cDNA products were analyzed by denaturing polyacrylamide gel electrophoresis followed by autoradiography.
Specific real-time RT-PCR was performed essentially as described previously (45). This method is based on reverse transcription using tagged primers to add a “tag” sequence at the 5′ end, followed by the hot-start method. Real-time PCR using the tagged primer as the forward primer and a segment-specific reverse primer ensures specificity for quantifying mRNAs and vRNAs. PCRs were performed in 96-well PCR plates, using SYBR green PCR master mix and the specific primers, in an ABI Prism 7000 sequence detection system (Applied Biosystems). The cycle threshold (CT) was determined with SDS software (Applied Biosystems). Serial dilutions of cDNA were used to ensure amplification.
To analyze primary transcription, cells were either control silenced or CHD1 silenced, infected 3 days later with the VIC strain (3 PFU/cell), and treated with cycloheximide (100 μg/ml). At 6 hpi, total RNA was extracted and was used to detect NS mRNA by real-time RT-PCR.
RNA synthesis.
Intracellular RNA synthesis was detected using a chemical method based on biosynthetic incorporation of the uridine analog 5-ethynyluridine (EU) into newly transcribed RNA. On average, EU is incorporated once every 35 uridine residues in total RNA. EU-labeled cellular RNA is detected rapidly and with high sensitivity using a copper(I)-catalyzed cycloaddition reaction (also termed click chemistry) with fluorescent azides (Click-iT Nascent RNA Capture kit; Life Technologies), followed by microscopic imaging. Alternatively, we measured bromouridine (BrU) incorporation in cell culture to label nascent RNA. Cells were cultured with 2.5 mM BrU (15 min), followed by immunofluorescence using a monoclonal anti-iododeoxyuridine (IdU)/bromodeoxyuridine (BrdU) antibody (Caltag Laboratories). We also detected RNA elongation by specific immunolabeling of phosphorylated Ser2 (Ser2P) of RNAP II using a monoclonal anti-H5 antibody (Abcam).
RESULTS
CHD1 interacts with the influenza virus polymerase complex.
We previously observed the interaction of influenza virus polymerase with the CHD6 chromatin remodeler (18). To characterize additional interactions of viral polymerase with chromatin components, we performed coimmunoprecipitation analysis of several CHD family members in A549 human respiratory epithelial cells infected with 3 PFU of influenza virus A/Victoria/3/75 (VIC)/cell at 4 h postinfection (hpi). Extracts of mock-infected or infected cells were coimmunoprecipitated using either a rabbit polyclonal antibody against CHD1, CHD3, CHD5, CHD6, or CHD9 or a control antibody (IgG), washed, and analyzed by Western blotting (25). The three polymerase subunits were associated with CHD6, and we observed a clear association with CHD1; viral polymerase subunits also showed some association with the CHD5 chromatin remodeler (Fig. 1A). Given the strong association of viral polymerase subunits with CHD1, we also examined CHD1 for colocalization with components of the viral RNP (the PB2 protein and NP). Cultures of A549 cells were infected with the VIC strain (3 PFU/cell); cells were fixed at 4 hpi and were analyzed by immunofluorescence microscopy. Single confocal sections and the colocalization mask that produces binary images showing only overlapping pixels (white spots) were used (Fig. 1B). In agreement with the observed association of CHD1 with the viral polymerase, we found CHD1 colocalization with PB2 and NP. Our results indicate that CHD1 interacts with the polymerase complex and colocalizes with viral RNP in infected cells.
FIG 1.

CHD1 interacts with the influenza virus polymerase complex. (A) A549 cells were either mock infected (−) or infected (+) with the VIC strain (3 PFU/cell, 4 h); cell extracts were obtained and were immunoprecipitated (IP) using CHD-specific or control (IgG) antibodies. Proteins were monitored in Western blots probed with appropriate antibodies. Input, A549 cell extracts. (B) A549 cells were infected with influenza virus (3 PFU/cell, 4 h); cells were fixed and were processed for immunofluorescence using anti-CHD1, anti-PB2, and anti-NP antibodies. DAPI was used to stain nuclei. The colocalization panels show the signals common to the two antibodies, obtained with the colocalization mask. Representative images from one of three experiments are shown.
Effect of CHD1 silencing on cell physiology.
Viral and cellular transcriptions are functionally associated to allow the cap-snatching process, and viral polymerase interacts with cellular RNAP II and other transcription-related factors. The data suggested that CHD1 plays a role in the influenza virus life cycle. Since CHD1 is a chromatin remodeler with an important function in mRNA transcription elongation (46, 47), we first tested the effect of CHD1 knockdown on cell physiology. For RNA interference (RNAi)-mediated CHD1-silencing experiments, we used lentiviruses expressing shRNAs specific for CHD1 (shCHD1.1, shCHD1.2, shCHD1.3) or a control that expresses irrelevant shRNA (shTT) (48, 49). Expression of the CHD1 shRNAs reduced CHD1 protein accumulation by 25 to 70% from that with shTT (Fig. 2A). An MTT assay (see Materials and Methods) of the HEK293T and A549 cell lines at 5 days postsilencing showed no effect of any of the silencers on cell viability (Fig. 2B).
FIG 2.

CHD1 silencing does not affect cell viability or RNA polymerase activity. HEK293T and A549 cells were infected with lentiviruses expressing CHD1 silencers or a control lentivirus (shTT). (A) Western blot analysis of CHD1 proteins. (B) The viability of transduced HEK293T and A549 cells was determined by the MTT assay measuring cell metabolic activity. (C) Quantification of EU and BrU incorporation as well as RNAP II staining in 300 A549 cells infected with lentiviruses expressing CHD1 silencers or a control lentivirus (shTT). The relative intensity found in each analysis was compared with the intensity of uninfected control cells. Bars represent means ± standard errors of the means; no statistical differences were found.
We used several approaches to analyze the effect of CHD1 silencing on overall de novo cellular transcription. The Click-iT RNA Alexa Fluor 488 imaging kit enabled the detection of newly synthesized RNA (50) using an alkyne-modified nucleoside, the compound 5-ethynyluridine (EU), which is incorporated into RNA but not DNA. We also analyzed bromouridine (BrU) incorporation and RNAP II-Ser2P levels. Control-silenced or CHD1-silenced A549 cells either were treated with EU or BrU or were left untreated (see Materials and Methods) and were processed for immunofluorescence detection of EU, BrU, or RNAP II-Ser2P, respectively. To quantify the effect of CHD1 silencing on cell transcription, we analyzed the label intensity in 300 cells for each condition and compared the relative intensity with that of untreated control cells (Fig. 2C). The absence of any significant differences among any of the signals indicated that CHD1 silencing does not have a general deleterious effect on RNA transcription.
Effect of CHD1 silencing on influenza virus polymerase activity.
To establish possible CHD1 function in influenza virus infection, we analyzed its role in viral polymerase activity. To reconstitute viral RNP, control-silenced or CHD1-silenced cells were transfected with plasmids pCMV-PB1, pCMV-PB2, pCMV-PA, pCMV-NP, and pHH-NSCAT, which encode VIC strain proteins PB1, PB2, PA, and NP and a negative-sense virus-like CAT RNA under the control of the polymerase I (Pol I) promoter (34, 51). In control cells, plasmid pCMV-PB1 was omitted. A GFP-expressing plasmid was added under each condition as a transfection control for normalization of the CAT accumulation used to measure the RNA replication/transcription activity of recombinant RNP (Fig. 3A). CAT accumulation decreased 30 to 60% in CHD1-silenced cells, concomitantly with the degree of CHD1 silencing (Fig. 2A). To exclude the possibility of a decrease in the accumulation of the RNP and/or in RNA polymerase II-driven expression of viral polymerase subunits, which could explain the decreased CAT activity, we analyzed the accumulation of viral polymerase subunits and β-actin by Western blotting; the accumulation of all these proteins was independent of CHD1 silencing (Fig. 3B). These data indicated that CHD1 is not essential for RNP accumulation but positively modulates viral polymerase activity. We used primer extension analyses (52) to examine the virus-like CAT positive- and negative-sense RNA levels produced by the reconstituted RNP in control-silenced and CHD1-silenced cells. We found 42% to 70% reductions in vRNA and mRNA levels when RNP were reconstituted in the different CHD1 knockdown cells (Fig. 3C and D), indicating that CHD1 stimulates viral RNA polymerase activity.
FIG 3.

CHD1 silencing reduces influenza virus RNP activity. (A) HEK293T cells were infected with lentiviruses expressing CHD1 silencers or a control lentivirus (shTT) and were used for CAT RNP reconstitution (see Materials and Methods). At 48 h postreconstitution, the amount of CAT protein in total-cell extracts was analyzed by ELISA. CAT activity was normalized in each case to the amount of GFP expression, which was used as a transfection control. Activity in control lentivirus-transfected cells was considered 100%. Mock, untransfected cells; negative control, the plasmid expressing PB1 was omitted. (B) Aliquots of samples for which results are shown in panel A were analyzed by Western blotting for RNP, GFP, and β-actin. (C) Aliquots of samples for which results are shown in panel A were subjected to primer extension analysis (see Materials and Methods). Asterisks indicate nonspecific bands in uninfected cells (−). (D) Quantification of data from panel C. Bars represent means ± standard errors of the means. Significance was determined using an unpaired Student t test with Welch's correction; 3 independent experiments were performed. **, P < 0.01; ***, P < 0.001.
CHD1 affects influenza virus transcription.
To analyze the relevance of CHD1 in virus RNA replication, we infected A549 cells with lentiviruses expressing the same CHD1 silencers or control shRNA, followed by infection with the VIC strain (3 PFU/cell). Total RNA was extracted at various times postinfection, and RT quantitative PCR (RT-qPCR) was used as described previously (45) to quantify viral genomic RNA (vRNA) and viral mRNA in the NP segment. Reductions in the levels of negative-sense (Fig. 4A) and positive-sense (Fig. 4B) viral RNA were detected after CHD1 silencing, corresponding to the degree of CHD1 reduction. These results indicated that CHD1 is needed for viral RNA replication but could not indicate a role for CHD1 in virus transcription. To clarify this question, we determined the levels of primary virus transcripts after the infection of CHD1- and control-silenced cells in the presence of cycloheximide to prevent viral protein synthesis and hence viral RNA replication (53). Accumulation of total NP transcripts, determined by RT-qPCR, showed that CHD1 silencing led to a reduction in primary transcription (Fig. 5A). We verified the inhibition of virus multiplication after cycloheximide treatment by determining virus mRNA levels in treated and untreated infected cells, which showed an approximately 10-fold reduction after drug treatment (Fig. 5B).
FIG 4.
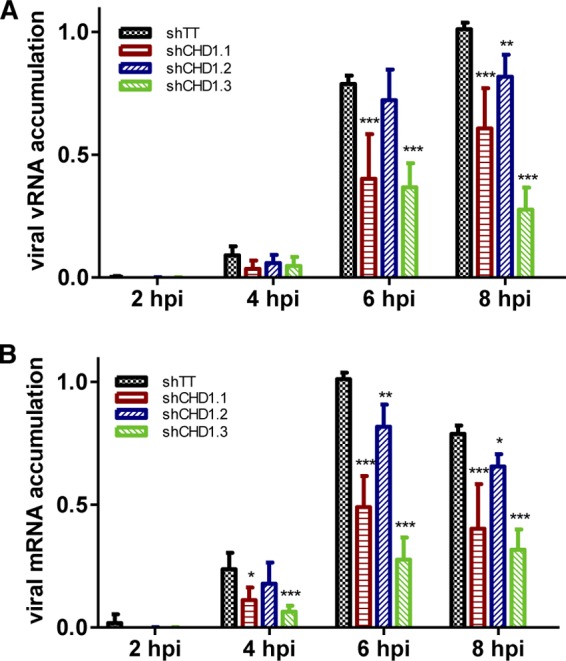
Influenza virus RNA transcription and replication in CHD1-downregulated cells. A549 cells were infected first with lentiviruses expressing CHD1 silencers or control shRNA and then with the VIC strain (3 PFU/cell); total RNA was extracted at various times postinfection and was used to quantify viral genomic RNA (vRNA) and the viral mRNA of the NP segment by RT-qPCR (see Materials and Methods). Bars represent means ± standard errors of the means. Significance was determined by an unpaired Student t test with Welch's correction; 3 technical replicates of 3 independent experiments were performed. *, P < 0.5; **, P < 0.01; ***, P < 0.001.
FIG 5.
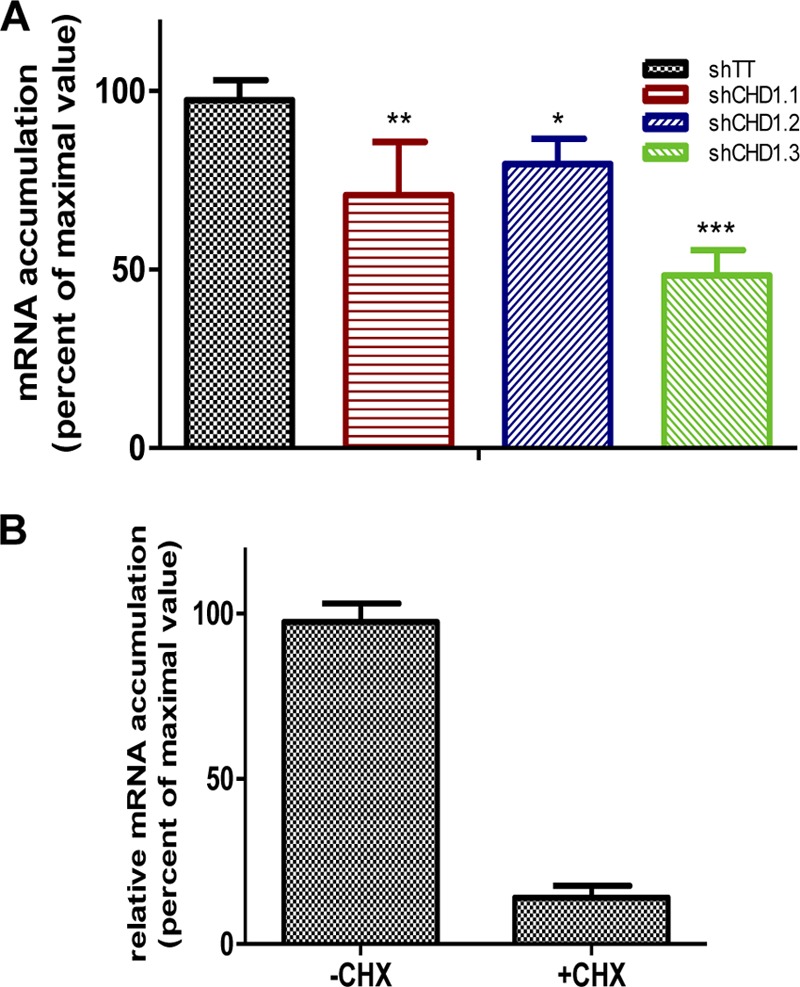
CHD1 downregulation affects influenza virus mRNA transcription. (A) CHD1-silenced and control-silenced A549 cells were treated with cycloheximide to prevent the synthesis of viral proteins. The cells were then infected with the VIC strain (3 PFU/cell); at 6 hpi, cell extracts were used for detection of the NP segment of viral mRNA by RT-qPCR (see Materials and Methods). (B) Detection of viral mRNA in influenza virus-infected cells that were either left untreated or treated with cycloheximide (CHX). Bars represent means ± standard errors of the means. Significance was determined by an unpaired Student t test with Welch's correction; 3 technical replicates of 3 independent experiments were performed. *, P < 0.5; **, P < 0.01; ***, P < 0.001.
CHD1 is specifically needed for influenza virus infection.
To determine whether CHD1 specifically modulates influenza virus infection, we evaluated the effect of CHD1 silencing on virus multiplication by infecting control- or CHD1-silenced A549 cells at a low multiplicity of infection (MOI) (10−3 PFU/cell) with the VIC strain (subtype H3N2), and determined virus titers by plaque assays on MDCK cells. We found reductions in viral titers of ∼0.5 to 1 log unit, depending on CHD1 levels, in CHD1-deficient cells relative to control cells (Fig. 6A). A concomitant decrease in viral protein accumulation (Fig. 6B) was also observed. To test whether CHD1 can be considered a general modulator of influenza virus multiplication, we performed similar experiments using influenza strains of a different subtype. Control- or CHD1-silenced A549 cells were infected at a low multiplicity with the laboratory-passaged influenza virus strain A/PR/8/34 (PR8) (Fig. 6C) or with a natural human isolate, the 2009 pandemic strain A/California/04/09 (CAL) (Fig. 6D), both of which belong to the H1N1 subtype. As with the VIC strain, these cells showed 0.5- to 1-log reductions in viral titers that correlated with the degree of CHD1 silencing, indicating that the CHD1 chromatin remodeler is a positive modulator of human influenza virus.
FIG 6.

CHD1 silencing reduces viral titers in multistep growth experiments in A549 cells. (A) CHD1-silenced or control-silenced A549 cells were first generated by lentivirus infection and then infected with the A/Victoria/3/75 (VIC) influenza virus strain (10−3 PFU/cell). Cell extracts were obtained at the indicated times postinfection, and virus titers were determined by plaque assays in MDCK cells. (B) Aliquots of samples for which results are shown in panel A were used for the detection of proteins by Western blotting. (C and D) A549 cells were treated as for panel A, except that they were infected with the influenza virus A/PR8/8/34 (PR8) strain (C) or the influenza virus A/California/04/09 (CAL) strain (D). Error bars represent standard errors of the means. Significance was determined by an unpaired Student t test with Welch's correction; 3 technical replicates of 3 independent experiments were performed. **, P < 0.01; ***, P < 0.001.
To ascertain the specificity of CHD1 in the modulation of the influenza virus life cycle, we studied the multiplication of two additional viruses in CHD1-silenced cells: vesicular stomatitis virus (VSV), another negative-stranded RNA virus, and adenovirus 5 (Ad5), a nuclear virus strongly dependent on cellular transcription and splicing machineries. A549 cell cultures were first CHD1 silenced or control silenced and then infected with VSV (Fig. 7A) or Ad5 (Fig. 7B). We determined the amounts of virus that had accumulated in the culture supernatant (VSV) or the infected cells (Ad5) by plaque assays on BHK21 (VSV) or HEK293T (Ad5) cells, as described previously (54). Ad5 multiplication was unaffected by CHD1 downregulation; for VSV replication, the silencer shCHD1.3 elicited an initial delay in virus multiplication, although cells infected with this lentivirus attained a viral titer similar to that of infected cells at 48 hpi. These results indicate that CHD1 is a particularly important host factor for influenza virus multiplication.
FIG 7.
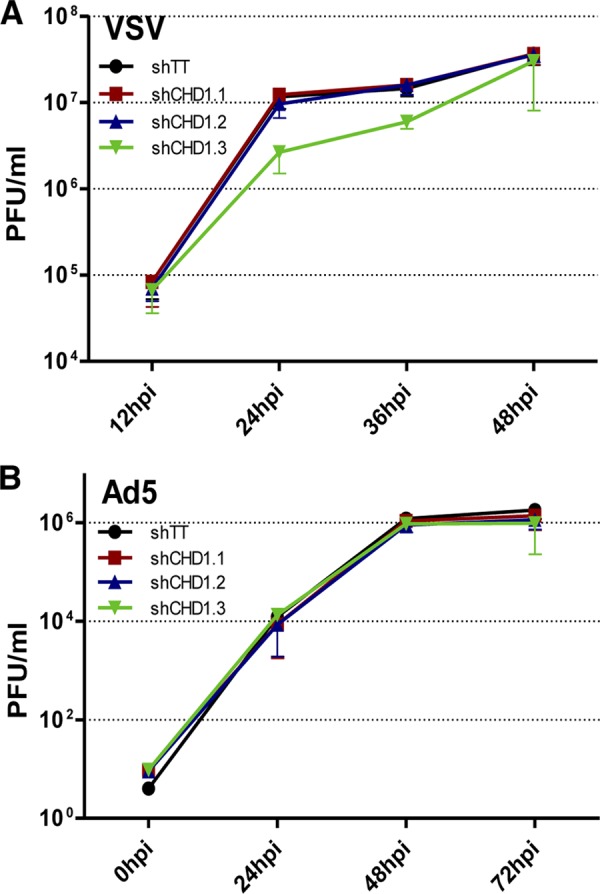
CHD1 does not control VSV or adenovirus replication. Cultures of A549 cells were first CHD1 silenced or control silenced and then infected with VSV (A) or Ad5 (B) (see Materials and Methods). Levels of virus accumulation in the culture supernatant (VSV) or in infected cells (Ad5) were determined by plaque assays on BHK21 (VSV) or HEK293T (Ad5) cells. Error bars represent standard errors of the means. No significant differences were found in 3 technical replicates of 3 independent experiments.
CHD1 is degraded in parallel with RNAP II during influenza virus infection.
CHD1 is a component of several transcription complexes, such as Mediator and RNA polymerase II-associated factor (PAF) (55, 56). Mediator is a multiprotein complex that acts as a transcriptional coactivator in all eukaryotes and is necessary for successful transcription of RNAP II-dependent genes in yeasts and mammals (56). Mediator associates with general transcription factors and with the C-terminal domain of the RNAP II holoenzyme, acting as a bridge between this enzyme and transcription factors (57). Previous data for CHD1-Mediator association support a model in which Mediator coordinates the assembly of the transcription preinitiation complex as well as CHD1 recruitment (29). CHD1 also interacts with hPaf1/PD2, a subunit of the human PAF complex, which is involved in the regulation of transcriptional elongation in pancreatic cancer cells (55). Influenza virus transcription requires ongoing cellular transcription to carry out the cap-snatching process, and the viral RNA polymerase binds to the C-terminal part of RNAP II (10). In spite of this viral-cellular polymerase interaction, RNAP II degradation begins in midinfection, once viral transcription is complete and de novo cell transcription is no longer needed (24, 25). This degradation is reflected as a shorter protein half-life and is proteasome independent (24). In vivo experiments indicate that RNAP II degradation correlates with pathogenicity in mice (27).
Since CHD1 associates with influenza virus polymerase (Fig. 1) and with the Mediator and PAF complexes, we investigated the possibility of CHD1–RNAP II association and tested whether CHD1 is degraded in parallel with RNAP II during influenza virus infection. In coimmunoprecipitation studies using CHD1-specific antibodies, we analyzed CHD1 association with RNAP II in mock- or virus-infected A549 cells at 4 hpi. We found CHD1 and RNAP II proteins in the immune complexes of uninfected and infected cells (Fig. 8A and B); immunofluorescence studies also indicated nuclear colocalization of these proteins (Fig. 8C).
FIG 8.

CHD1 interacts with RNA polymerase II. (A) A549 cell extracts were used in coimmunoprecipitation studies with a specific anti-RNAP II or anti-CHD1 antibody or a control antibody (IgG). Coimmunoprecipitated proteins were detected by Western blotting and probing with appropriate antibodies. IP, immunoprecipitation. (B) Quantification of data from panel A. RNAP II IP, amount of CHD1 immunoprecipitated with an anti-RNAP II antibody; CHD1 IP, amount of RNAP II immunoprecipitated with an anti-CHD1 antibody. Bars represent means ± standard errors of the means. No significant differences were found in 2 independent experiments. (C) A549 cells were used in immunofluorescence studies with anti-CHD1 and anti-RNAP II antibodies. The colocalization panel shows the signals common to the two antibodies, obtained with the colocalization mask.
We examined CHD1 accumulation in A549 cells infected with influenza virus at a high MOI. Starting at 6 hpi in strain VIC-infected cells, we observed reduced CHD1 accumulation, which paralleled that of RNAP II; both were almost undetectable at late times postinfection (Fig. 9). In addition, we evaluated possible variations in the amount of specific posttranslational methylated histone recognized by CHD1 during influenza virus infection. The H3K4me3 histone mark decreased in parallel with RNAP II and CHD1 in infected cells, whereas the amount of H3K27me3, a histone mark of inactive chromatin and of unmodified histone 3, remained unchanged (Fig. 9). These data indicate that influenza virus infection triggers strong inhibition of cellular mRNA expression by the host cell, which probably aids in reducing the antiviral response.
FIG 9.

CHD1 and H3K4me3 accumulation decreases in influenza virus-infected cells. (A) A549 cells were either mock infected or infected with the VIC strain (3 PFU/cell). At the indicated times postinfection, CHD1, RNAP II, PB1, PA, H3K4me3, H3K27me3, H3, and β-actin were monitored by Western blotting of total-cell extracts. (B) A549 cells were infected with influenza virus (3 PFU/cell); at different times postinfection, cells were fixed and were processed for immunofluorescence using antibodies against CHD1, H3K4me3, and NP. DAPI was used to stain nuclei. Images representative of three independent experiments are shown.
To test whether decreased CHD1 accumulation was due to virus-induced degradation or normal protein decay, we compared the half-lives of CHD1 in mock- and virus-infected A549 cells treated with cycloheximide at 6 hpi to halt de novo protein synthesis. At various times posttreatment, cell extracts were prepared, and CHD1 levels were analyzed by Western blotting. The level of CHD1 that accumulated immediately before addition of the drug was considered 100% (Fig. 10A). The estimated half-life of CHD1 was ∼9 h in mock-infected cells and ∼4 h in virus-infected cells (Fig. 10B), which showed that influenza virus infection triggers CHD1 degradation.
FIG 10.

CHD1 is degraded in influenza virus-infected cells. (A) Mock- and virus-infected A549 cells were treated with cycloheximide (CHX) at 6 hpi. At various times posttreatment, cell extracts were obtained, and the accumulation of CHD1, PB1, and PA proteins was determined by Western blotting. CHD1 protein accumulation prior to drug addition was considered 100%. hpt, hours posttreatment. (B) Quantification of the amounts of CHD1 in panel A. Error bars represent standard errors of the means. Significance was determined by an unpaired Student t test with Welch's correction; 3 independent experiments were performed. **, P < 0.01; ***, P < 0.001. (C) Cultured A549 cells were either infected with the VIC strain or mock infected, alone or with the proteasome inhibitor MG132 added 1 h before infection. At various times postinfection, levels of the PB1 and PA polymerase subunits, CHD1, and ubiquitin (UB) were analyzed by Western blotting.
We tested whether CHD1 degradation was proteasome independent, as is RNAP II degradation. Cultured A549 cells were infected with the VIC strain or remained uninfected, alone or with the addition (1 h preinfection) of the proteasome inhibitor MG132. We measured the accumulation of the PB1 and PA polymerase subunits and of CHD1 at different times postinfection (Fig. 10C). Under these conditions, CHD1 was almost totally degraded after infection, irrespective of the presence or absence of the proteasome inhibitor. To confirm the effectiveness of the drug, we probed the extracts with an anti-ubiquitin antibody and found marked accumulation of ubiquitinated proteins following MG132 treatment. These results indicate that, as is the case for RNAP II degradation (24), the ubiquitin-mediated proteasomal degradation pathway does not mediate the CHD1 degradation triggered by influenza virus infection.
Recombinant influenza viruses with alterations at amino acid position 550 in PA or 504 in PB2 determine the ability to degrade RNAP II (25). The hvPR8 strain (high virulence, RNAP II degradation inducer) bears PA 550L and PB2 504V, whereas the lvPR8 strain (low virulence, noninducer of RNAP II degradation) has PA 550I and PB2 504I. A defined combination of PA 550L and PB2 504V confers on the PR8 background the ability to degrade RNAP II, as it also does for a natural isolate, the pandemic CAL 2009 strain (27). Individual changes of PA 550L or PB2 504V to isoleucine attenuate the ability of these recombinant viruses to degrade RNAP II, and the double mutation (DM) eliminates this ability. The specific PA 550L–PB2 504V combination restores the ability of lvPR8DM to degrade RNAP II, and conversely, the double mutation of these amino acids to isoleucine inhibits the degradation abilities of CAL and hvPR8 (27).
To study whether the abilities of these virus strains to degrade CHD1 parallel their abilities to degrade RNAP II, we infected A549 cells with the VIC, hvPR8, or lvPR8 strain and evaluated CHD1 accumulation by Western blotting. CHD1 and RNAP II were degraded after VIC or hvPR8 infection but not in lvPR8-infected cells (Fig. 11A). We analyzed the phenotypes of single and double mutants with alterations in PA and PB2 subunits in the recombinant hvPR8, lvPR8, and CAL strains (Fig. 11B). In all cases, CHD1 and RNAP II showed similar degradation patterns: they remained stable in hvPR8DM, wild-type (WT) lvPR8, and CALDM, as in mock-infected cells; single mutants led to partial degradation; and WT hvPR8, lvPR8DM, and WT CAL caused total degradation (Fig. 11B). These data indicate that CHD1 and RNAP II are degraded concomitantly during influenza virus infections with different strains or recombinant viruses.
FIG 11.

CHD1 degradation parallels RNAP II degradation. (A) A549 cells were either mock infected or infected with the indicated viruses; at 12 hpi, CHD1, RNAP II, and the indicated proteins were monitored in Western blots of total-cell extracts. (B) A549 cells were either mock infected or infected with the indicated recombinant viruses; at 12 hpi, CHD1, RNAP II, and the indicated proteins were monitored by Western blotting. DM, recombinant viruses with double mutations in residues PA 550 and PB2 504. The results shown are representative of 3 independent experiments performed.
DISCUSSION
Members of the CHD (chromodomain-helicase-DNA binding) family of ATP-dependent chromatin remodelers have tandem chromodomains that recognize modified histones and are divided into subfamilies 1 (CHD1 and -2), 2 (CHD3 to -5), and 3 (CHD6 to -9) (19, 20). CHD1 protein is conserved from yeasts to humans; it has DNA-dependent ATPase activity (58) and interacts with components of the FACT, Paf1, and Spt4–Spt5 elongation complexes. In addition, CHD1 binds to H3K4me3 both in vitro (22) and in vivo (29). Subfamily 2 members CHD3/CHD4 are recruited to specific genes as part of the transcriptional repressor NuRD complex (59). NuRD links chromatin remodeling with histone deacetylation activity and is generally considered a transcriptional repressor (60).
Less is known of the molecular function of mammalian CHD protein subfamily 3. CHD6 activity remains mainly uncharacterized, but several studies support its function as a transcriptional activator. CHD6 is found at intranuclear sites of mRNA synthesis (43) and operates as a coactivator for the cellular Nrf2 transcription factor (61). Very little is known of CHD6 function, although reports suggest its involvement in processes such as human cancers (62–64). Mutations in the CHD7 gene are described as responsible for CHARGE syndrome, a complex neurological syndrome (65); since it interacts with CHD7, CHD8 might also be involved in CHARGE syndrome (66). The CHD8 tandem chromodomains bind specifically to histone H3 dimethylated at lysine 4 in vitro (67). CHD9 appears to play a role in regulating transcription during osteogenic cell differentiation (68). These data indicate that members of the CHD family might act as transcriptional activators and repressors and that their chromodomains recognize distinct posttranscriptional modifications of histone tails for the recruitment of specific genes.
The influenza virus paradox.
Viruses do not possess the full equipment needed to express their genomes; they must thus use host cell factors and compete for and manipulate the host cell to their own benefit. Influenza virus faces a challenge in that it requires active cellular transcription to provide 5′-capped oligonucleotides to the viral polymerase for viral transcription, but conversely, active cellular transcription machinery permits an efficient antiviral response. To overcome this problem, influenza virus induces a degradative process that affects central components of the cell transcription system, such as RNAP II itself (24, 26), the CHD6 chromatin remodeler (28), and the CHD1 chromatin remodeler discussed here. Once viral transcription is complete, these proteins are degraded, which appears to contribute to host cell shutoff, since inhibition of cellular transcription correlates with the degree of RNAP II degradation in various influenza virus strains (24, 25). The abilities of different viruses to degrade RNAP II, which correlate strictly with CHD1 degradation (Fig. 9 to 11), are linked to viral pathogenicity in mice (27), reinforcing the role of degradation in viral pathogenesis. The combined degradation of RNAP II and CHD1, both of which play major roles in gene expression, might be a virus-induced mechanism to evade the cell antiviral response. The reduction in H3K4me3, the histone mark recognized by CHD1 protein (Fig. 9), during infection supports an important function for H3K4me3 turnover in the control of the antiviral response elicited by influenza virus infection.
Possible role of CHD1 in influenza virus control.
Chromatin remodelers are thought to be essential for cellular transcription, since they maintain chromatin in an “open” or “closed” configuration and thus regulate the access of transcription factors and RNA polymerases to specific genes (69). The recruitment of chromatin remodelers to specific genes is mainly the result of remodeler binding to transcription factors that recognize specific sequences within the promoters, followed by the recognition of specific posttranslational histone modifications (70). CHD1 recruitment to the genes it regulates appears to be mediated by its association with transcription initiation complexes and with H3K4me3 near the beginning of active genes (29). This observation supports a model in which the combined action of transcription complexes and H3K4me3 targets CHD1 specifically to active genes. Histone methylation is fundamental in gene regulation, and the H3K4me3 modification is considered a general marker of actively transcribed genes, since about three-quarters of protein-coding genes have promoter-proximal nucleosomes enriched for H3K4me3 (71).
Here we show that CHD1 interacts with influenza virus polymerase (Fig. 1) and positively modulates the viral life cycle (Fig. 6) and viral RNA transcription (Fig. 5). Association of the viral polymerase with transcriptionally active chromatin regions, mediated by the CHD1–H3K4me3 interaction, could position the viral polymerase near the “open” chromatin sites where precursor mRNAs are being synthesized. This connection would provide a platform for an interaction that sustains the cap-snatching process necessary for viral initiation of mRNA transcription.
We previously described a distinct negative function of CHD6 in modulating influenza virus multiplication. CHD6 colocalizes preferentially with active chromatin markers in human lung epithelial cells (18), but it is not known whether it controls a large number of genes or a subpopulation of only a few specific genes, as suggested by transcriptome analysis in CHD6-silenced cells (our unpublished data). CHD6 is both degraded and recruited to histones with epigenetic marks of inactive chromatin in influenza virus-infected cells at late times postinfection (18). These actions could reduce cellular transcription activity, since they would allow the virus to hijack the infected-cell metabolism, to promote viral RNA replication that persists throughout infection (72), and/or to reduce the antiviral response.
H3K4me3 modification during influenza virus infection.
Influenza virus infection induces H3K4me3 addition and the removal of repressive histone marks (H3K27me3) in several interferon-stimulated genes (ISG), allowing the binding of activated transcription factors, such as STAT1 and IRF7, and permitting robust ISG expression (73). The importance of histone modification in influenza virus control has also been shown using viral proteins that mimic endogenous histone marks. The H3N2 subtype NS1 protein has a histone H3K4-like sequence (histone mimic) at its carboxyl terminus that is used by the virus to target the human PAF1 transcription elongation complex, leading to suppression of hPAF1C-mediated transcriptional control of inducible antiviral gene expression (74). The NXP2/MORC3 protein, a histone reader that recognizes H3K4me3 (75), interacts with the influenza virus polymerase complex and positively modulates viral transcription (76); this protein relocalizes partially to the cytoplasm late in influenza virus infection (76). Although NXP2/MORC3, unlike the CHD1 H3K4me3 reader, is not degraded during infection, its relocalization could inhibit its still-uncharacterized nuclear function.
In summary, since the influenza virus is not integrated into the host genome, its transcription mechanism requires early, precise functional association with the host transcription apparatus. Chromatin dynamics therefore determine the viral life cycle. Changes in chromatin structure also modulate the access of transcription complexes to specific genes needed to counteract the host cell antiviral response. Chromatin control of viral infection is thus a new area of research with potential targets for the development of antiviral therapies.
ACKNOWLEDGMENTS
We are indebted to J. Ortin, A. Falcón, and P. Gastaminza for criticism of the manuscript. We are grateful to E. Fodor for providing the primer extension protocol. We gratefully acknowledge the technical assistance of N. Zamarreño and the editorial assistance of C. Mark.
This work was funded by the Spanish Ministry of Economy and Competitiveness, Plan Nacional de Investigacion Científica, Desarrollo e Innovacion Tecnologica (BFU2011-26175 and BFU2014-57797-R), and the CIBER de Enfermedades Respiratorias. L. Marcos-Villar and A. Pazo were supported by the CIBER de Enfermedades Respiratorias. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Resa-Infante P, Jorba N, Coloma R, Ortin J. 2011. The influenza virus RNA synthesis machine: advances in its structure and function. RNA Biol 8:207–215. doi: 10.4161/rna.8.2.14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martín-Benito J, Ortin J. 2013. Influenza virus transcription and replication. Adv Virus Res 87:113–137. doi: 10.1016/B978-0-12-407698-3.00004-1. [DOI] [PubMed] [Google Scholar]
- 3.Plotch SJ, Bouloy M, Ulmanen I, Krug RM. 1981. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 23:847–858. doi: 10.1016/0092-8674(81)90449-9. [DOI] [PubMed] [Google Scholar]
- 4.Dias A, Bouvier D, Crepin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, Ruigrok RW. 2009. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458:914–918. doi: 10.1038/nature07745. [DOI] [PubMed] [Google Scholar]
- 5.Honda A, Okamoto T, Ishihama A. 2007. Host factor Ebp1: selective inhibitor of influenza virus transcriptase. Genes Cells 12:133–142. doi: 10.1111/j.1365-2443.2007.01047.x. [DOI] [PubMed] [Google Scholar]
- 6.Jorba N, Juarez S, Torreira E, Gastaminza P, Zamarreno N, Albar JP, Ortin J. 2008. Analysis of the interaction of influenza virus polymerase complex with human cell factors. Proteomics 8:2077–2088. doi: 10.1002/pmic.200700508. [DOI] [PubMed] [Google Scholar]
- 7.Landeras-Bueno S, Jorba N, Perez-Cidoncha M, Ortin J. 2011. The splicing factor proline-glutamine rich (SFPQ/PSF) is involved in influenza virus transcription. PLoS Pathog 7:e1002397. doi: 10.1371/journal.ppat.1002397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huarte M, Sanz-Ezquerro JJ, Roncal F, Ortín J, Nieto A. 2001. PA subunit from influenza virus polymerase complex interacts with a cellular protein with homology to a family of transcriptional activators. J Virol 75:8597–8604. doi: 10.1128/JVI.75.18.8597-8604.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodriguez A, Pére-González A, Nieto A. 2011. Cellular human CLE/c14orf166 protein interacts with influenza virus polymerase and is required for viral replication. J Virol 85:12062–12066. doi: 10.1128/JVI.00684-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engelhardt OG, Smith M, Fodor E. 2005. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J Virol 79:5812–5818. doi: 10.1128/JVI.79.9.5812-5818.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bukrinskaya AG, Vorkunova GK, Vorkunova NK. 1979. Cytoplasmic and nuclear input virus RNPs in influenza virus-infected cells. J Gen Virol 45:557–567. doi: 10.1099/0022-1317-45-3-557. [DOI] [PubMed] [Google Scholar]
- 12.Jackson DA, Caton AJ, McCready SJ, Cook PR. 1982. Influenza virus RNA is synthesized at fixed sites in the nucleus. Nature 296:366–368. doi: 10.1038/296366a0. [DOI] [PubMed] [Google Scholar]
- 13.López-Turiso JA, Martinez C, Tanaka T, Ortin J. 1990. The synthesis of influenza virus negative-strand RNA takes place in insoluble complexes present in the nuclear matrix fraction. Virus Res 16:325–337. doi: 10.1016/0168-1702(90)90056-H. [DOI] [PubMed] [Google Scholar]
- 14.Bui M, Wills EG, Helenius A, Whittaker GR. 2000. Role of the influenza virus M1 protein in nuclear export of viral ribonucleoproteins. J Virol 74:1781–1786. doi: 10.1128/JVI.74.4.1781-1786.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chase GP, Rameix-Welti MA, Zvirbliene A, Zvirblis G, Gotz V, Wolff T, Naffakh N, Schwemmle M. 2011. Influenza virus ribonucleoprotein complexes gain preferential access to cellular export machinery through chromatin targeting. PLoS Pathog 7:e1002187. doi: 10.1371/journal.ppat.1002187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takizawa N, Watanabe K, Nouno K, Kobayashi N, Nagata K. 2006. Association of functional influenza viral proteins and RNAs with nuclear chromatin and sub-chromatin structure. Microbes Infect 8:823–833. doi: 10.1016/j.micinf.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 17.Hu Y, Liu X, Zhang A, Zhou H, Liu Z, Chen H, Jin M. 12 September 2014. CHD3 facilitates vRNP nuclear export by interacting with NES1 of influenza A virus NS2. Cell Mol Life Sci doi: 10.1007/s00018-014-1726-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alfonso R, Lutz T, Rodriguez A, Chavez JP, Rodriguez P, Gutierrez S, Nieto A. 2011. CHD6 chromatin remodeler is a negative modulator of influenza virus replication that relocates to inactive chromatin upon infection. Cell Microbiol 13:1894–1906. doi: 10.1111/j.1462-5822.2011.01679.x. [DOI] [PubMed] [Google Scholar]
- 19.Flanagan JF, Blus BJ, Kim D, Clines KL, Rastinejad F, Khorasanizadeh S. 2007. Molecular implications of evolutionary differences in CHD double chromodomains. J Mol Biol 369:334–342. doi: 10.1016/j.jmb.2007.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yap KL, Zhou MM. 2011. Structure and mechanisms of lysine methylation recognition by the chromodomain in gene transcription. Biochemistry 50:1966–1980. doi: 10.1021/bi101885m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim Y, Minor W, Rastinejad F, Khorasanizadeh S. 2005. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature 438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- 22.Sims RJ III, Chen CF, Santos-Rosa H, Kouzarides T, Patel SS, Reinberg D. 2005. Human but not yeast CHD1 binds directly and selectively to histone H3 methylated at lysine 4 via its tandem chromodomains. J Biol Chem 280:41789–41792. doi: 10.1074/jbc.C500395200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clapier CR, Cairns BR. 2009. The biology of chromatin remodeling complexes. Annu Rev Biochem 78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez A, Pérez-Gonzalez A, Nieto A. 2007. Influenza virus infection causes specific degradation of the largest subunit of cellular RNA polymerase II. J Virol 81:5315–5324. doi: 10.1128/JVI.02129-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez A, Perez-Gonzalez A, Hossain MJ, Chen LM, Rolling T, Perez-Brena P, Donis R, Kochs G, Nieto A. 2009. Attenuated strains of influenza A viruses do not induce degradation of RNA polymerase II. J Virol 83:11166–11174. doi: 10.1128/JVI.01439-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vreede FT, Chan AY, Sharps J, Fodor E. 2010. Mechanisms and functional implications of the degradation of host RNA polymerase II in influenza virus infected cells. Virology 396:125–134. doi: 10.1016/j.virol.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Llompart CM, Nieto A, Rodriguez-Frandsen A. 2014. Specific residues of PB2 and PA influenza virus polymerase subunits confer the ability for RNA polymerase II degradation and virus pathogenicity in mice. J Virol 88:3455–3463. doi: 10.1128/JVI.02263-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alfonso R, Rodriguez A, Rodriguez P, Lutz T, Nieto A. 2013. CHD6, a cellular repressor of influenza virus replication, is degraded in human alveolar epithelial cells and mice [sic] lungs during infection. J Virol 87:4534–4544. doi: 10.1128/JVI.00554-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin JJ, Lehmann LW, Bonora G, Sridharan R, Vashisht AA, Tran N, Plath K, Wohlschlegel JA, Carey M. 2011. Mediator coordinates PIC assembly with recruitment of CHD1. Genes Dev 25:2198–2209. doi: 10.1101/gad.17554711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krogan NJ, Kim M, Ahn SH, Zhong G, Kobor MS, Cagney G, Emili A, Shilatifard A, Buratowski S, Greenblatt JF. 2002. RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomic approach. Mol Cell Biol 22:6979–6992. doi: 10.1128/MCB.22.20.6979-6992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Warner MH, Roinick KL, Arndt KM. 2007. Rtf1 is a multifunctional component of the Paf1 complex that regulates gene expression by directing cotranscriptional histone modification. Mol Cell Biol 27:6103–6115. doi: 10.1128/MCB.00772-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yao H, Pan J, Wu C, Shen H, Xie J, Wang Q, Wen L, Ma L, Wu L, Ping N, Zhao Y, Sun A, Chen S. 2015. Transcriptome sequencing reveals CHD1 as a novel fusion partner of RUNX1 in acute myeloid leukemia with t(5;21)(q21;q22). Mol Cancer 14:81. doi: 10.1186/s12943-015-0353-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Falcón A, Marión R, Zürcher T, Gomez P, Portela A, Nieto A, Ortín J. 2004. Defective RNA replication and late gene expression in temperature-sensitive influenza viruses expressing deleted forms of the NS1 protein. J Virol 78:3880–3888. doi: 10.1128/JVI.78.8.3880-3888.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Perales B, Sanz-Ezquerro JJ, Gastaminza P, Ortega J, Fernandez-Santarén J, Ortín J, Nieto A. 2000. The replication activity of influenza virus polymerase is linked to the capacity of the PA subunit to induce proteolysis. J Virol 74:1307–1312. doi: 10.1128/JVI.74.3.1307-1312.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. 1998. A third-generation lentivirus vector with a conditional packaging system. J Virol 72:8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, Weinberg RA, Novina CD. 2003. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Levitz SM, Diamond RD. 1985. A rapid colorimetric assay of fungal viability with the tetrazolium salt MTT. J Infect Dis 152:938–945. doi: 10.1093/infdis/152.5.938. [DOI] [PubMed] [Google Scholar]
- 38.Huarte M, Falcón A, Nakaya Y, Ortín J, García-Sastre A, Nieto A. 2003. Threonine 157 of influenza virus PA polymerase subunit modulates RNA replication in infectious viruses. J Virol 77:6007–6013. doi: 10.1128/JVI.77.10.6007-6013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones JC, Phatnani HP, Haystead TA, MacDonald JA, Alam SM, Greenleaf AL. 2004. C-terminal repeat domain kinase I phosphorylates Ser2 and Ser5 of RNA polymerase II C-terminal domain repeats. J Biol Chem 279:24957–24964. doi: 10.1074/jbc.M402218200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bárcena J, de la Luna S, Ochoa M, Melero JA, Nieto A, Ortín J, Portela A. 1994. Monoclonal antibodies against the influenza virus PB2 and NP polypeptides interfere with the initiation step of viral mRNA synthesis in vitro. J Virol 68:6900–6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.González S, Ortín J. 1999. Distinct regions of influenza virus PB1 polymerase subunit recognize vRNA and cRNA templates. EMBO J 18:3767–3775. doi: 10.1093/emboj/18.13.3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jorba N, Coloma R, Ortin J. 2009. Genetic trans-complementation establishes a new model for influenza virus RNA transcription and replication. PLoS Pathog 5:e1000462. doi: 10.1371/journal.ppat.1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lutz T, Stoger R, Nieto A. 2006. CHD6 is a DNA-dependent ATPase and localizes at nuclear sites of mRNA synthesis. FEBS Lett 580:5851–5857. doi: 10.1016/j.febslet.2006.09.049. [DOI] [PubMed] [Google Scholar]
- 44.Robb NC, Smith M, Vreede FT, Fodor E. 2009. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J Gen Virol 90:1398–1407. doi: 10.1099/vir.0.009639-0. [DOI] [PubMed] [Google Scholar]
- 45.Kawakami E, Watanabe T, Fujii K, Goto H, Watanabe S, Noda T, Kawaoka Y. 2011. Strand-specific real-time RT-PCR for distinguishing influenza vRNA, cRNA, and mRNA. J Virol Methods 173:1–6. doi: 10.1016/j.jviromet.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simic R, Lindstrom DL, Tran HG, Roinick KL, Costa PJ, Johnson AD, Hartzog GA, Arndt KM. 2003. Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. EMBO J 22:1846–1856. doi: 10.1093/emboj/cdg179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zentner GE, Tsukiyama T, Henikoff S. 2013. ISWI and CHD chromatin remodelers bind promoters but act in gene bodies. PLoS Genet 9:e1003317. doi: 10.1371/journal.pgen.1003317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burgui I, Yángüez E, Sonenber N, Nieto A. 2007. Influenza mRNA translation revisited: is the eIF4E cap-binding factor required for viral mRNA translation? J Virol 81:12427–12438. doi: 10.1128/JVI.01105-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Lucas S, Peredo J, Marion RM, Sanchez C, Ortin J. 2010. Human Staufen1 protein interacts with influenza virus ribonucleoproteins and is required for efficient virus multiplication. J Virol 84:7603–7612. doi: 10.1128/JVI.00504-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jao CY, Salic A. 2008. Exploring RNA transcription and turnover in vivo by using click chemistry. Proc Natl Acad Sci U S A 105:15779–15784. doi: 10.1073/pnas.0808480105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mena I, de la Luna S, Albo C, Martín J, Nieto A, Ortín J, Portela A. 1994. Synthesis of biologically active influenza virus core proteins using a vaccinia-T7 RNA polymerase expression system. J Gen Virol 75:2109–2114. doi: 10.1099/0022-1317-75-8-2109. [DOI] [PubMed] [Google Scholar]
- 52.Fodor E, Palese P, Brownlee GG, García-Sastre A. 1998. Attenuation of influenza A virus mRNA levels by promoter mutations. J Virol 72:6283–6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hay AJ, Lomniczi B, Bellamy AR, Skehel JJ. 1977. Transcription of the influenza virus genome. Virology 83:337–355. doi: 10.1016/0042-6822(77)90179-9. [DOI] [PubMed] [Google Scholar]
- 54.Aparicio O, Razquin N, Zaratiegui M, Narvaiza I, Fortes P. 2006. Adenovirus virus-associated RNA is processed to functional interfering RNAs involved in virus production. J Virol 80:1376–1384. doi: 10.1128/JVI.80.3.1376-1384.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dey P, Ponnusamy MP, Deb S, Batra SK. 2011. Human RNA polymerase II-association factor 1 (hPaf1/PD2) regulates histone methylation and chromatin remodeling in pancreatic cancer. PLoS One 6:e26926. doi: 10.1371/journal.pone.0026926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Allen BL, Taatjes DJ. 2015. The Mediator complex: a central integrator of transcription. Nat Rev Mol Cell Biol 16:155–166. doi: 10.1038/nrm3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Björklund S, Gustafsson CM. 2005. The yeast Mediator complex and its regulation. Trends Biochem Sci 30:240–244. doi: 10.1016/j.tibs.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 58.Tran HG, Steger DJ, Iyer VR, Johnson AD. 2000. The chromo domain protein Chd1p from budding yeast is an ATP-dependent chromatin-modifying factor. EMBO J 19:2323–2331. doi: 10.1093/emboj/19.10.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Torchy MP, Hamiche A, Klaholz BP. 22 March 2015. Structure and function insights into the NuRD chromatin remodeling complex. Cell Mol Life Sci doi: 10.1007/s00018-015-1880-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Le Guezennec X, Vermeulen M, Brinkman AB, Hoeijmakers WA, Cohen A, Lasonder E, Stunnenberg HG. 2006. MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol Cell Biol 26:843–851. doi: 10.1128/MCB.26.3.843-851.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nioi P, Nguyen T, Sherratt PJ, Pickett CB. 2005. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol Cell Biol 25:10895–10906. doi: 10.1128/MCB.25.24.10895-10906.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, He M, Li Z, Sun X, Jia W, Chen J, Yang S, Zhou F, Zhao X, Wan S, Ye R, Liang C, Liu Z, Huang P, Liu C, Jiang H, Wang Y, Zheng H, Sun L, Liu X, Jiang Z, Feng D, Wu S, Zou J, Zhang Z, Yang R, Zhao J, Xu C, Yin W, Guan Z, Ye J, Zhang H, Li J, Kristiansen K, Nickerson ML, Theodorescu D, Li Y, Zhang X, Li S, Wang J, Yang H, Cai Z. 2011. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet 43:875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mouradov D, Sloggett C, Jorissen RN, Love CG, Li S, Burgess AW, Arango D, Strausberg RL, Buchanan D, Wormald S, O'Connor L, Wilding JL, Bicknell D, Tomlinson IP, Bodmer WF, Mariadason JM, Sieber OM. 2014. Colorectal cancer cell lines are representative models of the main molecular subtypes of primary cancer. Cancer Res 74:3238–3247. doi: 10.1158/0008-5472.CAN-14-0013. [DOI] [PubMed] [Google Scholar]
- 64.Ali Hassan NZ, Mokhtar NM, Kok Sin T, Mohamed Rose I, Sagap I, Harun R, Jamal R. 2014. Integrated analysis of copy number variation and genome-wide expression profiling in colorectal cancer tissues. PLoS One 9:e92553. doi: 10.1371/journal.pone.0092553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wong MT, Scholvinck EH, Lambeck AJ, van Ravenswaaij-Arts CM. 18 February 2015. CHARGE syndrome: a review of the immunological aspects. Eur J Hum Genet doi: 10.1038/ejhg.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Batsukh T, Pieper L, Koszucka AM, von Velsen N, Hoyer-Fender S, Elbracht M, Bergman JE, Hoefsloot LH, Pauli S. 2010. CHD8 interacts with CHD7, a protein which is mutated in CHARGE syndrome. Hum Mol Genet 19:2858–2866. doi: 10.1093/hmg/ddq189. [DOI] [PubMed] [Google Scholar]
- 67.Rodríguez-Paredes M, Ceballos-Chavez M, Esteller M, Garcia-Dominguez M, Reyes JC. 2009. The chromatin remodeling factor CHD8 interacts with elongating RNA polymerase II and controls expression of the cyclin E2 gene. Nucleic Acids Res 37:2449–2460. doi: 10.1093/nar/gkp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shur I, Socher R, Benayahu D. 2006. In vivo association of CReMM/CHD9 with promoters in osteogenic cells. J Cell Physiol 207:374–378. doi: 10.1002/jcp.20586. [DOI] [PubMed] [Google Scholar]
- 69.Hall JA, Georgel PT. 2007. CHD proteins: a diverse family with strong ties. Biochem Cell Biol 85:463–476. doi: 10.1139/O07-063. [DOI] [PubMed] [Google Scholar]
- 70.Bartholomew B. 2014. Regulating the chromatin landscape: structural and mechanistic perspectives. Annu Rev Biochem 83:671–696. doi: 10.1146/annurev-biochem-051810-093157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. 2007. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shapiro GI, Gurney TJ, Krug RM. 1987. Influenza virus gene expression: control mechanisms at early and late times of infection and nuclear-cytoplasmic transport of virus-specific RNAs. J Virol 61:764–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Menachery VD, Eisfeld AJ, Schafer A, Josset L, Sims AC, Proll S, Fan S, Li C, Neumann G, Tilton SC, Chang J, Gralinski LE, Long C, Green R, Williams CM, Weiss J, Matzke MM, Webb-Robertson BJ, Schepmoes AA, Shukla AK, Metz TO, Smith RD, Waters KM, Katze MG, Kawaoka Y, Baric RS. 2014. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. mBio 5:e01174-14. doi: 10.1128/mBio.01174-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Marazzi I, Ho JS, Kim J, Manicassamy B, Dewell S, Albrecht RA, Seibert CW, Schaefer U, Jeffrey KL, Prinjha RK, Lee K, Garcia-Sastre A, Roeder RG, Tarakhovsky A. 2012. Suppression of the antiviral response by an influenza histone mimic. Nature 483:428–433. doi: 10.1038/nature10892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li X, Foley EA, Molloy KR, Li Y, Chait BT, Kapoor TM. 2012. Quantitative chemical proteomics approach to identify posttranslational modification-mediated protein-protein interactions. J Am Chem Soc 134:1982–1985. doi: 10.1021/ja210528v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ver LS, Marcos-Villar L, Landeras-Bueno S, Nieto A, Ortin J. 22 July 2015. The cellular factor NXP2/MORC3 is a positive regulator for influenza virus multiplication. J Virol doi: 10.1128/JVI.01530-15. [DOI] [PMC free article] [PubMed] [Google Scholar]