

ABSTRACT
The field of viral metagenomics has expanded our understanding of viral diversity from all three domains of life (Archaea, Bacteria, and Eukarya). Traditionally, viral metagenomic studies provide information about viral gene content but rarely provide knowledge about virion morphology and/or cellular host identity. Here we describe a new virus, Acidianus tailed spindle virus (ATSV), initially identified by bioinformatic analysis of viral metagenomic data sets from a high-temperature (80°C) acidic (pH 2) hot spring located in Yellowstone National Park, followed by more detailed characterization using only environmental samples without dependency on culturing. Characterization included the identification of the large tailed spindle virion morphology, determination of the complete 70.8-kb circular double-stranded DNA (dsDNA) viral genome content, and identification of its cellular host. Annotation of the ATSV genome revealed a potential three-domain gene product containing an N-terminal leucine-rich repeat domain, followed by a likely posttranslation regulatory region consisting of high serine and threonine content, and a C-terminal ESCRT-III domain, suggesting interplay with the host ESCRT system. The host of ATSV, which is most closely related to Acidianus hospitalis, was determined by a combination of analysis of cellular clustered regularly interspaced short palindromic repeat (CRISPR)/Cas loci and dual viral and cellular fluorescence in situ hybridization (viral FISH) analysis of environmental samples and confirmed by culture-based infection studies. This work provides an expanded pathway for the discovery, isolation, and characterization of new viruses using culture-independent approaches and provides a platform for predicting and confirming virus hosts.
IMPORTANCE Virus discovery and characterization have been traditionally accomplished by using culture-based methods. While a valuable approach, it is limited by the availability of culturable hosts. In this research, we report a virus-centered approach to virus discovery and characterization, linking viral metagenomic sequences to a virus particle, its sequenced genome, and its host directly in environmental samples, without using culture-dependent methods. This approach provides a pathway for the discovery, isolation, and characterization of new viruses. While this study used an acidic hot spring environment to characterize a new archaeal virus, Acidianus tailed spindle virus (ATSV), the approach can be generally applied to any environment to expand knowledge of virus diversity in all three domains of life.
INTRODUCTION
Our knowledge and understanding of archaeal viruses (viruses that infect Archaea) are limited. Only 117 archaeal viruses are described at some level, with only a few being characterized in any significant depth (1–4). Remarkably, these relatively few archaeal viruses have formed 16 new virus families (5) and have expanded our appreciation of virion morphology, diversity, and gene content (5–7). Known archaeal viruses infect just 16 of 98 known genera of Archaea (8), further highlighting our lack of knowledge of archaeal viruses. A recent study examining viral sequences within bacterial and archaeal genomes found 12,498 new viral sequences, further emphasizing our lack of knowledge of viruses in natural systems and the need for more archaeal virus-specific studies (9). Undoubtedly, many archaeal viruses remain to be discovered, but discovery has been limited to primarily culture-based approaches.
In recent years, viral metagenomics has emerged as a culture-independent approach for exploring viral diversity in natural environments (10, 11). In marine environments, viral metagenomic studies have significantly advanced our understanding of marine viral ecology (12). These advances include the discovery of the dominance of temperate viruses and fitness advantages of multiple viral replication strategies (13); the discovery of ecological drivers of viral community composition that help explain the high viral diversity (74, 75); and the establishment of core, flexible, and niche-containing gene sets (76). Additionally, knowledge of genomes from cultured ocean phages has been used to identify and track viruses in viral metagenomes (14–16). These findings demonstrate the value of viral metagenomics at the gene, community, and population levels to inform viral ecology. Viral metagenomic studies of acidic hot springs (17–19) and hypersaline environments (20, 21) have led to the discovery of partial and full genomes of new archaeal viruses as well as viral groups formed by the clustering of related metagenomic contigs and estimates of total virus community diversity (18, 22).
One limitation of traditional viral metagenomic approaches is that they usually provide information about viral gene content but lack information on virion morphology and host identity. However, advances over the past several years have provided promising new tools for use in linking viral metagenomic sequences to cellular hosts (12), using digital PCR (23), viral tagging (24), single-cell genomics (25–27), and knowledge derived from the clustered regularly interspaced short palindromic repeat (CRISPR)/Cas system (20, 28). Even with these expanded tools, only a fraction of viral metagenomic sequences have been linked to hosts, and even fewer have been linked to a specific virus particle morphology.
In this study, we used viral metagenomic data to identify a new archaeal virus, designated Acidianus tailed spindle virus (ATSV), from Alice Spring, a high-temperature acidic hot spring in the Crater Hills area of Yellowstone National Park (YNP). We were able to determine the complete ATSV genome, identify its virion morphology, and determine its host. All this was accomplished by using culture-independent approaches, in effect closing the “viral metagenomic loop” from fragmented environmental viral sequence data to complete-genome sequencing, virion isolation, host identification, and confirmation by culturing.
MATERIALS AND METHODS
Viral metagenomics.
An acidic hot spring in the Crater Hills area of YNP, Alice Spring (CHAS) (CHAN0041; 82°C; pH 2.5; 44°39.179′N, 110°20.090′W), was chosen as a sampling site. A hot spring water sample was collected in January 2008. Sample collection, virus purification, DNA extraction, amplification by multiple-displacement amplification, GS FLX 454 sequencing by the University of Illinois Sequencing Center, and assembly were described previously (18).
Selection of the target viral metagenomic contig.
A viral metagenome contig of interest was identified based on several criteria. First, viral contigs with lengths of >5 kb were chosen. Next, contigs were searched against the NCBI RefSeq protein database by using BLASTX to identify viral hallmark protein signatures (29). Specifically, a contig containing a putative virus major coat protein (MCP) was chosen as a candidate for virus tracking and purification.
Major coat protein cloning, expression, and purification and polyclonal antiserum production.
The gene encoding the likely virion MCP was PCR amplified from CHAS total community viral DNA by using a nested-PCR approach (30). The PCR primers introduced a Shine-Dalgarno sequence, an N-terminal 6×His tag, and attB sites to facilitate homologous recombination into the Gateway pDONR201 vector (Invitrogen by Life Technologies, Grand Island, NY). All primer sequences are available upon request. After confirmation of the correct product by DNA sequencing, the entry clone was recombined into the pDEST14 expression vector (Invitrogen). The likely MCP was expressed by autoinduction. A starter culture of Escherichia coli BL21(DE3)/pLysS cells containing the MCP gene in pDEST14 was used to inoculate 500 ml of ZYP-0.8G plasmid growth medium (31). After 20 h of growth at 37°C with shaking, the cells were harvested by centrifugation at 5,500 × g for 10 min. The cell pellet was resuspended in 5 ml/g of cell pellet of lysis buffer (20 mM Tris and 400 mM NaCl [pH 8.0]) with 0.1 mM phenylmethylsulfonyl fluoride (PMSF), lysed with three passages through a Microfluidizer (Microfluidics Corp., Westwood, MA), and clarified by centrifugation at 22,000 × g for 20 min. The supernatant was incubated at 65°C for 10 min, cooled on ice, and clarified by centrifugation at 22,000 × g for 20 min. The supernatant was applied to a gravity column containing a 1.5-ml bed volume of Ni-nitrilotriacetic acid (NTA) agarose, washed with 8 column volumes of wash buffer (20 mM Tris, 400 mM NaCl, 10 mM imidazole [pH 8.0]), and eluted in a solution containing 10 mM Tris, 50 mM NaCl, and 20 mM imidazole (pH 8.0). The protein was applied to a calibrated Superdex-75 size exclusion column (GE Healthcare, Little Chalfont, United Kingdom) equilibrated with 10 mM Tris (pH 8.0) and 50 mM NaCl. Purified protein was stored at −20°C.
Protein purified by size exclusion was diluted to 0.1 mg/ml in 1× phosphate-buffered saline (PBS) (pH 7.5), followed by 1:1 dilution with Hunter's adjuvant, and 100 μl of the mixture was injected intramuscularly into rabbits. Serum containing polyclonal antibodies to MCP was purified with a protein A antibody purification column (Pierce by Life Technologies, Grand Island, NY).
Quantitative PCR assay development.
Quantitative PCR (qPCR) primers were designed to target a 202-bp internal region of the MCP gene. Purified plasmid pDONR201 containing the MCP gene was quantitated by using a Qubit fluorimeter (Qiagen, Valencia, CA) and was used to create qPCR standards ranging from 9 × 104 to 9E1011 copies/ml (1 fg/2 μl to 10 ng/2 μl). Quantitative PCR was performed by using SsoFast EvaGreen supermix (Bio-Rad, Hercules, CA) on a Rotor-gene Q real-time PCR machine (Qiagen).
Virus purification.
Approximately 11 liters of CHAS hot spring water was collected (2 September 2013), and cells were removed by in-line filtration through a 0.4-μm polycarbonate filter (Millipore, Billerica, MA). An iron precipitation method was used to concentrate viral particles (18, 32). Briefly, the pH of the hot spring sample was raised to pH 4.0 to induce the formation of virus-trapping iron clusters, and the resulting precipitate was collected by filtration onto a 0.8-μm polycarbonate filter (Millipore). The iron precipitate was resolubilized in 50 ml of 250 mM ascorbic acid (pH 2.5), and the sample was extensively dialyzed with 5 mM glycine (pH 2.5). Virus particles were subjected to isopycnic centrifugation using CsCl gradients at 169,000 × g for 26 h in a Beckman SW41 rotor and an ultracentrifuge. Gradient fractions were screened for the presence of viral DNA by qPCR. Positive fractions were pooled, loaded onto a second CsCl gradient, and centrifuged at 238,000 × g for 5 h in a Beckman MLN-80 near-vertical rotor. Gradient fractions were screened for the virus by using qPCR, and positive fractions were concentrated with a SpinX 100,000-molecular-weight-cutoff (MWCO) filter concentrator (Corning, Corning, NY). Virus particles were stained with 1.5% uranyl acetate and imaged on a Leo912AB transmission electron microscope.
Sequencing and genome assembly.
DNA was extracted from the CsCl-purified virus-enriched sample by using the Purelink viral DNA/RNA extraction kit (Invitrogen). One hundred nanograms of viral DNA was sequenced by the University of Illinois Sequencing Center using the Illumina MiSeq v3 system with paired-end reads (2 by 300 nucleotides [nt]). The sequencing reads were assembled by using the Mira assembly program (version 4.0.4) (33). Genome assembly statistics are available upon request.
Genome annotation.
Open reading frames (ORFs) were predicted by using a combination of Glimmer (34, 35), the Geneious ORF calling program (35), and hand curation. Homology of ORFs to known proteins was determined by searching the NCBI RefSeq database using BLASTX (January 2015 release) (29). Additional protein homology prediction was performed by using HHpred, a server for remote protein homology detection based on the pairwise comparison profile of hidden Markov models (36). The genome was searched for repeat regions by using Repfind (37), PALINDROME (38), and e-inverted (38).
Western blot analysis.
The CsCl-purified CHAS virus sample was separated on a 15% SDS-PAGE gel and transferred onto a nitrocellulose membrane. The membrane was washed with Tris-buffered saline–0.05% Tween 20 (TBST) and blocked for 30 min with 5% blotting-grade blocker (Bio-Rad) in TBST. Purified polyclonal MCP antibodies were added at a 1:10,000 dilution and incubated for 4 h, followed by overnight incubation with horseradish peroxidase (HRP)-linked goat anti-rabbit secondary antibodies at a 1:10,000 dilution. The Western blot was developed by using the Opti-CN colorimetric Western development kit (Bio-Rad).
Identification of MCP in environmental samples using mass spectrometry.
CsCl-purified CHAS virus samples were separated on a 15% SDS-PAGE gel and stained with Gelcode blue stain (Fisher Scientific, Pittsburgh, PA). Major bands were excised, in-gel digested with trypsin (39), and analyzed on an Agilent 6520 quadrupole time of flight (Q-TOF) mass analyzer equipped with an Agilent 1290 μHPLC instrument. Peaks were compared against an in-house database containing ORFs from 2008-2010 CHAS viral metagenomic data sets by using the Mascot ion/ion search program (Matrix Science, Boston, MA).
Host identification using the CRISPR/Cas system.
The virus genome was searched against the CRISPRfinder spacer database (40), which contains spacer sequences from cellular CRISPR loci from all reported NCBI bacterial and archaeal genomes, using BLAST. The resulting CRISPR spacer sequence library was assembled onto the viral genome by using the Geneious assembler (35).
Viral FISH, a dual virus-host labeling method using CARD-FISH.
Dual viral and cellular fluorescence in situ hybridization (viral FISH), a dual virus-host labeling method using the catalyzed reporter deposition fluorescence in situ hybridization (CARD-FISH) method, was adapted from methods described previously by Allers et al. (41), using virus and host probes linked to digoxigenin (DIG). The design of the 16S rRNA host probes was described previously by Munson-McGee et al. (42). Four 250-bp probes to the new virus genome were created by using PolyPro (43) and were designed to all have a matching hybridization temperature for a given formamide concentration (67.7 to 70°C at 35% formamide). Actual temperatures and formamide concentrations were empirically optimized. Primers specific for the ends of each probe were used to amplify the full probe sequence from purified virus DNA, and probes were labeled with DIG by using the PCR DIG probe synthesis kit (Roche). Cell fixation and hybridization were performed according to protocols described previously by Allers et al. (41), with the following modifications: (i) cells were fixed with 1% paraformaldehyde for 1 h at room temperature and washed 3 times with 1× PBS; (ii) fixed samples were placed into wells of glass slides, air dried, and dehydrated with ethanol; (iii) wells were covered with permeabilization solution (50 mM glucose, 20 mM Tris [pH 7.5], 10 mM EDTA, and 0. 2% Tween 20) for 1 h on ice and washed with 1× PBS for 5 min and with H2O for 1 min; (iv) 20% formamide was used for probe hybridization; (v) for CARD amplification, all samples were overlaid with a solution containing 1× PBS, 10% dextran sulfate, 0.1% blocking reagent (Roche, Nutley, NJ), 2 M NaCl, 0.0015% H2O2, and 0.33 μg/μl Alexa 488- or Alexa 594-labeled tyramides and incubated at 37°C for 15 min for cellular probes and 45 min for viral probes; and (vi) samples were embedded with Vectashield containing 1 μg ml−1 4′,6-diamidino-2-phenylindole (DAPI). Samples were imaged on a Leica TBS SP8 confocal microscope fitted with a 63× oil immersion lens, and images were collected sequentially by using the Leica DAPI, Alexa 488, and Alexa 594 channels. Adobe Photoshop was used to merge images and to modify brightness and contrast for clarity of presentation.
Acidianus sp. virus infection.
Cultures of an Acidianus strain isolated from CHAS (99% 16S rRNA gene identity to Acidianus hospitalis W1) were kindly provided by M. J. Amenabar and E. S. Boyd (Montana State University) (M. J. Amenabar and E. S. Boyd, unpublished data). The strain, designated here Acidianus sp. strain CHAS, was grown anaerobically with colloidal elemental sulfur using an 80:20 H2-CO2 headspace in synthetic base salts medium (44). The pH of the medium was 2.0, and the cultures were incubated at 80°C. For virus infection, CHAS hot spring water was filtered through a 0.6-μm polycarbonate filter (Millipore) to remove cells and concentrated 250 times with a 100,000-MWCO SpinX filter concentrator (Corning, Corning, NY). One milliliter of concentrated virus was injected into a 17-ml Acidianus sp. culture after 72 h of growth. Samples were taken every 4 to 8 h for 62 h postinoculation. DNA from each sample was extracted by using the Purelink viral DNA/RNA extraction kit (Invitrogen) and used to track virus and host growth by using qPCR (see below for details). The remaining sample was dialyzed into 5 mM glycine (pH 2.5) for transmission electron microscopy (TEM) imaging. At 62 h postinfection, the culture was passaged into fresh medium, and samples were taken every 4 to 8 h postinoculation for 136 h and processed as described above.
Acidianus sp. CHAS qPCR assay.
For detection and quantification of Acidianus sp. CHAS during growth, primers were designed for a 200-bp fragment of the Acidianus hospitalis W1 beta lactamase domain gene (GenBank accession number AEE93525). The product was amplified from Acidianus sp. CHAS genomic DNA and cloned into the pCR2.1 TOPO vector (Invitrogen). The purified plasmid was used to create qPCR standards from 9 × 104 to 9E1011 copies/ml. Quantitative PCR was performed with SsoFast EvaGreen supermix (Bio-Rad) on a Rotor-gene Q real-time PCR machine (Qiagen), using the primers described above.
Nucleotide sequence accession number.
The complete sequence of the ATSV genome has been submitted to GenBank under accession number KU645528.
RESULTS
Identification of a virus-like contig from a hot spring viral metagenome.
A gene encoding a putative viral major coat protein (MCP) was identified on an 8,655-bp contig assembled from the January 2008 CHAS viral metagenome (sequencing and assembly statistics are available upon request) by BLAST analysis of the NCBI RefSeq database (17). The contig was found to contain several ORFs with homology to Sulfolobus tengchongensis spindle-shaped virus 1 (STSV1), a large spindle-shaped archaeal virus previously isolated from a hot spring in Tengchong, China (45). One ORF within the contig was homologous to the STSV1 MCP. The 135-amino-acid (408-bp) ORF had 59% amino acid identity and an E value of 4e−41 to the STSV1 MCP. A quantitative PCR (qPCR) assay was developed by using primers and standards for this MCP gene in order to track the purification of the new virus directly from environmental hot spring samples. The qPCR assay was linear in the range of 104 to 1011 genomes/ml.
Virus purification directly from CHAS hot spring water.
Virus was purified directly from CHAS hot spring environmental samples by using the MCP gene qPCR assay to track the virus. Virus particle separation on CsCl gradients resulted in a single peak with a density of 1.28 to 1.34 g/cm3, with a maximum peak height observed at 1.32 g/cm3, which is within the range of known virus densities (46). Examination of the CsCl gradient peak viral fraction by TEM showed a large, spindle-shaped virion with a tail extending from one end (Fig. 1). Occasionally, a second short tail extending from the opposite end of the spindle-shaped head was observed. The spindle-shaped head averaged 169 nm (±50 nm) in length and 98 nm (±30 nm) in width, with a tail with a highly variable length extending from one end. Tail lengths ranged from 35 to 720 nm and averaged 243 nm, and tails were 19 nm (±9 nm) in width. Unlike Acidianus two-tailed virus (ATV), in which the spindle head contracts as the tails extend, there was no correlation between the spindle volume and the tail length in the isolated virus particles (data not shown). Based on the virion morphology and its cellular host (described below), we named this new virus Acidianus tailed spindle virus (ATSV).
FIG 1.

Transmission electron micrographs of large spindle-shaped virus particles with variable tail lengths isolated on cesium chloride density gradients. Gradient fractions containing the large spindle-shaped virus particles also contained the MCP gene originally identified in the metagenomic data set by quantitative PCR analysis. Bars, 100 nm.
Viral genome assembly.
DNA from the ATSV-enriched sample was sequenced by using paired-end MiSeq Illumina technology. A circular double-stranded DNA (dsDNA) genome of 70,812 bp with 80× average coverage was assembled (Fig. 2). This assembly was confirmed by PCR using PCR primers spanning the majority of the assembled genome.
FIG 2.

Genome map of the ATSV 70,812-bp circular dsDNA genome showing 96 open reading frames (ORFs). ORF colors indicate BLAST homology to large tailed spindle virus proteins (green), bacterial or archaeal proteins (purple), other archaeal virus proteins (blue), or the major coat protein (yellow) or no homology (gray). Red, putative origin of replication; alternating dark purple/orange, interspersed repeat region; cyan, viral CRISPR locus.
Genome properties and annotation.
The ATSV genome has a GC content of 37.4%. Eighty-nine percent of the genome is coding. A total of 96 ORFs were identified, encoding proteins with predicted molecular masses ranging from 4.3 to 248.8 kDa (46 to 2254 amino acids) (Fig. 2). The ORFs use ATG (79 ORFs), TTG (8), or GTG (9) as the start codon (see Data Set S1 in the supplemental material). Most ORFs (95%) appear to be preceded by putative promoters with high AT content that are 5 to 21 bp long and start 17 to 50 bp before the putative start codon. Additionally, 52 ORFs are preceded by a Shine-Dalgarno sequence 5 to 15 bp before the start codon (see Data Set S1 in the supplemental material). Genes are predicted to be transcribed on both strands, with 53 genes in the forward direction and 43 in the reverse direction.
BLASTX was used to analyze ATSV gene content (see Data Set S1 in the supplemental material). ATSV has homology to members of the large tailed spindle viruses (ATSV, STSV1 and -2, ATV, and Sulfolobus monocaudavirus 1 [SMV1]) (47, 53). Twenty-one ORFs show significant homology (>1 × 10−5) to proteins from the above-mentioned viruses. ATSV is most similar to STSV1 and STSV2, a close relative of STSV1, with 18 ORFs showing significant homology (see Data Set S1 in the supplemental material). The homologous ORFs show various levels of amino acid identity to the large tailed spindle viruses, ranging from 27 to 74%. Some of these ORFs have a putative function that can be predicted, including a glycosyltransferase (D339), an integral membrane protein (B530), the MCP (D135), two ATPases (F518 and D331), a thymidylate synthase (B293), and an integrase (C242) (see Data Set S1 in the supplemental material). Interestingly, the ATSV genome lacks most of the DNA-modifying proteins present in STSV1 and STSV2 (45). Several of the ATSV ORFs have homologs in other archaeal viruses, including 7 ORFs that make up a core set of genes carried by all or most large spindle virus genomes (ATSV, STSV1 and -2, ATV, and SMV1) (see Data Set S1 in the supplemental material) (48). These ORFs include the MCP (D135), a viral integrase (C242), 2 ATPases (F518 and D331), and 3 genes of unknown functions (F737, D1241, and B2246).
The ATSV genome reveals gene content with BLAST homology to Archaea (including a 7-kDa binding protein, a CopG transcriptional regulator, and two transposases), Bacteria [including a poly(A) polymerase], or both Archaea and Bacteria (including a methyltransferase, phosphoadenyl-sulfate reductase, phosphatase, MoxR ATPase, and AAA+ ATPase) (see Data Set S1 in the supplemental material). Additionally, all ATSV ORFs were compared to the archaeal (ArCOG), bacterial (COG), and phage (POG) Clusters of Orthologous Genes databases (49–51) by using BLASTX with an E value cutoff of 1e−4. Twenty-two ORFs had significant hits in ArCOGs, and 15 ORFs had significant hits in bacterial COGs, 14 of which had corresponding ArCOGs (data are available upon request). C450, the putative poly(A) polymerase, showed a hit with a bacterial COG but not an ArCOG.
Two ATSV ORFs, E429 and D194, are likely derived from transposable elements (see Data Set S1 in the supplemental material). E429 has homology to a Sulfolobus islandicus TnpA transposase protein (94% amino acid identity; E value = 3e−130), a serine recombinase. D194 has homology to an Acidianus hospitalis W1 TnpB transposase (92% amino acid identity; E value = 0) of unknown function. Both proteins belong to the IS607 transposase family and are often found next to each other in archaeal genomes. Both ORFs have dozens of high-identity homologs (>80% amino acid identity) in transposases from Sulfolobus, Acidianus, and Metallosphaera species. While mobile elements are absent in most known crenarchaeal viruses, the large spindle viruses ATV, SMV1, and STSV2 all contain at least two transposases (3, 52, 53), making them a feature common to the archaeal large spindle viruses.
Additionally, a 342-bp nonautonomous mobile element containing ORF D57 (genome positions 17249 to 17563) was found to have 93% identity to SMN1, a Sulfolobus islandicus REN1H1 nonautonomous mobile element (54). This mobile element is found in most sequenced S. islandicus genomes, with as many as 13 copies per genome. The virus genome also contains the consensus target sequence of TTT/A, into which the mobile element inserts (54). Interestingly, SMV1 contains two copies of SMN2, a related mobile element (52).
HHpred was used to determine more distal putative functions of uncharacterized ATSV ORF gene products (36). Eighteen ORF gene products were found to have homology to protein structures in the Protein Data Bank (PDB) with a probability of 80% or higher (see Data Set S1 in the supplemental material). These ORFs have a wide variety of putative functions, including several DNA binding proteins, an oligosaccharyltransferase, and a Sulfolobus spindle-shaped virus adaptor protein.
HHpred also revealed several ORFs with homology to proteins that are rare or absent in archaeal viruses (see Data Set S1 in the supplemental material). These proteins include a flavin adenine dinucleotide (FAD)-linked sulfhydryl oxidase, a flavorubredoxin, both a toxin and an antitoxin, and a potential self-regulating multidomain protein. This multidomain protein (C581) contains a leucine-rich repeat (LRR) domain at the N terminus (>99% probability) and a region homologous to human CHMP3, an ESCRT-III protein, at the C terminus (90.5% probability). The two domains are separated by a region of high serine and threonine content, suggesting that it serves as a posttranslational regulatory region. Finally, E355 has low homology by BLAST (E value = 0.002) but very-high-probability homology by HHpred to multiple proteins of the ParB family of chromosome and plasmid partitioning proteins. ParA and ParB proteins have been observed in bacteriophage genomes, including P1, along with a toxin/antitoxin system that aids in the passing of episomal prophages to daughter cells using an addiction system (55, 56).
Analysis of intergenic regions.
The ATSV genome contains multiple repeat-rich intergenic regions. The first repeat region, located between positions 9232 and 10146 in the virus genome, contains a long string of interspersed repeats, with 24 repeated 38-bp sequences (Fig. 3A). Within each 38-bp repeat, there are two conserved regions (13 bp and 8 bp) separated by two unconserved regions of 9 bp each (Fig. 3B). This type of repeat locus is unique due to its short length, high number of repeats, and strict conservation of repeat length. Similar repeat structures have not been seen in other viruses, including the other large spindle viruses. The function of this unusual repeat locus is not known.
FIG 3.

ATSV intergenic repeat locus. (A) Schematic of the locus. Purple triangles, repeat 1 (TGCACAXXAGCAA consensus); orange triangles, repeat 2 (TCAATAA consensus). Repeats 1 and 2 are each followed by 9 unconserved nucleotides. Gray arrows indicate surrounding ORFs. (B) MUSCLE (73) alignment of interspersed repeat loci. (C) Schematic of the 645-bp putative origin of viral replication. Red triangles, inverted repeats; purple and gray arrows, surrounding ORFs. (D) Sequences of the 5 perfect inverted repeats. Genome positions are indicated on the left.
A second 645-bp-long intergenic region contains 5 inverted repeats (Fig. 3C and D). By Z curve analysis (57), the intergenic region corresponds to an MK disparity peak (A and C bases in excess of G and T bases). A 115-bp stretch within this region also has high AT content (bp 219 to 333; 79.1% AT content). This combination of palindromic repeats, base pair disparity, and high AT content points to a potential origin of viral genome replication (57). Other archaeal viruses, including STSV1 and APSV1 (45, 58), also have putative origins of replication annotated based on intergenic regions that contain similar features.
Identification of a short CRISPR locus within the ATSV genome.
A CRISPR locus was found within the ATSV genome (genome positions 11703 to 11967), containing three complete direct repeats (DRs) and one partial DR matching an A. hospitalis W1 Ahos-40 CRISPR locus (genome positions 1562889 to 1565199) (Fig. 4A). Compared to the A. hospitalis W1 Ahos-40 consensus DR, the first DR of the viral CRISPR locus is truncated, and the other 3 DRs contain 1 to 2 mismatches (Fig. 4B). The spacer regions between the DRs are of various lengths, and the CRISPR spacers do not have significant hits in the NCBI database. This viral CRISPR locus is found to be conserved in viral contigs related to ATSV in viral metagenomes from CHAS and NL10, a related hot spring (data not shown), suggesting that the viral CRISPR locus is essential to the virus. In rare cases, other viruses encode CRISPR loci, sometimes with an intact system with adjacent Cas genes (56, 59–61). In this case, no ATSV ORFs have homology to Cas genes. The function of the ATSV CRISPR locus is unknown.
FIG 4.

Putative viral CRISPR locus. (A) Schematic of the intergenic region containing the CRISPR locus. Direct repeats (DR) are in cyan and surrounding ORFs are in gray. (B) ClustalW alignment of the DR sequences from the A. hospitalis Ahos-40 CRISPR locus and the ATSV short CRISPR locus.
Linking the viral genome to virus particle morphology.
Two methods, mass spectrometry and Western blot analysis, were used to link the large spindle-shaped virus particles to the viral genome. Purified virus was displayed on SDS-PAGE gels, and the dominant band was excised from the gel, digested with trypsin by in-gel digestion, and analyzed by liquid chromatography-mass spectrometry (LC-MS) (Fig. 5A). The resulting in silico peptide masses were compared to peptide masses from a database of viral ORFs, including the 96 ORFs of the assembled viral genome, by using the Mascot tandem mass spectrometry (MS/MS) ion search engine. A significant peptide match to D135, the putative MCP, was identified with a Mascot score of 50 (Fig. 5B). In a parallel experiment, Western blot analysis was performed by using polyclonal antibodies to the heterologously expressed D135 MCP. The expected 14-kDa dominant band was recognized by polyclonal antibodies to D135, further confirming the link between the large spindle-shaped, long tailed virus morphology; the putative MCP; and the assembled virus genome (Fig. 5C).
FIG 5.
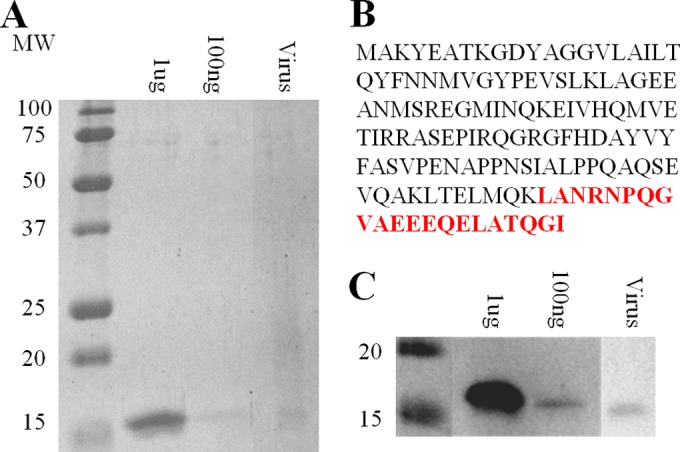
Identification and analysis of the ATSV MCP. (A) SDS-PAGE analysis of ATSV-enriched cesium-purified virus. Known amounts of the heterologously expressed D135 MCP shown are for comparison. MW, molecular weight (in thousands). (B) ATSV D135 MCP amino acid sequence. The peptide identified by mass spectrometry is indicated in red. (C) Parallel Western blotting using primary antibodies to the heterologously expressed D135 protein.
ATSV host identification using the CRISPR/Cas system.
A bioinformatic search of cellular CRISPR/Cas systems was used to identify possible ATSV hosts. Each cellular CRISPR locus contains short 20- to 50-bp DNA sequences (termed CRISPR spacer DNA) derived from invading elements, including viruses and plasmids. The CRISPR spacer DNA sequences therefore provide a record of a cell's past viral infections. CRISPR spacers from reported genomes were identified by using CRISPRfinder (40), extracted, and assembled onto the 70.8-kb genome. Acidianus hospitalis W1, an archaeal species originally isolated from the Crater Hills thermal area of YNP (62), has 10 CRISPR spacer matches to the 70.8-kbp genome, with 7 of these being perfect matches (data are available upon request), making A. hospitalis W1 a likely host. The CRISPR spacer matches are distributed across the virus genome. Additional CRISPR spacer matches with 2 to 5 mismatches were found for Sulfolobus islandicus REY15A, S. islandicus L.D.8.5., Metallosphaera sedula DSM 5348, and Sulfolobus solfataricus 98-2, indicating that the virus could have a broader host range.
Dual virus and host labeling by CARD-FISH (viral FISH).
A dual virus-host fluorescence labeling technique using CARD-FISH was used to confirm that A. hospitalis W1 is a host for ATSV. Independent hybridization of horseradish peroxidase-conjugated DNA probes to the A. hospitalis 16S rRNA gene and of DIG-labeled DNA probes to the viral genome allowed signal amplification of a fluorescently labeled tyramide (red for the virus and green for the host). Probes were hybridized to concentrated, fixed environmental cells directly obtained from the CHAS hot spring. Colocalization of the two colors was used as an indication of ATSV infection. It was found that A. hospitalis cells colocalized with ATSV genome probes, indicating that A. hospitalis is present in the hot spring, and that a subset of the A. hospitalis cells contains viral DNA (Fig. 6). Controls using ATSV probes in combination with probes to other archaeal species found in CHAS (Stygiolobus sp. and Vulcanisaeta sp.) did not show colocalization.
FIG 6.

Viral FISH analysis of dual ATSV and Acidianus hospitalis fluorescence labeling by CARD-FISH. (A) A. hospitalis 16S rRNA-labeled cells. (B) ATSV-labeled cells. (C) Overlay of DAPI-stained cells (blue), A. hospitalis 16S rRNA-labeled cells (green), and ATSV-labeled cells (red). Examples of cells with colocalization of ATSV and A. hospitalis are indicated by red boxes.
ATSV replication in Acidianus hospitalis cultures.
Based on the results of CRISPR spacer sequence matches and viral FISH analysis, we tested the ability of ATSV to establish virus replication in an Acidianus strain (Acidianus sp. CHAS) isolated from CHAS that was closely related at the level of 16S rRNA gene homology to A. hospitalis W1. After infection of an Acidianus sp. CHAS culture with filtered, concentrated virus obtained directly from a CHAS environmental sample, a 3-fold increase in the level of ATSV DNA was measured by qPCR, and long-tailed, large spindle-shaped virus particles, similar to those seen in CsCl-purified environmental virus samples, were visible by TEM (Fig. 7A). The initial virus-infected culture was grown until 62 h postinfection and passaged into fresh medium. Virus and host were measured over a 136-h time course. The passaged cultured showed an 84- to 97-fold increase in virus production for two replicates (Fig. 7B). The peak level of viral DNA was seen after the peak and decline in the levels of cellular DNA, indicating the possibility of a lytic life cycle. Virus particles were again observed by TEM. No other virus-like particle morphologies were observed.
FIG 7.

Acidianus sp. strain CHAS infection by ATSV. (A) TEM images of viruses from Acidianus sp. CHAS cultures infected with filtered, concentrated CHAS hot spring water show large tailed spindle-shaped virus particles. Bars, 100 nm. (B) Growth of ATSV and Acidianus sp. CHAS in a passage of primary Acidianus sp. CHAS infection. ATSV (gray) and Acidianus sp. CHAS (black) DNA signals were measured by qPCR.
DISCUSSION
We report here the development of a workflow beginning with environmental viral metagenomic data sets to isolate and initially characterize a new archaeal virus directly from environmental samples. The developed workflow includes bioinformatic identification of a virus MCP, qPCR assays to track virus purification directly from environmental samples, mass spectrometry to link isolated virions to genomes, and analysis of the cellular CRISPR/Cas loci and viral FISH assays for host identification. Through this workflow that is not strictly dependent on traditional culture-dependent virus isolation techniques, we have isolated and characterized ATSV, a new large spindle-shaped archaeal virus from a high-temperature acidic hot spring located in YNP.
Our data support assigning ATSV as a new member of the archaeal large spindle viruses that include ATV, STSV1, STSV2, and SMV1. These viruses share the common characteristics of a large, spindle-shaped body; a variable-length tail extending from one or both ends of the main virion body; and a set of 7 core gene products, including the major coat protein, an integrase, multiple ATPases, and several uncharacterized proteins with features such as coiled-coil motifs, repeats, and transmembrane domains. Our data support the inclusion of ATSV into a new family of archaeal viruses with the proposed name Fusellocaudaviridae and into a superfamily named Magnusfuselloviridae with STSV1, STSV2, SMV1, and ATV.
Analysis of the ATSV 70.8-kb circular dsDNA genome, one of the largest archaeal virus genomes to date, reveals many remarkable features. Hidden Markov model-based homology searches using HHpred, which searches alignments instead of individual proteins in order to find more distal hits, expand our understanding of possible ATSV gene functions. The most striking result comes from C581, a large, 3-domain protein. Despite low sequence homology (13%), the N-terminal 188 residues revealed 100 high-probability (>99%) hits to LRR domains. LRR proteins have multiple functions, but many function in protein-protein or other protein-ligand interactions (63, 64). The top HHpred score (99.9%) is for the LRR domain of Arabidopsis thaliana FLS2 (65). The A. thaliana LRR domain was shown to interact with a second LRR domain of the BAK1 protein, a serine/threonine kinase. In the case of ATSV, the LRR domain is followed by a string of 233 amino acid residues made up almost exclusively of serines (98 residues), threonines (73 residues), prolines (27 residues), and glutamines (16 residues). Typically, such serine/threonine-rich regions serve as regulatory regions controlled by phosphorylation. The genome of the ATSV cellular host A. hospitalis has 9 annotated serine/threonine kinases and 2 homoserine kinases, which could potentially phosphorylate this region (62). Finally, the C terminus of C581 is predicted by HHpred analysis to be an ESCRT-III-related protein. A 53-residue section in the middle of the 140-residue C-terminal domain has 90.5% and 89% probability matches to two versions of the human charged multivesicular protein 3 (CHMP3) crystal structure (PDB accession numbers 5GRD and 3FRT). CHMP3 is an ESCRT-III protein that complexes with other CHMP proteins and the VPS4 ATPase to aid in membrane budding and scission (66). In eukaryotes, the ESCRT system has many functions, including multivesicular body formation, cytokinesis, macroautophagy, and virus budding. ESCRT-I and -II systems are involved in membrane budding, while ESCRT-III proteins are involved in membrane scission (66). Most Archaea, including A. hospitalis, have an ESCRT-III system, including a CHMP3 homolog that has been shown to be essential for cell division by forming filaments that constrict the cell (67, 68). The region of CHMP3 with homology to C581 is the dimerization region and has a helix-turn-helix motif. While it is unlikely that this 53-residue region is sufficient to functionally mimic ESCRT-aided cell division, we speculate that it could act as an adaptor protein (or as a transdominant negative effector) interacting with the host cell division machinery, affecting cell division, and creating a biochemical environment for virus replication. Both the N-terminal LRR region and the serine/threonine-rich middle region of the protein have the potential for protein-protein interactions, so it is possible that the ESCRT-III domain of C581 directs a protein complex to the area of cell division. While both eukaryotic and archaeal viruses have been shown to hijack the ESCRT system to aid in virus budding and egress (66, 69), this would be the first instance of a virus encoding its own ESCRT homolog. Future studies are needed to confirm interactions with ESCRT machinery, identify cellular and viral binding partners, and elucidate the role of possible posttranslational phosphorylation modifications of the serine/threonine-rich region.
In addition to its interesting gene content, ATSV contains three intergenic regions, all with different types of repeat structures. One region of interest is a short CRISPR locus (Fig. 4). Although it is not common, CRISPR loci have been identified in other viruses. Five Vibrio cholerae ICP-1-related phages were found to carry functional CRISPR/Cas systems, with 2 CRISPR loci and 6 cas genes (60). The majority of spacers have 100% identity to a phage-inhibitory chromosomal island, which protects the bacteria from phage infection. The phage CRISPR/Cas system was shown to successfully degrade the island, overcoming the host antiviral defense. Some of the CRISPR spacers matched separate viral contigs, indicating a possible mechanism for virus-virus competition. In the case of ATSV, the spacers do not match other known viruses or the host DNA. We speculate that the CRISPR spacer could be targeted to an as-yet-unknown competing virus or, alternatively, that it acts as an RNA decoy to prevent the CRISPR/Cas system from targeting the ATSV genome.
A second 645-bp intergenic region (Fig. 3C and D) may function as an origin of viral genome replication. Like STSV1, the region has high base pair disparity (MK disparity) and a series of inverted repeats. MK disparity peaks are seen at the origins of replication for several archaeal species, including Pyrococcus abyssi GE5 and Sulfolobus acidocaldarius DSM 639 (57). The intergenic region also contains 5 stem-loop structures. Stem-loops are important for DNA replication in many organisms, both for protein target recognition and for access to single-stranded DNA for replication initiation (57).
The final ATSV intergenic region contains 24 repeats, each containing two short conserved regions (7 bp and 13 bp), each separated by an unconserved region (9 bp and 10 bp) (Fig. 3A and B). The high number of repeats and the layout of the locus have not been previously observed. While similar to CRISPR loci in that repeats are separated by unconserved spacers, the significantly shorter lengths of ATSV repeats and unconserved regions (7 to 10 bp for ATSV versus 20 to 50 bp for CRISPRs) and the alternative of two types of repeats make the ATSV locus quite different from CRISPR loci. The function of this region is not known.
ATSV encodes a tyrosine recombinase-type integrase with 85% identity to the SMV1 integrase, 75% identity to the ATV integrase, and 43% identity to the STSV1 and STSV2 integrases. While ATV has been shown to integrate into the host chromosome, STSV1 and STSV2 have not been found to integrate. Given the higher homology to and similar length as the ATV integrase, it is possible that ATSV might integrate into the host chromosome and exist in a lysogenic state. Combined with the observation of a host peak signal before the virus peak signal (suggesting lysis), it is possible that, similar to members of the Fuselloviridae (70), integration is not essential but instead is one of two modes of the virus life cycle.
The tools and experimental approaches described in this work could be generally applied to the discovery and initial characterization of viruses from diverse environments. The extension of analysis beyond viral metagenomic data sets alone to include other techniques to allow complete genome sequencing, virus isolation, and host identification directly from environmental samples greatly extends the utility of viral metagenomic data sets. Even though many of the tools and approaches described here have been used by others in isolation (21, 28, 41, 71, 72), no other studies to our knowledge have combined them to identify and initially characterize a new virus starting with only viral metagenomic data. To our knowledge, this is the first example of linking a virus genome to both particle morphology and a host with confirmation by culturing, effectively closing the viral metagenomic loop.
Supplementary Material
ACKNOWLEDGMENTS
We thank Megan Maddio for her work on protein expression and Jennifer Wirth and Cassia Wagner for critical reading of the text. We also thank Stacey Gunther and the Yellowstone National Park Research Resource Office for their help in facilitating sampling. Research was conducted in Yellowstone National Park under the conditions of permit YELL-2013-SCI-5090.
This work was funded by National Science Foundation award DEB-4W4596 to M.J.Y. and by NASA award NNA15BB02A to E.S.B.
We declare no conflict of interest.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03098-15.
REFERENCES
- 1.Rice G, Tang L, Stedman K, Roberto F, Spuhler J, Gillitzer E, Johnson JE, Douglas T, Young M. 2004. The structure of a thermophilic archaeal virus shows a double-stranded DNA viral capsid type that spans all domains of life. Proc Natl Acad Sci U S A 101:7716–7720. doi: 10.1073/pnas.0401773101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prangishvili D, Arnold HP, Gotz D, Ziese U, Holz I, Kristjansson JK, Zillig W. 1999. A novel virus family, the Rudiviridae: structure, virus-host interactions and genome variability of the Sulfolobus viruses SIRV1 and SIRV2. Genetics 152:1387–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prangishvili D, Vestergaard G, Haring M, Aramayo R, Basta T, Rachel R, Garrett RA. 2006. Structural and genomic properties of the hyperthermophilic archaeal virus ATV with an extracellular stage of the reproductive cycle. J Mol Biol 359:1203–1216. doi: 10.1016/j.jmb.2006.04.027. [DOI] [PubMed] [Google Scholar]
- 4.Martin A, Yeats S, Janekovic D, Reiter WD, Aicher W, Zillig W. 1984. SAV 1, a temperate u.v.-inducible DNA virus-like particle from the archaebacterium Sulfolobus acidocaldarius isolate B12. EMBO J 3:2165–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dellas N, Snyder JC, Bolduc B, Young MJ. 2014. Archaeal viruses: diversity, replication, and structure. Annu Rev Virol 1:399–425. doi: 10.1146/annurev-virology-031413-085357. [DOI] [PubMed] [Google Scholar]
- 6.DiMaio F, Yu X, Rensen E, Krupovic M, Prangishvili D, Egelman EH. 2015. A virus that infects a hyperthermophile encapsidates A-form DNA. Science 348:914–917. doi: 10.1126/science.aaa4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prangishvili D, Koonin EV, Krupovic M. 2013. Genomics and biology of rudiviruses, a model for the study of virus-host interactions in Archaea. Biochem Soc Trans 41:443–450. doi: 10.1042/BST20120313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dellas N, Lawrence CM, Young MJ. 2013. A survey of protein structures from archaeal viruses. Life 3:118–130. doi: 10.3390/life3010118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roux S, Hallam SJ, Woyke T, Sullivan MB. 22 July 2015. Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. eLife doi 10.7554/eLife.08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosario K, Breitbart M. 2011. Exploring the viral world through metagenomics. Curr Opin Virol 1:289–297. doi: 10.1016/j.coviro.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 11.Mokili JL, Rohwer F, Dutilh BE. 2012. Metagenomics and future perspectives in virus discovery. Curr Opin Virol 2:63–77. doi: 10.1016/j.coviro.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brum JR, Sullivan MB. 2015. Rising to the challenge: accelerated pace of discovery transforms marine virology. Nat Rev Microbiol 13:147–159. doi: 10.1038/nrmicro3404. [DOI] [PubMed] [Google Scholar]
- 13.Brum JR, Hurwitz BL, Schofield O, Ducklow HW, Sullivan MB. 2016. Seasonal time bombs: dominant temperate viruses affect Southern Ocean microbial dynamics. ISME J 10:437–449. doi: 10.1038/ismej.2015.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holmfeldt K, Solonenko N, Shah M, Corrier K, Riemann L, Verberkmoes NC, Sullivan MB. 2013. Twelve previously unknown phage genera are ubiquitous in global oceans. Proc Natl Acad Sci U S A 110:12798–12803. doi: 10.1073/pnas.1305956110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang I, Oh HM, Kang D, Cho JC. 2013. Genome of a SAR116 bacteriophage shows the prevalence of this phage type in the oceans. Proc Natl Acad Sci U S A 110:12343–12348. doi: 10.1073/pnas.1219930110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y, Temperton B, Thrash JC, Schwalbach MS, Vergin KL, Landry ZC, Ellisman M, Deerinck T, Sullivan MB, Giovannoni SJ. 2013. Abundant SAR11 viruses in the ocean. Nature 494:357–360. doi: 10.1038/nature11921. [DOI] [PubMed] [Google Scholar]
- 17.Bolduc B, Shaughnessy DP, Wolf YI, Koonin EV, Roberto FF, Young M. 2012. Identification of novel positive-strand RNA viruses by metagenomic analysis of Archaea-dominated Yellowstone hot springs. J Virol 86:5562–5573. doi: 10.1128/JVI.07196-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolduc B, Wirth JF, Mazurie A, Young MJ. 2015. Viral assemblage composition in Yellowstone acidic hot springs assessed by network analysis. ISME J 9:2162–2177. doi: 10.1038/ismej.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schoenfeld T, Patterson M, Richardson PM, Wommack KE, Young M, Mead D. 2008. Assembly of viral metagenomes from Yellowstone hot springs. Appl Environ Microbiol 74:4164–4174. doi: 10.1128/AEM.02598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Emerson JB, Thomas BC, Andrade K, Heidelberg KB, Banfield JF. 2013. New approaches indicate constant viral diversity despite shifts in assemblage structure in an Australian hypersaline lake. Appl Environ Microbiol 79:6755–6764. doi: 10.1128/AEM.01946-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santos F, Yarza P, Parro V, Briones C, Anton J. 2010. The metavirome of a hypersaline environment. Environ Microbiol 12:2965–2976. doi: 10.1111/j.1462-2920.2010.02273.x. [DOI] [PubMed] [Google Scholar]
- 22.Lima-Mendez G, Van Helden J, Toussaint A, Leplae R. 2008. Reticulate representation of evolutionary and functional relationships between phage genomes. Mol Biol Evol 25:762–777. doi: 10.1093/molbev/msn023. [DOI] [PubMed] [Google Scholar]
- 23.Tadmor AD, Ottesen EA, Leadbetter JR, Phillips R. 2011. Probing individual environmental bacteria for viruses by using microfluidic digital PCR. Science 333:58–62. doi: 10.1126/science.1200758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng L, Ignacio-Espinoza JC, Gregory AC, Poulos BT, Weitz JS, Hugenholtz P, Sullivan MB. 2014. Viral tagging reveals discrete populations in Synechococcus viral genome sequence space. Nature 513:242–245. doi: 10.1038/nature13459. [DOI] [PubMed] [Google Scholar]
- 25.Bhattacharya D, Price DC, Bicep C, Bapteste E, Sarwade M, Rajah VD, Yoon HS. 2013. Identification of a marine cyanophage in a protist single-cell metagenome assembly. J Phycol 49:207–212. doi: 10.1111/jpy.12028. [DOI] [PubMed] [Google Scholar]
- 26.Roux S, Hawley AK, Beltran MT, Scofield M, Schwientek P, Stepanauskas R, Woyke T, Hallam SJ, Sullivan MB. 2014. Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta-genomics. eLife 3:e03125. doi: 10.7554/eLife.03125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez-Garcia M, Santos F, Moreno-Paz M, Parro V, Anton J. 2014. Unveiling viral-host interactions within the ‘microbial dark matter. Nat Commun 5:4542. doi: 10.1038/ncomms5542. [DOI] [PubMed] [Google Scholar]
- 28.Tyson GW, Banfield JF. 2008. Rapidly evolving CRISPRs implicated in acquired resistance of microorganisms to viruses. Environ Microbiol 10:200–207. [DOI] [PubMed] [Google Scholar]
- 29.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 30.Kraft P, Kummel D, Oeckinghaus A, Gauss GH, Wiedenheft B, Young M, Lawrence CM. 2004. Structure of D-63 from Sulfolobus spindle-shaped virus 1: surface properties of the dimeric four-helix bundle suggest an adaptor protein function. J Virol 78:7438–7442. doi: 10.1128/JVI.78.14.7438-7442.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Studier FW. 2005. Protein production by auto-induction in high-density shaking cultures. Protein Expr Purif 41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 32.John SG, Mendez CB, Deng L, Poulos B, Kauffman AKM, Kern S, Brum J, Polz MF, Boyle EA, Sullivan MB. 2011. A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environ Microbiol Rep 3:809–809. doi: 10.1111/j.1758-2229.2011.00301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chevreux B, Wetter T, Suhai S. 1999. Genome sequence assembly using trace signals and additional sequence information. GCB '99: German Conference on Bioinformatics http://www.bioinfo.de/isb/gcb99/talks/chevreux. [Google Scholar]
- 34.Salzberg SL, Delcher AL, Kasif S, White O. 1998. Microbial gene identification using interpolated Markov models. Nucleic Acids Res 26:544–548. doi: 10.1093/nar/26.2.544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Betley JN, Frith MC, Graber JH, Choo S, Deshler JO. 2002. A ubiquitous and conserved signal for RNA localization in chordates. Curr Biol 12:1756–1761. doi: 10.1016/S0960-9822(02)01220-4. [DOI] [PubMed] [Google Scholar]
- 38.Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European molecular biology open software suite. Trends Genet 16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 39.Maaty WSA, Ortmann AC, Dlakic M, Schulstad K, Hilmer JK, Liepold L, Weidenheft B, Khayat R, Douglas T, Young MJ, Bothner B. 2006. Characterization of the archaeal thermophile Sulfolobus turreted icosahedral virus validates an evolutionary link among double-stranded DNA viruses from all domains of life. J Virol 80:7625–7635. doi: 10.1128/JVI.00522-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grissa I, Vergnaud G, Pourcel C. 2007. CRISPRFinder: a Web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res 35:W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Allers E, Moraru C, Duhaime MB, Beneze E, Solonenko N, Barrero-Canosa J, Amann R, Sullivan MB. 2013. Single-cell and population level viral infection dynamics revealed by phageFISH, a method to visualize intracellular and free viruses. Environ Microbiol 15:2306–2318. doi: 10.1111/1462-2920.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Munson-McGee JH, Field EK, Bateson M, Rooney C, Stepanauskas R, Young MJ. 2015. Nanoarchaeota, their Sulfolobales host, and Nanoarchaeota virus distribution across Yellowstone National Park hot springs. Appl Environ Microbiol 81:7860–7868. doi: 10.1128/AEM.01539-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moraru C, Moraru G, Fuchs BM, Amann R. 2011. Concepts and software for a rational design of polynucleotide probes. Environ Microbiol Rep 3:69–78. doi: 10.1111/j.1758-2229.2010.00189.x. [DOI] [PubMed] [Google Scholar]
- 44.Boyd ES, Jackson RA, Encarnacion G, Zahn JA, Beard T, Leavitt WD, Pi Y, Zhang CL, Pearson A, Geesey GG. 2007. Isolation, characterization, and ecology of sulfur-respiring Crenarchaea inhabiting acid-sulfate-chloride-containing geothermal springs in Yellowstone National Park. Appl Environ Microbiol 73:6669–6677. doi: 10.1128/AEM.01321-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiang XY, Chen LM, Huang XX, Luo YM, She QX, Huang L. 2005. Sulfolobus tengchongensis spindle-shaped virus STSV1: virus-host interactions and genomic features. J Virol 79:8677–8686. doi: 10.1128/JVI.79.14.8677-8686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thurber RV, Haynes M, Breitbart M, Wegley L, Rohwer F. 2009. Laboratory procedures to generate viral metagenomes. Nat Protoc 4:470–483. doi: 10.1038/nprot.2009.10. [DOI] [PubMed] [Google Scholar]
- 47.Erdmann S, Bauer SL, Garrett RA. 2014. Inter-viral conflicts that exploit host CRISPR immune systems of Sulfolobus. Mol Microbiol 91:900–917. doi: 10.1111/mmi.12503. [DOI] [PubMed] [Google Scholar]
- 48.Hochstein R, Bollschweiler D, Engelhardt H, Lawrence CM, Young M. 2015. Large tailed spindle viruses of Archaea: a new way of doing viral business. J Virol 89:9146–9149. doi: 10.1128/JVI.00612-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Makarova KS, Sorokin AV, Novichkov PS, Wolf YI, Koonin EV. 2007. Clusters of orthologous genes for 41 archaeal genomes and implications for evolutionary genomics of archaea. Biol Direct 2:33. doi: 10.1186/1745-6150-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. 2001. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res 29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kristensen DM, Waller AS, Yamada T, Bork P, Mushegian AR, Koonin EV. 2013. Orthologous gene clusters and taxon signature genes for viruses of prokaryotes. J Bacteriol 195:941–950. doi: 10.1128/JB.01801-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Erdmann S, Shah SA, Garrett RA. 2013. SMV1 virus-induced CRISPR spacer acquisition from the conjugative plasmid pMGB1 in Sulfolobus solfataricus P2. Biochem Soc Trans 41:1449–1458. doi: 10.1042/BST20130196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Erdmann S, Chen B, Huang XX, Deng L, Liu C, Shah SA, Bauer SL, Sobrino CL, Wang HN, Wei YL, She QX, Garrett RA, Huang L, Lin LB. 2014. A novel single-tailed fusiform Sulfolobus virus STSV2 infecting model Sulfolobus species. Extremophiles 18:51–60. doi: 10.1007/s00792-013-0591-z. [DOI] [PubMed] [Google Scholar]
- 54.Berkner S, Lipps G. 2007. An active nonautonomous mobile element in Sulfolobus islandicus REN1H1. J Bacteriol 189:2145–2149. doi: 10.1128/JB.01567-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodionov O, Yarmolinsky M. 2004. Plasmid partitioning and the spreading of P1 partition protein ParB. Mol Microbiol 52:1215–1223. doi: 10.1111/j.1365-2958.2004.04055.x. [DOI] [PubMed] [Google Scholar]
- 56.Bellas CM, Anesio AM, Barker G. 2015. Analysis of virus genomes from glacial environments reveals novel virus groups with unusual host interactions. Front Microbiol 6:656. doi: 10.3389/fmicb.2015.00656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao F. 2014. Recent advances in the identification of replication origins based on the Z-curve method. Curr Genomics 15:104–112. doi: 10.2174/1389202915999140328162938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mochizuki T, Yoshida T, Tanaka R, Forterre P, Sako Y, Prangishvili D. 2010. Diversity of viruses of the hyperthermophilic archaeal genus Aeropyrum, and isolation of the Aeropyrum pernix bacilliform virus 1, APBV1, the first representative of the family Clavaviridae. Virology 402:347–354. doi: 10.1016/j.virol.2010.03.046. [DOI] [PubMed] [Google Scholar]
- 59.Minot S, Sinha R, Chen J, Li HZ, Keilbaugh SA, Wu GD, Lewis JD, Bushman FD. 2011. The human gut virome: inter-individual variation and dynamic response to diet. Genome Res 21:1616–1625. doi: 10.1101/gr.122705.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Seed KD, Lazinski DW, Calderwood SB, Camilli A. 2013. A bacteriophage encodes its own CRISPR/Cas adaptive response to evade host innate immunity. Nature 494:489–491. doi: 10.1038/nature11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garcia-Heredia I, Martin-Cuadrado A-B, Mojica FJM, Santos F, Mira A, Anton J, Rodriguez-Valera F. 2012. Reconstructing viral genomes from the environment using fosmid clones: the case of haloviruses. PLoS One 7:e33802. doi: 10.1371/journal.pone.0033802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.You XY, Liu C, Wang SY, Jiang CY, Shah SA, Prangishvili D, She QX, Liu SJ, Garrett RA. 2011. Genomic analysis of Acidianus hospitalis W1 a host for studying crenarchaeal virus and plasmid life cycles. Extremophiles 15:487–497. doi: 10.1007/s00792-011-0379-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kobe B, Deisenhofer J. 1994. The leucine-rich repeat—a versatile binding motif. Trends Biochem Sci 19:415–421. doi: 10.1016/0968-0004(94)90090-6. [DOI] [PubMed] [Google Scholar]
- 64.Bella J, Hindle KL, McEwan PA, Lovell SC. 2008. The leucine-rich repeat structure. Cell Mol Life Sci 65:2307–2333. doi: 10.1007/s00018-008-8019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun YD, Li L, Macho AP, Han ZF, Hu ZH, Zipfel C, Zhou JM, Chai JJ. 2013. Structural basis for flg22-induced activation of the Arabidopsis FLS2-BAK1 immune complex. Science 342:624–628. doi: 10.1126/science.1243825. [DOI] [PubMed] [Google Scholar]
- 66.Schmidt O, Teis D. 2012. The ESCRT machinery. Curr Biol 22:R116–R120. doi: 10.1016/j.cub.2012.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Samson RY, Bell SD. 2009. Ancient ESCRTs and the evolution of binary fission. Trends Microbiol 17:507–513. doi: 10.1016/j.tim.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 68.Dobro MJ, Samson RY, Yu ZH, McCullough J, Ding HJ, Chong PLG, Bell SD, Jensen GJ. 2013. Electron cryotomography of ESCRT assemblies and dividing Sulfolobus cells suggests that spiraling filaments are involved in membrane scission. Mol Biol Cell 24:2319–2327. doi: 10.1091/mbc.E12-11-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Snyder JC, Samson RY, Brumfield SK, Bell SD, Young MJ. 2013. Functional interplay between a virus and the ESCRT machinery in Archaea. Proc Natl Acad Sci U S A 110:10783–10787. doi: 10.1073/pnas.1301605110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Clore AJ, Stedman KM. 2007. The SSV1 viral integrase is not essential. Virology 361:103–111. doi: 10.1016/j.virol.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 71.Minot S, Bryson A, Chehoud C, Wu GD, Lewis JD, Bushman FD. 2013. Rapid evolution of the human gut virome. Proc Natl Acad Sci U S A 110:12450–12455. doi: 10.1073/pnas.1300833110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anderson RE, Sogin ML, Baross JA. 2014. Evolutionary strategies of viruses, bacteria and archaea in hydrothermal vent ecosystems revealed through metagenomics. PLoS One 9:e109696. doi: 10.1371/journal.pone.0109696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hurwitz BL, Westveld AH, Brum JR, Sullivan MB. 2014. Modeling ecological drivers in marine viral communities using comparative metagenomics and network analyses. Proc Natl Acad Sci U S A 111:10714–100719. doi: 10.1073/pnas.1319778111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brum JR, Ignacio-Espinoza JC, Roux S, Doulcier G, Acinas SG, Alberti A, Chaffron S, Cruaud C, de Vargas C, Gasol JM, Gorsky G, Gregory AC, Guidi L, Hingamp P, Iudicone D, Not F, Ogata H, Pesant S, Poulos BT, Schwenck SM, Speich S, Dimier C, Kandels-Lewis S, Picheral M, Searson S; Tara Oceans Coordinators, Bork P, Bowler C, Sunagawa S, Wincker P, Karsenti E, Sullivan MB. 2015. Global patterns and ecological drivers of ocean viral communities. Science 348:1261498. doi: 10.1126/science.1261498. [DOI] [PubMed] [Google Scholar]
- 76.Hurwitz BL, Brum JR, Sullivan MB. 2015. Depth-stratified functional and taxonomic niche specialization in the ‘core’ and ‘flexible’ Pacific Ocean Virome. ISME J 9:472–484. doi: 10.1038/ismej.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.