

ABSTRACT
Previous studies in our laboratory have identified equine CXCL16 (EqCXCL16) to be a candidate molecule and possible cell entry receptor for equine arteritis virus (EAV). In horses, the CXCL16 gene is located on equine chromosome 11 (ECA11) and encodes a glycosylated, type I transmembrane protein with 247 amino acids. Stable transfection of HEK-293T cells with plasmid DNA carrying EqCXCL16 (HEK-EqCXCL16 cells) increased the proportion of the cell population permissive to EAV infection from <3% to almost 100%. The increase in permissiveness was blocked either by transfection of HEK-EqCXCL16 cells with small interfering RNAs (siRNAs) directed against EqCXCL16 or by pretreatment with guinea pig polyclonal antibody against EqCXCL16 protein (Gp anti-EqCXCL16 pAb). Furthermore, using a virus overlay protein-binding assay (VOPBA) in combination with far-Western blotting, gradient-purified EAV particles were shown to bind directly to the EqCXCL16 protein in vitro. The binding of biotinylated virulent EAV strain Bucyrus at 4°C was significantly higher in HEK-EqCXCL16 cells than nontransfected HEK-293T cells. Finally, the results demonstrated that EAV preferentially infects subpopulations of horse CD14+ monocytes expressing EqCXCL16 and that infection of these cells is significantly reduced by pretreatment with Gp anti-EqCXCL16 pAb. The collective data from this study provide confirmatory evidence that the transmembrane form of EqCXCL16 likely plays a major role in EAV host cell entry processes, possibly acting as a primary receptor molecule for this virus.
IMPORTANCE Outbreaks of EVA can be a source of significant economic loss for the equine industry from high rates of abortion in pregnant mares, death in young foals, establishment of the carrier state in stallions, and trade restrictions imposed by various countries. Similar to other arteriviruses, EAV primarily targets cells of the monocyte/macrophage lineage, which, when infected, are believed to play a critical role in EVA pathogenesis. To this point, however, the host-specified molecules involved in EAV binding and entry into monocytes/macrophages have not been identified. Identification of the cellular receptors for EAV may provide insights to design antivirals and better prophylactic reagents. In this study, we have demonstrated that EqCXCL16 acts as an EAV entry receptor in EAV-susceptible cells, equine monocytes. These findings represent a significant advance in our understanding of the fundamental mechanisms associated with the entry of EAV into susceptible cells.
INTRODUCTION
Equine arteritis virus (EAV) is a single-stranded, positive-sense RNA virus in the family Arteriviridae, genus Arterivirus, order Nidovirales. Besides EAV, the family Arteriviridae contains four other viruses: porcine reproductive and respiratory syndrome virus (PRRSV), simian hemorrhagic fever virus (SHFV), lactate dehydrogenase-elevating virus (LDV) of mice, and wobbly possum disease virus (WPDV), which is the most recently identified member of the Arteriviridae (1–3). Arteriviruses primarily target monocyte/macrophage lineage cells in their respective hosts, with disease outcomes being highly variable, in that they range from persistent asymptomatic infections to respiratory disease, reproductive failure (abortion), and even fatal hemorrhagic fever (4–7). EAV is the causative agent of equine viral arteritis (EVA) in horses, in which clinical signs can range from an asymptomatic infection to a flu-like illness in adult horses, abortion in pregnant mares, and interstitial pneumonia in neonatal foals (8, 9). Furthermore, in a variable percentage of stallions (10 to 70%), EAV can establish persistent infection in the reproductive tract, from which it is shed in semen for extended periods of time; carrier stallions are widely accepted to be the natural reservoir of the virus (9, 10). EAV infects equine endothelial cells, monocytes, macrophages, and a small subpopulation of CD3+ T cells (11–13). In addition, the virus can replicate in a number of other mammalian cell types (including some human cells), suggesting that it may be capable of using more than one receptor molecule to gain entry into cells (13).
In general, the process of viral entry into target cells is initiated by binding to a specific host cell receptor molecule(s) on the plasma membrane (14–18). This interaction is a major determinant of viral tropism and pathogenesis. Currently, the cellular receptor(s) for EAV is not known, although previous studies have implicated the involvement of a heparin-like molecule in binding to rabbit kidney (RK-13) cells (19, 20). Interestingly, a recent genome-wide association study (GWAS) identified a region in equine chromosome 11 (ECA11; positions 49572804 to 49643932) with potential involvement in EAV infection and pathogenesis (8). Several genes within this region (e.g., CXCL16, HRNE, RABEP1, ARRB2) have structural properties that could enable them to participate in either the cell surface attachment or endocytosis of EAV. However, pathway analysis using Ingenuity Pathway Analysis (Ingenuity Systems Inc., Redwood City, CA) software and the PANTHER classification system (www.pantherdb.org) revealed that one of the candidate receptor molecules, equine CXCL16 (EqCXCL16), has scavenger receptor properties in common with CD163, an entry receptor of PRRSV (11, 14, 21, 22). Although the molecules are not structurally identical (23), the utilization of functionally comparable membrane-associated proteins by EAV and PRRSV represents a potentially interesting parallel between these two very closely related viruses, and as such, we hypothesized that EqCXCL16 could be one of the cellular receptors for EAV. This equine molecule has not been studied extensively; however, there is a considerable amount of published information concerning human CXCL16 (huCXCL16) (24, 25), which is a member of the CXC chemokine family. The human variant of this protein (huCXCL16) possesses a single transmembrane domain along with an intracellular SH2 binding domain and is expressed in both membrane-bound and soluble forms (26, 27). While soluble huCXCL16 can function as a chemokine, the membrane-bound form has scavenger receptor activity for phosphatidylserine and oxidized lipoprotein (SR-PSOX) (26, 28, 29). huCXCL16 is also involved in viral infections, arthritis, atherosclerosis, and the metastasis of certain cancers (25, 26, 30, 31). The in silico analysis outlined in this report indicated that EqCXCL16 has a structural organization and functional properties very similar to those of its human counterpart, including the existence of membrane-bound and soluble forms. In this study, we unequivocally confirm that the transmembrane form of EqCXCL16 functions as a cellular receptor for initiating EAV infection in susceptible cell types.
MATERIALS AND METHODS
Cells.
Equine pulmonary artery endothelial cells (EECs) were maintained in Dulbecco's modified Eagle's medium (DMEM; Mediatech, Herndon, VA) with sodium pyruvate, 10% fetal bovine serum (FBS; HyClone Laboratories, Inc., Logan, UT), 100 U/ml of penicillin, 100 μg/ml streptomycin, and 200 mM l-glutamine (32–34). High-passage-number rabbit kidney cells (HP-RK-13 [KY] P399-409 cells; originally derived from CCL-37 cells [American Type Culture Collection {ATCC}, Manassas, VA]) and baby hamster kidney (BHK-21) cells (CCL-10; ATCC) were propagated in Eagle's minimal essential medium with 10% ferritin-supplemented bovine calf serum (HyClone Laboratories, Inc., Logan, UT), 100 U/ml of penicillin, and 100 μg/ml of streptomycin (Gibco, Carlsbad, CA). Human embryonic kidney (HEK-293T) cells (CRL-3216; ATCC) were propagated in DMEM with 10% ferritin-supplemented bovine calf serum (HyClone Laboratories, Inc., Logan, UT) and 100 U/ml of penicillin and 100 μg/ml streptomycin (Gibco, Carlsbad, CA).
Viruses.
Two strains of EAV, the virulent Bucyrus strain (VBS; VR-796; ATCC) (5, 6) and recombinant EAV VBS expressing mCherry (EAV sVBSmCherry) (35), were used in this study. Both viruses were propagated in EECs to generate high-titer working stocks as previously described (36, 37). Briefly, EECs infected with each virus were frozen at −80°C when a 90 to 100% cytopathic effect (CPE) was observed. Cell lysates were clarified by centrifugation (500 × g) at 4°C for 15 min, followed by ultracentrifugation (Beckman Coulter, Miami, FL) at 121,600 × g through a 20% sucrose cushion in NET buffer (150 mM NaCl, 5 mM EDTA, 50 mM Tris-HCl, pH 7.5) at 4°C for 4 h to pellet the virus. Purified preparations of each strain of EAV were resuspended in phosphate-buffered saline (PBS; pH 7.4) and frozen at −80°C. Virus stocks were titrated by standard plaque assay in RK-13 cells, and titers were expressed as the number of PFU per milliliter (38).
Horses.
Horses used for collection of blood samples were maintained at pasture on the University of Kentucky Department of Veterinary Science's Main Chance Farm, Lexington, KY. Blood samples were collected aseptically using Vacutainer tubes containing anticoagulant. This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Research Council (39). The animal protocol involving horses was approved by the University of Kentucky Institutional Animal Care and Use Committee (IACUC; protocol number 2013-1098).
Sequencing of equine CXCL16 gene.
DNA samples from a Thoroughbred horse (horse TB10) and a standardbred horse (horse ST22), identified for the susceptibility of their CD3+ T cells to in vitro infection with EAV, were isolated for sequencing. Briefly, genomic DNA (gDNA) was obtained from purified peripheral blood mononuclear cells (PBMCs) from each animal using a Puregene whole-blood extraction kit (Qiagen, Valencia, CA) following the manufacturer's instructions. DNA quality and concentration were assessed using a NanoDrop spectrophotometer (Thermo Scientific, Wilmington, DE) at an absorbance ratio of the optical density (OD) at 260 nm (OD260)/OD280. Approximately 6 μg of gDNA from each animal was sent to BGI Americas (Davis, CA) for library construction and sequencing. Libraries with 500-bp inserts were prepared and sequenced as paired-end reads on an Illumina HiSeq 2000 sequencer (2 times 100-bp sequences) to generate coverage of approximately 30 times. Reads were mapped to the horse genome reference sequence (Ecab 2.0) using CLC Workbench (version 8.0.1; CLC bio, Boston, MA). Consensus sequences for each horse were analyzed for the EqCXCL16 annotated gene using the reference sequence.
Cloning and expression of EqCXCL16 in Escherichia coli.
Primers for the cloning of native equine CXCL16 were designed by use of the Primer (version 3) program and the horse genome sequences obtained as part of this study. The sequence encoding the entire EqCXCL16 without the first 16 amino acid residues from the signal sequences was amplified from cDNA made from mRNA extracted from equine monocytes using a Smart cDNA synthesis kit (Clontech Laboratories Inc., Mountain View, CA). The cDNA amplification was carried out according to a standard laboratory PCR protocol using forward primer cx16-15F (5′-GCGCTCGAGGCGTTGCTGACTCTGCAAGG-3′) and reverse primer cx16-15R (5′-GCGGATCCGCACTGCCACTGTAACTGAT-3′) (IDT, Coralville, IA). For cloning purposes, the forward and the reverse primers were flanked with XhoI and BamHI restriction endonuclease recognition sequences, respectively, which are underlined in the sequences given above. The amplified EqCXCL16 fragment (amino acids [aa] 17 to 247) was run on a 1% agarose gel (E-Gel EX; Life Technologies, Grand Island, NY) and purified with a commercial kit (Zymoclean gel recovery kit; Zymo Research, Irvine, CA). The purified EqCXCL16 fragment was digested with the XhoI and BamHI restriction enzymes and ligated (Rapid DNA ligation kit; Thermo Scientific, Rockford, IL) into the bacterial expression vector pET15b (EMD Millipore Novagen, Temecula, CA) following digestion with the same restriction enzymes. The resultant plasmid construct (p15-16A) was used to transform E. coli NovaBlue (EMD Millipore Novagen, Temecula, CA) using a TransformAid kit (Thermo Scientific, Rockford, IL). Recombinant plasmid p15-16A (aa 17 to 247) was isolated from ampicillin-resistant clones using a ZR plasmid miniprep kit (Zymo Research, Irvine, CA). In order to express recombinant protein, the plasmid was transformed into E. coli BL21(DE3) cells (EMD Millipore Novagen, Temecula, CA). For large-scale EqCXCL16 protein (aa 17 to 247) production, 500-ml cultures of E. coli BL21(DE3)/p15-16A were grown overnight at 37°C in MagicMedia medium supplemented with 50 μg/ml ampicillin. Following centrifugation at 6,000 × g for 15 min, the cell pellet was resuspended in buffer A (50 mM sodium phosphate, 6 M guanidine-HCl, 300 mM NaCl, pH 7.0) and subjected to several short cycles of sonication to reduce viscosity. The lysate was centrifuged at 16,000 × g for 30 min to remove the debris. The His-tagged recombinant EqCXCL16 protein (aa 17 to 247) was purified from the supernatant by affinity chromatography using Talon Superflow metal affinity resin (Clontech Laboratories Inc., Mountain View, CA) in combination with a fast performance liquid chromatography apparatus (Amersham Pharmacia Biotech Inc., Piscataway, NJ). The columns were equilibrated and washed with buffer A, while proteins were eluted using buffer B (45 mM sodium phosphate, 5.4 M guanidine-HCl, 270 mM NaCl, 150 mM imidazole, pH 7.0). Eluted recombinant protein was dialyzed against PBS using Slide-A-Lyser dialysis cassettes (Thermo Scientific, Rockford, IL). The purity and integrity of recombinant EqCXCL16 (aa 17 to 247) were evaluated by subjecting it to electrophoresis on a 4 to 20% gradient gel-SDS followed by staining with Coomassie brilliant blue solution (Bio-Rad Laboratories, Hercules, CA). The protein concentration was determined with a bicinchoninic acid (BCA) protein assay (Thermo Scientific, Rockford, IL), using bovine serum albumin as the standard.
Generation of antibodies (Abs) to EqCXCL16.
Protein-specific rabbit antipeptide sera to EqCXCL16 were generated by immunizing rabbits with two synthetic peptides. Specifically, the nucleotide sequence of EqCXCL16 was used to deduce the amino acid sequence of the protein. Peptides were designed according to the predicted antigenic regions of EqCXCL16 using Thermo Scientific Antigen Profiler software (Thermo Scientific, Rockford, IL) and the Geneious (version 6.1.4) program (Biomatters Ltd., Auckland, New Zealand). One peptide (peptide A; 39-RFPSDSPPAVHIMKHFRK-56; Fig. 1A) was located within the amino-terminal ectodomain of the EqCXCL16 protein, whereas the other peptide (peptide B; 174-HSLVSGPEAGENQKQLKEHV-193; Fig. 1A) was located toward the end of the ectodomain. Adult New Zealand White rabbits were immunized with keyhole limpet hemocyanin (KLH)-conjugated synthetic peptide antigens according to a standard immunization protocol. Two New Zealand White rabbits were immunized per peptide (rabbits PA7505 and PA7506 for peptide A and rabbits PA7509 and PA7510 for peptide B). Each KLH-conjugated peptide (0.25 mg) was emulsified with Freund's complete adjuvant and administered into 4 quarter sites intramuscularly on day 1, followed by booster immunization with 0.1 mg of each peptide with incomplete Freund's adjuvant on days 14, 42, 56, and 103. Serum samples were collected on day 0 (preimmunization) and on days 28, 56, 72, and 117 postimmunization. A polyclonal antibody (pAb) against the whole EqCXCL16 molecule minus the first 16 signal sequences (aa 17 to 247)(Gp anti-EqCXCL16 pAb) was generated by immunization of guinea pigs with recombinant protein expressed in E. coli. Briefly, each guinea pig was immunized with 0.1 mg of recombinant EqCXCL16 protein antigen emulsified with Freund's complete adjuvant at 4 quarter sites intramuscularly on day 1, followed by booster immunizations with 50 μg of recombinant EqCXCL16 protein with incomplete Freund's adjuvant on days 21, 42, 62, and 90. Blood samples were collected on day 0 (preimmunization) and on days 56 and 104 postimmunization. The rabbit antipeptide sera and polyclonal guinea pig sera were initially tested by enzyme-linked immunosorbent assay (ELISA) using antigen consisting of either the respective peptides or the EqCXCL16 recombinant protein. The antisera were further characterized by indirect immunofluorescence assay (IFA) and Western blot (WB) analyses. The animal protocol involving rabbits and guinea pigs was approved by the Thermo Scientific, Rockford, IL, IACUC (NIH OLAW assurance number, A3669-01; USDA research license registration number, 23-R-0089; PHS assurance number, A3669-01).
FIG 1.

Amino acid sequence and schematic representation of the EqCXCL16 protein. (A) The amino acid sequence of the EqCXCL16 protein showing the signal peptide, amino-terminal ectodomain, transmembrane domain, and carboxyl-terminal cytoplasmic endodomain. The two antigenic regions that were targeted to generate EqXCXCL16 protein-specific rabbit antipeptide sera are boxed (peptide A, amino acids 39 to 56; peptide B, amino acids 174 to 193). (B) The EqCXCL16 protein is a type I transmembrane protein that is predicted to comprise an N-terminal chemokine domain, a glycosylated (Asn-143) mucin-like stalk, a transmembrane domain, and a carboxyl-terminal cytoplasmic domain.
ELISA.
Threefold serial dilutions (starting from 300 ng to 1 ng) of purified recombinant EqCXCL16 protein in 0.1 M bicarbonate/carbonate buffer, pH 9.6, were used to coat the 96-well ELISA plates (Corning, Tewksbury, MA). The plates were incubated overnight at 4°C and washed twice with wash buffer (PBS, pH 7.4; Life Technologies, Grand Island, NY) containing 0.05% Tween 20 (Bio-Rad, Hercules CA), and the reactions were blocked with 200 μl of blocking solution (5% nonfat dry milk in wash buffer) at room temperature (RT) for 1 h. Rabbit antipeptide sera against EqCXCL16 diluted 1:1,000 in blocking solution were added. The plates were then incubated for 1 h at RT. Then, the plates were washed four times in wash buffer and incubated with anti-rabbit IgG conjugated to horseradish peroxidase (HRP) for 1 h at RT. After washing four times in wash buffer, SureBlue tetramethylbenzidine microwell peroxidase substrate (Kirkegaard & Perry, Gaithersburg, MD) was added to each well and the plates were incubated for 30 min before the reaction was stopped with 100 μl of 0.18 M sulfuric acid (Mallinckrodt, Dublin, Ireland). The OD values at 450 and 650 nm were read using a Synergy H1MD microplate reader (BioTek Instruments Inc., Winooski, VT), with the OD at 650 nm of the reference sample being subtracted from the peak OD at 450 nm to eliminate the background.
Plasmids and transient transfection.
For expression of the EqCXCL16 protein in eukaryotic cells, a codon-optimized full-length EqCXCL16 sequence was commercially synthesized and cloned into the pJ609 plasmid, into which the puromycin resistance gene was incorporated, by DNA2.0 (Menlo Park, CA). This molecular construct was identified as pJ609-EqCXCL16 and used to transform E. coli DH10B cells (Life Technologies, Grand Island, NY). The recombinant plasmid DNA was purified using a commercial kit (Plasmid Plus maxiprep kit; Qiagen, Valencia, CA) for transfection of mammalian cells. The initial characterization of the rabbit antipeptide sera and the polyclonal guinea pig sera by IFA and WB analysis was performed in BHK-21 cells transiently transfected with plasmid pJ609-EqCXCL16 or pJ609-GFP (as a negative control) DNA. Briefly, BHK-21 cells were seeded either in 8-well Thermo Scientific Lab-Tek chamber slides (5 × 104 cells/well; Fisher Scientific, Pittsburgh, PA) or in 6-well plates (1 × 106 cells/well) and were transfected with 200 ng or 3 μg of plasmid DNA per well, respectively, using the Lipofectamine 3000 reagent (Life Technologies, Grand Island, NY) according to the manufacturer's recommendations. All IFA and WB assays were performed at 24 h posttransfection.
Establishment of cells stably expressing the EqCXCL16 protein (stable HEK-EqCXCL16 cells).
HEK-293T cells seeded in 6-well plates (2 × 106 cells/well) were transfected with 3 μg of plasmid pJ609-EqCXCL16 DNA using Lipofectamine 3000 (Life Technologies, Grand Island, NY) following the manufacturer's instructions. At 24 h posttransfection, the medium was replaced with fresh medium containing 4 μg/ml of puromycin (Clontech Laboratories Inc., Mountain View, CA) and the cells were incubated at 37°C in a CO2 incubator. This process of replacing the old culture medium with fresh medium was repeated every other day until only the puromycin-resistant colonies were selected. These puromycin-resistant cells were cloned by limiting dilution in 96-well plates and screened by IFA, after which clones showing the highest level of EqCXCL16 protein expression were frozen in commercial cell-freezing medium (Life Technologies, Grand Island, NY) and stored in liquid nitrogen until needed. At every 5th passage and up to the 50th serial passage, cells were analyzed by IFA using Gp anti-EqCXCL16 pAb to confirm the expression of EqCXCL16. All the experiments with HEK-293T cells expressing EqCXCL16 (HEK-EqCXCL16 cells) were performed within passage levels 5 to 10.
Confocal microscopy and IFA.
Stable HEK-EqCXCL16 cells or transiently transfected BHK-21 cells in 8-well Thermo Scientific Lab-Tek chamber slides were washed in cold PBS (pH 7.4) and fixed in 4% paraformaldehyde (PFA; Sigma-Aldrich, St. Louis, MO) for 30 min at RT. The cells were then stained for confocal microscopy and IFA as described elsewhere (40, 41). Briefly, following fixation, the cells were washed 5 times in ice-cold 10 mM glycine (Sigma-Aldrich, St. Louis, MO) in PBS, pH 7.4 (PBS-glycine), and were then permeabilized with 0.2% saponin (Sigma-Aldrich, St. Louis, MO) in PBS or left untreated with detergent where examination of surface staining was required. All cells were washed again in 10 mM PBS-glycine and blocked with 5% normal goat serum (MP Biomedicals, Santa Ana, CA) for 30 min at RT prior to incubation with specific primary antibodies (1:100 dilution) for 1 h at 37°C in a humidified chamber. After washing in 10 mM PBS-glycine, the cells were incubated with anti-rabbit or anti-guinea pig IgG secondary antibodies conjugated with Alexa Fluor 488 (AF488; 1:200 dilution) for 1 h at 37°C in a humidified chamber maintained in total darkness. After washing, the slides were mounted in Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA). The slides were observed either under a Leica TSP SP5 confocal microscope in an environmental chamber at the University of Kentucky HSRB imaging facility or with an inverted fluorescence microscope (ECLIPSE Ti; Nikon, Melville, NY).
SDS-PAGE and Western immunoblotting.
Cells were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer (Santa Cruz Biotechnology, Dallas, TX) containing Halt protease and phosphatase inhibitor cocktails (Thermo Scientific, Rockford, IL). The solubilized proteins were mixed with Pierce lane marker reducing 5× sample buffer containing 100 mM dithiothreitol (DTT; Thermo Scientific, Rockford, IL) and heated for 5 min at 95°C. Samples were resolved in an SDS-polyacrylamide gel (5% stacking gel and 12% resolving gel) at 200 V for 45 min and then transferred onto a polyvinylidene difluoride (PVDF) membrane at 100 V for 1 h using a Trans-Blot transfer system (Bio-Rad, Hercules, CA). The membrane was blocked with 5% nonfat milk powder (Bio-Rad, Hercules, CA) in Tris-buffered saline with Tween 20 (TBS-T; 10 mM Tris-HCl [pH 7.6], 150 mM NaCl, 0.1% Tween 20) for 1 h at RT and incubated with primary Abs (rabbit PA7506 anti-EqCXCL16 [1:500], rabbit PA7509 anti-EqCXCL16 [1:500], mouse monoclonal anti-EAV GP5 [monoclonal antibody {MAb} 6D10, 1:2,000], or guinea pig anti-EqCXCL16 polyclonal [1:1,000] Abs) diluted in 5% bovine serum albumin (Sigma-Aldrich, St. Louis, MO) in TBS-T overnight at 4°C. On the following day, the membranes were washed with TBS-T and then incubated with anti-rabbit, anti-mouse, or anti-guinea pig IgG, as appropriate, conjugated with HRP (1:3,000; Cell Signaling, Danvers, MA) for 1 h at RT. The membranes were washed again, and antibody binding was visualized with an enhanced chemiluminescence (ECL) detection system using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific, Rockford, IL).
Deglycosylation of EqCXCL16 protein.
EqCXCL16 proteins expressed in BHK-21 cells were deglycosylated using peptide-N-glycosidase F (PNGase F; New England BioLabs, Ipswich, MA) following the manufacturer's instructions. Briefly, BHK-21 cells transiently expressing EqCXCL16 were lysed in RIPA lysis buffer at 24 h posttransfection. Twenty micrograms of protein lysate and 1 μl of 10× glycoprotein denaturing buffer in a 10-μl total reaction volume were mixed, and the mixture was incubated at 95°C for 5 min. After they were briefly chilled, the denatured proteins were mixed with 2 μl of 10× GlycoBuffer 2 (New England Biolabs, Ipswich, MA), 2 μl of 10% NP-40, and 1 μl of PNGase F enzyme in a total volume of 20 μl. The mixture was incubated overnight at 37°C. The deglycosylated protein lysates were analyzed by Western blotting using rabbit PA7509 anti-EqCXCL16 Ab.
Isolation of PBMCs and flow cytometry.
Isolation of PBMCs from the peripheral blood of horses (n = 9) was performed as described previously (12) with some modifications (IACUC protocol number 2013-1098; University of Kentucky, Lexington, KY). Briefly, blood (20 ml) was collected aseptically using Vacutainer tubes containing 0.1 ml of 15% EDTA solution (Covidien, Dublin, Ireland). PBMCs were isolated from the buffy coat fraction by centrifugation through Ficoll-Paque Plus (Amersham Biosciences, Piscataway, NJ) at 500 × g for 30 min at 25°C. The PBMC layer was collected and washed twice with Hanks balanced salt solution (pH 7.4; Life Technologies, Grand Island, NY) by centrifugation at 300 × g for 10 min to eliminate the platelets. The cells were resuspended in RPMI 1640 medium with 2 mM GlutaMAX medium and 25 mM HEPES (Life Technologies, Grand Island, NY) without FBS and counted using a Countess automated cell counter (Life Technologies, Grand Island, NY). PBMCs (2 × 106) were infected with EAV sVBSmCherry at a multiplicity of infection (MOI) of 2.0 in a minimal volume (150 μl) of serum-free RPMI 1640 medium in a treated cell culture plate (24 well) and incubated at 37°C for 1 h in a CO2 (5%) incubator. The negative control was mock-infected PBMCs, which were cultured under conditions identical to those described above. After 1 h of incubation, prewarmed complete RPMI 1640 medium (1 ml) supplemented with 10% FBS, 50 μM 2-mercaptoethanol, 100 U/ml penicillin, and 100 μg/ml streptomycin was added to each culture, and the plates were reincubated at 37°C in 5% CO2 for another 36 h. PBMCs were harvested from the plates using a cell scraper, and up to 1 × 106 cells in 100 μl of flow buffer (PBS supplemented with 10% normal goat serum and 0.1% sodium azide) were incubated on ice for 30 min with rabbit PA7509 anti-EqCXCL16 Ab (1:2,000 dilution). The cells were washed and probed with anti-rabbit IgG conjugated to Alexa Fluor 488 (Life Technologies, Grand Island, NY) for 20 min. The cells were then washed and incubated with anti-CD14 (clone 105; Bettina Wagner, Cornell University, Ithaca, NY) conjugated to Alexa Fluor 647 for 30 min on ice. After washing, the cells were resuspended in flow buffer and acquisition was performed on a Sony Synergy SY3200 cell sorter system (Sony Biotechnology, San Jose, CA). Cells were gated for monocytes and then evaluated with two-color plots using WinList software (Verity Software House, Topsham, ME). When monocytes were infected with EAV sVBSmCherry, two-color plots of infected monocytes were generated using mCherry-gated cells.
Enrichment of equine CD14+ cells.
CD14+ monocytes were enriched indirectly with magnetic microbeads according to the manufacturer's protocol (Miltenyi Biotec, Bergisch Gladbach, Germany). Briefly, PBMCs were blocked with 5% normal mouse serum (Innovative Research, Novi, MI) and then incubated with anti-equine CD14 clone 105 conjugated with Alexa Fluor 647 (Bettina Wagner, Cornell University, Ithaca, NY) for 30 min. After washing with magnetically activated cell sorting (MACS) buffer, the cells were incubated with anti-Alexa Fluor 647 microbeads for 20 min. The cells were then washed with MACS buffer, applied to an LS column, and washed three times, and the bound CD14+ cells were eluted with MACS buffer in the absence of the magnet, washed, and counted using a Countess automated cell counter (Life Technologies, Carlsbad, CA). The purity of the CD14+ cells was confirmed by flow cytometric analysis. These cells were used for the experiment in which the cell surface EqCXCL16 protein was blocked using polyclonal guinea pig antiserum (Gp anti-EqCXCL16 pAb).
Blocking cell surface EqCXCL16 by Gp anti-EqCXCL16 pAb.
Approximately 1 × 104 stable HEK-293T cell transfectants expressing the EqCXCL16 protein (HEK-EqCXCL16 cells) were seeded per well of a 96-well plate and incubated for 18 h at 37°C before being treated with 2-fold serial dilutions (1:10 to 1:160) of guinea pig anti-EqCXCL16 pAb. After 2 h of incubation at 37°C, the cells were infected with EAV sVBSmCherry at an MOI of 1.0 in the presence of anti-EqCXCL16 antibody. At 12 h postinfection (hpi), the cells were analyzed for mCherry expression using an inverted fluorescence microscope. Similar antibody blocking experiments were conducted on enriched equine CD14+ monocytes; these were seeded in 96-well plates (1 × 104 cells/well) and incubated for 24 h at 37°C. Subsequently, these cells were incubated with Gp anti-EqCXCL16 pAb for another 2 h at 37°C before they were infected with EAV sVBSmCherry. At 24 hpi, the cells were analyzed for mCherry expression with an inverted fluorescence microscope.
siRNA-mediated downregulation of EqCXCL16 expression in HEK-EqCXCL16 cells.
Stable HEK-EqCXCL16 cells seeded (1 × 105 cells/well) in 24-well plates were transfected with small interfering RNA (siRNA) directed against EqCXCL16 using the Lipofectamine RNAiMax reagent (Life Technologies, Grand Island, NY). These consisted of the following two Silencer Select siRNA duplexes, designed using the GeneAssist custom siRNA builder (Life Technologies, Grand Island, NY): siRNA1, for which the sense sequence was 5′-GGC CGG AGA AAA UCA GAA ATT-3′ and the antisense sequence was 5′-UUU CUG AUU UUC UCC GGC Ctc-3′, and siRNA2, for which the sense sequence was 5′-AGA ACC UGA UUC ACG CGA ATT-3′ and the antisense sequence was 5′-UUC GCG UGA AUC AGG UUC UTG-3′). A commercially available nonspecific scrambled siRNA duplex was used as a negative control (Life Technologies, Grand Island, NY). At 30 h posttransfection, cells were infected with EAV sVBSmCherry. At 18 hpi, cells were stained with Gp anti-EqCXCL16 pAb and analyzed by fluorescence microscopy for the expression of mCherry and the EqCXCL16 protein. In a separate experiment, cells stably expressing the EqCXCL16 protein were transfected with siRNA duplexes targeting EqCXCL16 mRNA or scrambled siRNA. At 30 h posttransfection, cells were infected with EAV sVBSmCherry, and after 18 h of additional incubation, the cells were lysed in RIPA lysis buffer and analyzed for viral nonstructural protein 1 (nsp-1) expression, as well as for EqCXCL16 expression, by a WB assay.
Virus overlay protein-binding assay (VOPBA) and far-Western blotting.
Approximately 100 μg of total protein lysate from naive HEK-293T cells or stable HEK-EqCXCL16 cells was separated in 12% SDS-polyacrylamide gels and transferred onto a PVDF membrane for far-WB analysis following a modification of a previously published protocol (42). The bound proteins were then denatured and gradually renatured on the membrane by sequential incubation with 6 M, 3 M, 1 M, and 0.1 M guanidine-HCl in freshly prepared AC buffer (100 mM NaCl, 20 mM Tris [pH 7.5], 10% glycerol, 0.5 mM EDTA, 0.1% Tween 20, 2% nonfat dry milk, 5 mM DTT) for 30 min at RT or with AC buffer only in the absence of guanidine-HCl overnight at 4°C. The membrane was blocked with 5% nonfat dry milk in TBS-T (Tris-buffered saline with 0.1% Tween 20), overlaid with purified wild-type EAV VBS (15 μg/ml), and incubated overnight at 4°C. On the next day, the membrane was washed vigorously (3 washes of 10 min each) and incubated with mouse monoclonal Ab (anti-GP5; MAb 6D10) directed against the EAV GP5 envelope glycoprotein. Monoclonal antibody binding was detected by the ECL detection system using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific, Rockford, IL).
Labeling of EAV with biotin and EAV binding assay.
EAV VBS was purified by ultracentrifugation (at 121,600 × g for 4 h) through a 20% sucrose cushion, and the protein concentration was determined using a BCA protein assay (Thermo Scientific, Rockford, IL). About 2 mg of purified EAV was biotinylated using EZ-Link sulfo-NHS-biotin (Thermo Scientific, Rockford, IL) following the manufacturer's protocol. Excess unbound biotin was removed by filtering through a Zeba desalt spin column (molecular weight cutoff, 7,000; Thermo Scientific, Rockford, IL) equilibrated in PBS (pH 7.4). Naive HEK-293T and HEK-EqCXCL16 cells were washed in cold PBS (pH 7.4) and removed from the culture dish using a nonenzymatic cell dissociation solution (Cellstripper; Mediatech Inc., Manassas, VA). Cells were resuspended in cold PBS (pH 7.4) containing 2% FBS (PBS-F), centrifuged at 1,000 × g for 5 min at 4°C, and incubated with biotinylated EAV at an MOI of 100 on ice for 2 h in total darkness. Excess EAV was removed by washing 3 times in cold PBS-F. Subsequently, the cells were stained with streptavidin-fluorescein isothiocyanate (FITC; 1:100) and incubated at 4°C for 30 min in total darkness. Cells were washed in PBS-F at 1,000 × g for 5 min at 4°C, transferred onto glass microscope slides using a Shandon CytoSpin III cytocentrifuge with a Shandon single cytofunnel with white filter cards (Thermo Scientific, Rockford, IL), and incubated with DAPI solution to permit the visualization of cell nuclei. Cells were then analyzed using a Nikon inverted fluorescence microscope, and the percentage of cells bound to EAV was calculated.
Antibodies.
Mouse EAV anti-GP5 and mouse EAV anti-nsp-1 MAbs (MAb 6D10 and MAb 12A4, respectively) have been described previously (43, 44). Goat anti-rabbit IgG (H+L)-HRP and goat anti-mouse IgG (H+L)-HRP were purchased from Cell Signaling Technology (Danvers, MA). Goat anti-guinea pig IgG (H+L)-HRP, goat anti-rabbit IgG (H+L) conjugated to Alexa Fluor 488, and goat anti-guinea pig IgG conjugated to AF488 were purchased from Life Technologies (Grand Island, NY). Streptavidin conjugated to FITC was purchased from Southern Biotech (Birmingham, AL). The monoclonal anti-equine CD14 Ab (clone 105) was purchased from Bettina Wagner (Cornell University, Ithaca, NY) (45).
Nucleotide sequence accession numbers.
The sequences of the complete EqCXCL16 genes from two horses have been submitted to the Sequence Read Archive and can be found under BioSample/experiment accession numbers SAMN03838869/SRX1097022 and SAMN03838868/SRX1097492.
RESULTS
Sequencing of equine CXCL16 gene.
Previous studies in our laboratory have implicated an association between EqCXCL16 and EAV during infection of CD3+ T cells from some horses (11). Therefore, we sequenced the complete EqCXCL16 gene from two horses and found that the nucleotide sequences were identical. The EqCXCL16 gene is located on equine chromosome 11 (ECA11), spanning nucleotide positions 49746891 to 49750292, and is predicted to comprise 6 exons encoding a 247-amino-acid protein with an approximate molecular mass of 27.2 kDa. As no information on the structure of the EqCXCL16 protein was available, we analyzed these sequences using the Simple Modular Architecture Research Tool (SMART), a web resource (http://smart.embl.de/) for the identification of proteins, the annotation of protein domains, and exploration of protein domain architecture. This analysis suggested that EqCXCL16 has an organization very similar to that of its human counterpart (huCXCL16), in that it is predicted to comprise a signal sequence (aa 1 to 23) along with three structural domains that include an N-terminal ectodomain (aa 24 to 202), a transmembrane domain (aa 203 to 224), and a C-terminal domain (aa 225 to 247) (Fig. 1A and B). Although EqCXCL16 possesses only 54% predicted amino acid sequence identity with huCXCL16, the projected similarities in overall domain structure indicate that the N-terminal region probably consists of a chemokine domain followed by a glycosylated mucin-like stalk (Fig. 1B). Furthermore, the equine protein also likely possesses scavenger receptor activity for phosphatidylserine and oxidized lipoprotein (SR-PSOX)/CXC. Following analysis, the sequences from the two horses were used in subsequent experiments to design a codon-optimized variant of EqCXCL16 for transfection of mammalian cell lines, to predict B-cell epitopes (peptides A and B) for antiserum production, to design PCR amplification primers for cloning of EqCXCL16, and to design siRNAs.
Generation of EqCXCL16-specific antipeptide and polyclonal antisera.
Since reagents to detect EqCXCL16 were not available, we generated a specific antiserum by immunization of rabbits with EqCXCL16 peptide A (aa residues 39 to 56) or peptide B (aa residues 174 to 193) and by inoculation of guinea pigs with recombinant EqCXCL16 antigen (aa residues 17 to 247) produced in E. coli. These reagents were screened by ELISA using E. coli-produced recombinant EqCXCL16 protein as the coating antigen. Although detectable antibodies against EqCXCL16 were present in serum collected from all rabbits at 56 days postimmunization, the titers against peptide B were higher than those against peptide A (Fig. 2A). Serum collected prior to the first immunization (prebleed serum) produced no detectable reactions against the recombinant EqCXCL16 antigen (Fig. 2A). These results were corroborated using IFA and WB analysis on BHK-21 cells transiently transfected with pJ609-EqCXCL16 (Fig. 2B, rows a and b, and C, panels a and c). IFA and WB analysis results similar to those described above were obtained with serum from EqCXCL16-immunized guinea pigs (Fig. 2B, row c, and C, panel e). On the basis of bioinformatics predictions of an N-linked glycosylation site at amino acid residue 143 (Asn-143), it is considered likely that differential glycosylation is responsible for the double band (approximate molecular mass, 30 kDa) observed with all preparations of the EqCXCL16 antiserum (Fig. 2C, panels a, c, and e). This was confirmed by the disappearance of the upper band from the doublet (Fig. 2D) observed following peptide-N-glycosidase F (PNGase F) treatment of an EqCXCL16 protein lysate produced in BHK-21 cells.
FIG 2.


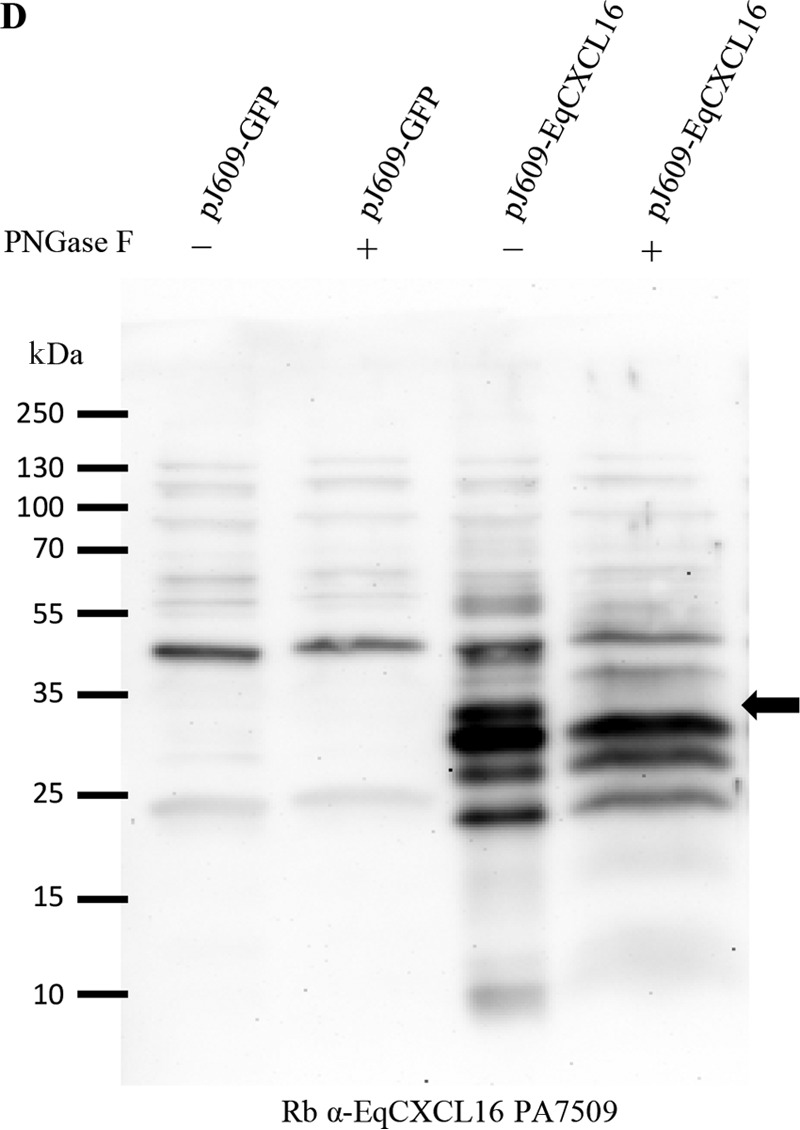
Characterization of EqCXCL16-specific rabbit antipeptide sera and polyclonal guinea pig sera by ELISA, IFA, and WB analysis. (A) An in-house ELISA was used to characterize the rabbit antipeptide sera generated against EqCXCL16 (Rb α-EqCXCL16). Various concentrations (1 to 300 ng) of recombinant EqCXCL16 expressed from E. coli were coated on a 96-well plate and then probed with antisera generated against peptides A and B (rabbit PA7506 anti-EqCXCL16 and rabbit PA7509 anti-EqCXCL16, respectively). Broken lines, prebleed (Pb) sera from the respective rabbits. (B) BHK-21 cells were transiently transfected with plasmids containing EqCXCL16 cDNA, and at 24 h posttransfection, cells were fixed in 4% PFA. Cells were stained with rabbit antipeptide sera against EqCXCL16 protein (row a, rabbit PA7506 serum against peptide A; row b, rabbit PA7509 serum against peptide B) or Gp anti-EqCXCL16 pAb (row c) or the corresponding prebleed sera. Cells were then stained with secondary antibody conjugated to AF488 and analyzed by inverted fluorescence microscopy. (C) BHK-21 cells that had been transfected with either pJ609-GFP (control) or pJ609-EqCXCL16 were lysed in RIPA cell lysis buffer and were analyzed in the WB assay using rabbit PA7506 anti-EqCXCL16 Ab (against peptide A) (a), rabbit PA7506 prebleed serum (b), rabbit PA7509 anti-EqCXCL16 serum (against peptide B) (c), rabbit PA7509 prebleed serum (d), Gp anti-EqCXCL16 pAb (e), and guinea pig prebleed serum (f). Arrows, the presence (a, c, e) or absence (b, d, f) of the EqCXCL16 protein in the WB membrane. (D)Protein lysates from BHK-21 cells transfected with either plasmid pJ609-GFP (control) or plasmid pJ609-EqCXCL16 were deglycosylated using PNGase F. The protein lysates were analyzed by a WB assay using rabbit PA7509 anti-EqCXCL16 antiserum. Arrow, EqCXCL16 bands.
Role of EqCXCL16 in EAV infection of equine monocytes.
As a prerequisite to determining if EqCXCL16 plays a role in EAV infection, experiments were conducted to investigate if this molecule was expressed on equine monocytes, as these are one of the major host cell targets for this virus. Flow cytometric analysis following dual staining of equine PBMCs with rabbit anti-EqCXCL16 antibody (peptide B, rabbit PA7509) and antibody against monocyte-specific marker CD14 demonstrated that both antibodies bound to only 36% of the cells, suggesting that EqCXCL16 is expressed by only a subpopulation of equine monocytes (Fig. 3A). The specificity of these reactions was confirmed by the fact that less than 1% of equine monocytes (Fig. 3A) bound to prebleed serum from the same rabbit (rabbit PA7509). In subsequent experiments involving infection of equine PBMCs with EAV sVBSmCherry followed by staining at 36 hpi with rabbit anti-EqCXCL16 and anti-CD14 antibodies, it was found that while mCherry expression was detectable in just 15% of equine monocytes (CD14+ and EAV-positive cells), 71% of these infected monocytes expressed EqCXCL16 on their surface (CD14+, EAV-positive, and EqCXCL16-positive cells) (Fig. 3B). This suggests a very strong association between EqCXCL16 surface expression and infection of equine CD14+ monocytes by EAV.
FIG 3.

Equine monocytes expressing CXCL16 were susceptible to EAV infection. (A) PBMCs (2 × 106) were stained with anti-CD14 antibody and rabbit PA7509 anti-EqCXCL16 serum (right) or prebleed serum (left). The plots were generated using WinList software with cells gated for monocytes on the basis of their scatter. The upper right quadrant in the right panel shows that 36% of equine CD14+ monocytes express EqCXCL16 on their surface. (B) Equine PBMCs (2 × 106) were infected with EAV sVBSmCherry at an MOI of 2.0 and harvested after 36 h in culture. The cells were stained for anti-CD14 and rabbit PA7509 anti-EqCXCL16 peptide Ab. The cells were gated for monocytes (upper left) and plotted for EqCXCL16 and CD14 expression (upper right). Approximately 34% of the cells were positive for CD14 (CD14+) and EqCXCL16 (EqCXCL16+) (green boxes). All the cells that were mCherry positive (15%; green box, lower left) were also plotted for EqCXCL16 and CD14 expression (lower right), with the quadrants being in the same location that they were in the upper right panel. Of these, 15% of mCherry-positive cells, approximately 71% of the cells, also expressed EqCXCL16. SC, scatter.
Effect of pretreatment with Gp anti-EqCXCL16 polyclonal antisera on EAV infection of purified equine monocytes.
Since EAV was found to preferentially infect EqCXCL16-expressing CD14+ monocytes (EqCXCL16+ CD14+ cells), we investigated the effect of the Gp anti-EqCXCL16 pAb treatment on infection of equine monocytes with EAV sVBSmCherry. Equine monocytes enriched (>85% purity) by selection with anti-CD14 antibody-conjugated magnetic microbeads were incubated with Gp anti-EqCXCL16 pAb for 2 h at 37°C prior to infection with EAV sVBSmCherry. Evaluation of mCherry expression at 24 hpi by fluorescence microscopy demonstrated a significant reduction in the number of Gp anti-EqCXCL16 pAb-treated infected CD14+ cells compared to the number of infected CD14+ cells incubated without antibody or with preimmunization guinea pig serum (Fig. 4). Interestingly, this observation is consistent with EqCXCL16 acting as an initial receptor-binding molecule for EAV.
FIG 4.

Effect of polyclonal guinea pig antibody-mediated blocking on EAV infection of purified CD14+ monocytes. Equine monocytes were purified, and 105 cells were plated in a 96-well plate in complete RPMI 1640 medium. After 24 h of incubation in RPMI 1640 medium, cells were further incubated either without antibody (a) or with Gp anti-EqCXCL16 antiserum (b) for 2 h at 37°C. As a negative control, monocytes were also treated with prebleed guinea pig serum from the same guinea pig (c). Cells were then infected with EAV sVBSmCherry, and at 24 hpi, cells were analyzed by inverted immunofluorescence microscopy for the evaluation of mCherry expression.
Role of EqCXCL16 as the putative EAV receptor.
Preliminary results from our laboratory demonstrated that EAV infects only a very small minority of the HEK-293T cell population (<3%). To investigate if constitutive expression of the EqCXCL16 molecule could increase the susceptibility of HEK-293T cells to infection with EAV, we first established a stable cell line expressing EqCXCL16. The HEK-293T cells were transfected with pJ609-EqCXCL16 plasmid DNA, and stable transfectants (HEK-EqCXCL16 cells) were selected using puromycin and analyzed for EqCXCL16 expression using confocal microscopy and WB analysis with Gp anti-EqCXCL16 pAb. Confocal microscopy revealed that the stably transfected HEK-293T cells expressed EqCXCL16 on the outer plasma membrane. This conclusion was arrived at on the basis of the fact that the cells were not permeabilized by detergent treatment during the staining procedure (Fig. 5A, row a). In contrast, HEK-EqCXCL16 cells did not show any staining with prebleed serum from the same guinea pig (Fig. 5A, row b), demonstrating the specificity of the Gp anti-EqCXCL16 pAb reagent. Furthermore, mock-transfected HEK-293T cells also failed to react with Gp anti-EqCXCL16 pAb (Fig. 5A, row c). These results were confirmed by WB analysis, where a band having an approximate molecular mass of 30 kDa, which is the molecular mass predicted for EqCXCL16, was observed, using the Gp anti-EqCXCL16 pAb, in cell lysates from HEK-EqCXCL16 cells but not those from naive HEK-293T cells (Fig. 5B, panel a). In contrast, there was no visible band (molecular mass, 30 kDa) in HEK-EqCXCL16 cell lysates stained with the prebleed guinea pig serum (Fig. 5B, panel b). Expression of EqCXCL16 was maintained for more than 50 serial passages (screened by IFA; data not shown), suggesting that these equine sequences are tolerated in HEK-293T cells and not subject to strong negative selective pressure. Following initial characterization, HEK-EqCXCL16 cells were infected with EAV sVBSmCherry at an MOI of 1.0. In contrast to naive HEK-293T cells, almost all cells in the population were found to express mCherry at 12 hpi (Fig. 6A, rows a and b). Similar results were seen with EAV sVBSmCherry used at an MOI of 0.1 and 2.0 (data not shown). Also, the data revealed that there was a significant difference in the intensity of mCherry expression in stable HEK-EqCXCL16 cells compared to that in naive HEK-293T cells, suggesting an enhanced level of viral gene expression in the stable cells (Fig. 6B). To confirm the effect of EqCXCL16 on EAV gene expression, naive and stable cells were infected with EAV, and at 18 hpi cells were analyzed by either IFA or Western blot assay using virus-specific antibodies. Indeed, the IFA data using an anti-nsp-1 MAb (MAb 12A4) demonstrated a significant increase in the level of expression of EAV nonstructural protein nsp-1 in the stable HEK-EqCXCL16 cells (Fig. 7A, row a) compared to that in naive HEK-293T cells infected with EAV sVBSmCherry (Fig. 7A, row b). The WB analysis demonstrated that despite similar levels of loading of cell lysate material in terms of actin amounts (Fig. 7B), EAV nsp-1 was readily detectable in infected HEK-EqCXCL16 cells but not in naive HEK-293T cells (Fig. 7B, panels a and b). Collectively, these results demonstrated that the constitutive expression of EqCXCL16 transformed HEK-293T cells from predominantly nonpermissive to permissive with respect to infection with EAV.
FIG 5.

Establishment of the stable HEK-293T cell line expressing the EqCXCL16 protein (HEK-EqCXCL16 cells). HEK-293T cells were transfected with plasmid pJ609-EqCXCL16 and selected by puromycin treatment. (A) Stable HEK-EqCXCL16 cells were surface stained with Gp anti-EqCXCL16 Ab (row a) or guinea pig prebleed serum (row b) as the primary antibody and analyzed by confocal microscopy to confirm the expression of the EqCXCL16 protein. Naive HEK-293T cells were also stained with Gp anti-EqCXCL16 Ab (row c). (B) Stable HEK-EqCXCL16 cells were lysed in RIPA cell lysis buffer, and equal amounts of lysates were analyzed by a WB assay using Gp anti-EqCXCL16 Ab (a) or guinea pig prebleed serum (b). Arrows, the presence (a) or absence (b) of the EqCXCL16 protein in the WB membrane.
FIG 6.

Effect of EqCXCL16 expression on EAV infection of stable HEK-EqCXCL16 cells. (A) Naive HEK-293T cells (row a) or stable HEK-EqCXCL16 cells expressing the EqCXCL16 protein (row b) were infected with EAV sVBSmCherry at an MOI of 1.0, and at 12 hpi, the cells were fixed with 4% PFA. Cells were analyzed by inverted immunofluorescence microscopy for mCherry expression. The same fields of cells were also analyzed by phase-contrast microcopy. Compared to the level of mCherry expression in naive HEK-293T cells, a significant increase in the level of mCherry expression was found in the stable HEK-EqCXCL16 cells (middle column). (B) The intensity of mCherry expression was quantitated using Nikon NIS-Elements AR (version 4.13.00) software. The data represent the means ± standard deviations from 3 independent experiments, and data were considered significant at a P value of <0.001 by Student's t test (***).
FIG 7.

Effect of EqCXCL16 expression on EAV gene expression. (A) Stable HEK-EqCXCL16 cells (row a) or naive HEK-293T cells (row b) were infected with EAV sVBSmCherry at an MOI of 1.0. At 18 hpi, cells were fixed with 4% PFA and stained with an anti-nsp-1 MAb (MAb 12A4) conjugated with AF488. Cells were analyzed by inverted fluorescence microscopy to evaluate the expression of EAV nsp-1. There was a significant increase in the level of EAV nsp-1 expression in stable HEK-EqCXCL16 cells compared to that in naive HEK-293T cells (middle panels and merged panels). (B) EAV sVBSmCherry-infected naive HEK-293T cells or stable HEK-EqCXCL16 cells were lysed in RIPA lysis buffer and analyzed for expression of the nsp-1 gene using an anti-nsp-1 MAb (MAb 12A4). Arrows, the location of the EAV nsp-1 (a) or β-actin (b) protein.
Anti-EqCXCL16 polyclonal antibodies block EAV infection of HEK-EqCXCL16 cells.
To investigate the potential for a direct association between EAV and EqCXCL16, HEK-EqCXCL16 cells were preincubated for 2 h at 37°C with 2-fold serial dilutions (from 1:10 to 1:160) of Gp anti-EqCXCL16 pAb prior to infection with EAV sVBSmCherry at an MOI of 1.0. Cells treated with similar 2-fold dilutions of prebleed guinea pig sera were considered negative controls. The outcome of this experiment confirmed that Gp anti-EqCXCL16 pAbs blocked EAV sVBSmCherry infection in HEK-EqCXCL16 cells, while untreated HEK-EqCXCL16 cells could still be successfully infected with the virus (Fig. 8a to f). The results also demonstrated that the inhibitory effect of pAb on EAV infection was dose dependent, since after a certain dilution (1:160), the Gp anti-EqCXCL16 pAb no longer blocked EAV infection (Fig. 8f). On the other hand, prebleed guinea pig sera could not block EAV sVBSmCherry infection of HEK-EqCXCL16 cells (Fig. 8g to k) even at the highest concentration tested (1:10 dilution). The Gp anti-EqCXCL16 pAb mediated blocking of cell surface EqCXCL16, and the resultant inhibition of EAV infection strongly suggests that EqCXCL16 is a receptor for EAV binding.
FIG 8.

Effect of polyclonal antibody-mediated blocking of EqCXCL16 on EAV infection. Stable HEK-EqCXCL16 cells were treated with either DMEM (a), 2-fold serial dilutions (from 1:10 to 1:160) of Gp anti-EqCXCL16 pAb (b to f), or prebleed guinea pig sera (g to k) for 2 h at 37°C. Cells were infected with EAV sVBSmCherry at an MOI of 1.0 in the presence of Gp anti-EqCXCL16 pAb or prebleed guinea pig sera. At 12 hpi, cells were fixed and analyzed by inverted fluorescence microscopy for the evaluation of mCherry expression. In the absence of antibody, stable cells infected with EAV sVBSmCherry expressed mCherry (a), whereas antibody treatment inhibited mCherry expression (b to e). The prebleed sera, however, did not inhibit mCherry expression (g to k).
Blocking EAV replication by siRNA-mediated downregulation of EqCXCL16 cell surface expression.
HEK-EqCXCL16 cells were transfected with Silencer Select siRNA duplexes (siRNA1 or siRNA2) designed to inhibit the expression of EqCXCL16. Nontransfected cells or HEK-EqCXCL16 cells transfected with a scrambled siRNA served as controls (Fig. 9). At 48 h posttransfection, immunofluorescence staining of cells with Gp anti-EqCXCL16 pAb revealed that both siRNA1 and siRNA2 were able to significantly reduce the surface expression of EqCXCL16 (Fig. 9A, rows c and d) compared with that for nontransfected and scrambled siRNA-transfected HEK-EqCXCL16 cells (Fig. 9A, rows a and b). More importantly, when HEK-EqCXCL16 cells were transfected with either siRNA1 or siRNA2 for 30 h prior to infection with EAV sVBSmCherry, they became completely refractory to virus infection, and this was clearly evident by the almost complete absence of detectable mCherry expression (Fig. 9A, rows c and d). In contrast, mCherry expression was readily detectable in equivalently infected nontransfected and scrambled siRNA-transfected HEK-EqCXCL16 cells (Fig. 9A, rows a and b). These results were further confirmed by WB analysis using equivalent amounts of cell lysates from HEK-EqCXCL16 cells transfected with either siRNA1 or scrambled siRNA and then subsequently infected (at 30 h posttransfection) with EAV sVBSmCherry (Fig. 9B). In addition to an apparent reduction in EqCXCL16 expression, transfection with siRNA1 almost completely eliminated EAV nsp-1 compared with the amount of EAV nsp-1 in the scrambled siRNA-transfected controls (Fig. 9B, panels a to c). Similar results were obtained with EAV-infected HEK-EqCXCL16 cells transfected with siRNA2 (data not shown). These experiments with siRNA provide additional support for the concept that the presence of EqCXCL16 is essential during early events in the EAV infection process and that this molecule can act as a putative host cell surface receptor.
FIG 9.

Effect of siRNA-mediated knockdown of EqCXCL16 on EAV infection. (A) Stable HEK-EqCXCL16 cells were transfected with two different siRNAs (siRNA1[row c] and siRNA2 [row d]) directed against EqCXCL16 mRNA. Mock-transfected cells (row a) or cells transfected with scrambled siRNA (row b) were considered negative controls. At 30 h posttransfection, cells were infected with EAV sVBSmCherry at an MOI of 1.0 for 18 h. Cells were fixed, stained with Gp anti-EqCXCL16 pAb, and analyzed using an inverted fluorescence microscope for the evaluation of mCherry and EqCXCL16 expression. Green staining reflects the surface expression of EqCXCL16, while red staining indicates mCherry expression. In the presence of EqCXCL16 (rows a and b), mCherry expression can be visualized. In contrast, when cells were transfected with siRNAs, mCherry expression was suppressed (rows c and d). (B) Stable HEK-EqCXCL16 cells were transfected with siRNA1, and at 30 h posttransfection, the cells were infected with EAV sVBSmCherry. After 18 h of incubation, the cells were lysed and analyzed by a WB assay using nsp-1 (a), β-actin (b), and rabbit anti-EqCXCL16 (c) Abs. Arrows, locations of nsp-1 (a), β-actin (b), and EqCXCL16 (c).
Binding of EAV with EqCXCL16 in vitro.
If EqCXCL16 functions as a primary receptor for EAV rather than an accessory protein, there should be detectable binding with the viral envelope glycoproteins. This possibility was investigated using a combination of the virus overlay protein-binding assay (VOPBA) and far-WB techniques. In the far-WB technique, protein-protein interactions are detected in vitro by electrophoresis followed by immobilization of the prey protein on a PVDF membrane and then incubation of the membrane with purified bait protein. Binding interactions between these molecules can be inferred if the bait protein (detected by protein-specific antisera) is found to occupy the same position on the membrane as the prey protein. In our experiments, total lysates from HEK-EqCXCL16 cells and naive HEK-293T cells were transferred onto a PVDF membrane (prey molecules) after protein separation by SDS-PAGE and sequentially denatured and renatured on the membrane by treatment with different concentrations of AC buffer containing guanidine-HCl. The membranes were incubated with purified EAV (the bait protein), which was detected using anti-GP5 MAb 6D10, which specifically recognizes EAV envelope protein GP5. The anti-GP5 MAb reacted strongly with immobilized cell lysates from HEK-EqCXCL16 cells but not those from naive HEK-293T cells (Fig. 10a). Moreover, the anti-GP5 MAb bound to the same position (Fig. 10a) on the membrane where the EqCXCL16 band was detected (30 kDa). For comparison, the migration of EqCXCL16 detected by the anti-EqCXCL16 pAb was shown in the same membrane after anti-GP5 MAb was stripped off and the membrane was reprobed with anti-EqCXCL16 pAb (Fig. 10c). To exclude the possibility of nonspecific interactions between the anti-GP5 MAb and cell lysate proteins, a control experiment in which the incubation step with purified EAV was omitted was performed. In this case, no reactivity was detected at a position corresponding to the migration of a 30-kDa protein (Fig. 10b). These data establish the fact that there is direct binding between the virus particle and the EqCXCL16 protein.
FIG 10.

Analysis of a direct in vitro interaction between purified EAV and EqCXCL16 protein by a combination of VOPBA and far-WB analysis. Stable HEK-EqCXCL16 or naive HEK-293T cell lysates were separated on a 12% SDS-polyacrylamide gel and transferred onto a PVDF membrane. The proteins in the membrane were denatured and renatured using sequentially decreasing concentrations of guanidine-HCl. The membranes were blocked and incubated either with purified EAV VBS at a concentration of 15 μg/ml in protein-binding buffer (a) or with protein-binding buffer only without purified EAV VBS (b). After they were washed, the membranes were incubated with anti-GP5 MAb 6D10 and developed using the ECL method. (a) Binding of EAV VBS to the EqCXCL16 protein (arrow). (c) Antibodies were stripped off the membrane shown in panel a and reprobed with Gp anti-EqCXCL16. As indicated by the arrow in panel c, EqCXCL16 was detected at the same position on the membrane where EAV GP5 was detected in panel a.
EqCXCL16 can act as an EAV binding receptor rather than an accessory molecule.
To investigate if EqCXCL16 can function as a receptor for EAV, a binding assay was performed at 4°C to prevent viral internalization using naive HEK-293T and HEK-EqCXCL16 cells. Stable as well as naive HEK-293T cells were incubated with biotinylated EAV VBS (MOI of 100) at 4°C for 2 h, streptavidin-FITC was added, and the cells were visualized by inverted fluorescence microscopy. It was found that EAV binding was significantly higher in stable HEK-EqCXCL16 cells than naive HEK-293T cells (Fig. 11A). The data revealed that expression of EqCXCL16 produced a significant (P < 0.01) increase in the level of binding of biotinylated EAV VBS to HEK-293T cells (Fig. 11B). This indicated that EqCXCL16 could act as a binding receptor for EAV rather than as an accessory molecule.
FIG 11.

Role of EqCXCL16 in the attachment of EAV to host cells. Equal numbers (2 × 106) of naive HEK-293T cells or stable HEK-EqCXCL16 cells were washed and resuspended in cold PBS (pH 7.4) in 2% FBS (PBS-F) and then incubated with biotinylated EAV-VBS on ice for 2 h in the dark. After adsorption, excess EAV was removed by washing in cold PBS-F, and the cells were then stained with streptavidin-FITC. After 3 washings, the cells were resuspended in PBS-F, 50 μl of cells was cytospun onto a slide, and DAPI solution was added to the cells. (A) Cells were analyzed using an inverted fluorescence microscope. The green dots in the IFA images reflect the binding of EAV-VBS. Compared to the level of binding seen in row a, row b shows a significant reduction in the level of EAV VBS binding to naive HEK-293T cells, as indicated by fewer green dots. (B) A total of 300 cells were counted using Nikon NIS-Elements AR (version 4.13.00) software, and the percentage of stable HEK-EqCXCL16 cells or naive HEK-293T cells that showed EAV binding was calculated. Data represent the means ± standard deviations, and data were considered significant at a P value of <0.01 by Student's t test (**).
DISCUSSION
Viruses utilize multiple different attachment factors, receptors, and entry mediators to gain entry into their target host cells (46). Although it has been demonstrated that complex interactions between the major and minor envelope proteins determine EAV tropism (12), the host cell molecules targeted by these viral proteins have not been identified. All that is known about the early events associated with EAV infection is that virus yields are reduced but not eliminated by pretreatment of host cells with heparinase (19, 20) and that the virus probably enters via classical, pH-dependent, clathrin pit-mediated endocytosis rather than by an endocytosis-independent mechanism (46–48). In this report, we present evidence that the transmembrane form of EqCXCL16 can function as a binding receptor for EAV. Completion of these experiments required the development and characterization of new reagents, including EqCXCL16-specific antisera, and establishment of a stable HEK-293T cell line (HEK-EqCXCL16 cells) that constitutively expressed the transmembrane form of the protein. The EAV construct encoding mCherry as a reporter gene (EAV sVBSmCherry) has been shown to have the same phenotypic characteristics as the parental strain EAV VBS in relation to virus growth and cellular tropism (35).
Equine monocytes are one of the primary target cells during natural EAV infection, and they play a significant role in the pathogenesis of EVA (37). However, studies in our laboratories have demonstrated that only a subpopulation of CD14+ monocytes become infected with EAV (L. Chelvarajan, Y. Y. Go, R. F. Cook, S. P. Mondal, S. Sarkar, P. J. Henney, F. Marti, D. W. Horohov, P. J. Timoney, and U. B. R. Balasuriya, submitted for publication) (12). On the basis of the results of this study, it is apparent that a majority of EAV-infected monocytes include a subpopulation that expresses EqCXCL16 (CD14+ CXCL16+). Furthermore, when enriched CD14+ equine monocytes were preincubated with Gp anti-EqCXCL16 pAb, EAV infection was significantly blocked, as evidenced by a reduction in the number of cells expressing the mCherry protein. Collectively, these results indicate that EqCXCL16 plays an important role during the initial stage of EAV infection of the susceptible CD14+ monocyte. This conclusion was strongly supported by evidence obtained with HEK-293T cells demonstrating that the constitutive expression and translocation of EqCXCL16 to the plasma membrane (determined by confocal microscopy) significantly enhanced the susceptibility of these human cells to infection with EAV. Moreover, this enhanced susceptibility in HEK-EqCXCL16 cells was effectively eliminated both by preincubation with EqCXCL16-specific polyclonal antisera and by transfection with siRNAs designed to downregulate expression of this equine protein. Although these results demonstrate that EqCXCL16 is important early in the EAV infection process, they do not prove that the molecule functions as a cellular receptor. Subsequent experimental data based on the results of VOPBA and far-Western comparative blot analysis of cell lysates prepared from HEK-EqCXCL16 and naive HEK-293T cells showed that purified EAV associates directly with EqCXCL16 even when immobilized on a PVDF membrane. The potential biological significance of this direct binding interaction was further strengthened by the fact that larger amounts of biotinylated EAV remained bound to HEK-EqCXCL16 cells than to naive HEK-293T cells when the cells were incubated at 4°C. Taken together, these experimental results are consistent with the fact that the transmembrane form of EqCXCL16 functions as an EAV receptor rather than an accessory protein that helps with virus adsorption to the cell surface. Studies performed on heparinase-treated cells (19, 20) suggest that an association with heparin is involved in infection of cells with EAV, although this probably functions as an attachment factor mediating nonspecific interactions that facilitate the concentration of virus particles on the cell surface prior to actual receptor binding. It is possible that the background levels of biotinylated EAV VBS binding at 4°C observed in the naive HEK-293T cells were the result of interactions with heparin sulfate.
Although EqCXCL16 may be important for EAV entry in some cell populations, it is almost certainly not the only receptor used by this virus. EAV has a relatively broad host cell tropism, in that it can infect a wide variety of common laboratory cell lines, including but not exclusively baby hamster kidney (BHK-21), African green monkey kidney (Vero, MA-104), rabbit kidney (RK-13), and human kidney cells (13). Obviously, these nonequine species-derived cell lines do not express EqCXCL16. Furthermore, not all CD14+ equine monocytes that can be infected with EAV express detectable levels of EqCXCL16, and infection of the equine monocyte population is only partially blocked by preincubation with Gp anti-EqCXCL16 pAb. These results suggest that even in the case of equine cells, EAV can use a molecule(s) other than EqCXCL16 as an entry receptor(s). It should be noted that the use of multiple receptors among the arteriviruses is not without precedent, as at least six different cellular molecules have been implicated as receptors for PRRSV (14, 49–53). These include sialoadhesin, heparan sulfate, CD151, CD163, dendritic cell-specific intercellular adhesion molecule 3 grabbing nonintegrin (DC-SIGN; CD209), and vimentin, although the first two probably function as attachment factors rather than specific cellular receptors.
To the best of our knowledge, there have been no other descriptions of CXCL16 being used as a virus host cell receptor. Although both HIV and the severe acute respiratory syndrome coronavirus have been reported to interact specifically with human CXCL16, these interactions influence pathogenesis rather than determine host cell attachment (26, 54). While utilization of the CXCL16 protein is currently unique to EAV, the use of scavenger receptors as host cell receptors is not. For example, studies suggest that CD163 can act as a receptor for both PRRSV and African swine fever virus (55, 56). Interestingly, both CD163 and CXCL16 are type 1 membrane proteins expressed in monocytes, macrophages, and endothelial cells (57, 58). The transmembrane form of CD163 is cleaved by the disintegrin-like metalloproteinases ADAM10 and ADAM17 (28, 59, 60). Similarly, human CXCL16 is also cleaved by ADAM10 to release soluble chemokine. However, the overall structure of these molecules varies considerably, with CXCL16 being classified as a class G scavenger receptor on the basis of the domain properties outlined above, whereas CD163 is a class I scavenger receptor consisting of proline-serine-threonine-rich (PST) domains coupled to multiple cysteine-rich domains (23).
In summary, unequivocal evidence based on EqCXCL16 expression in a subpopulation of virus-susceptible CD14+ monocytes coupled with results from a comprehensive series of experiments involving protein-specific antiserum and siRNA reagents conducted in HEK-EqCXCL16 cells confirm that EAV uses EqCXCL16 as a host cell receptor. At this stage, it is not known if EqCXCL16 acts as a primary receptor, if it can function alone, or if additional host cell-specified molecules are required. It is likely that heparin sulfate may be involved as an attachment factor enabling the concentration of EAV particles on the cell surface. Although EqCXCL16 is clearly a host cell receptor, it is almost certainly not the only receptor used by this virus.
ACKNOWLEDGMENTS
We thank Jennifer Strange and Greg Bauman, Flow Cytometry Core Facility, Department of Microbiology, Immunology and Molecular Genetics, College of Medicine, University of Kentucky (UK), for their assistance with flow cytometry.
This work was supported by Agriculture and Food Research Initiative competitive grant no. 2013-68004-20360 from the United States Department of Agriculture, National Institute of Food and Agriculture (USDA-NIFA).
REFERENCES
- 1.Snijder EJ, Meulenberg JJ. 1998. The molecular biology of arteriviruses. J Gen Virol 79(Pt 5):961–979. doi: 10.1099/0022-1317-79-5-961. [DOI] [PubMed] [Google Scholar]
- 2.Snijder EJ, Kikkert M, Fang Y. 2013. Arterivirus molecular biology and pathogenesis. J Gen Virol 94:2141–2163. doi: 10.1099/vir.0.056341-0. [DOI] [PubMed] [Google Scholar]
- 3.Snijder EJ, Kikkert M. 2013. Arteriviruses, p 859–879. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Cavanagh D. 1997. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch Virol 142:629–633. [PubMed] [Google Scholar]
- 5.Bryans JT, Crowe ME, Doll ER, McCollum WH. 1957. Isolation of a filterable agent causing arteritis of horses and abortion by mares; its differentiation from the equine abortion (influenza) virus. Cornell Vet 47:3–41. [PubMed] [Google Scholar]
- 6.Bryans JT, Doll ER, Knappenberger RE. 1957. An outbreak of abortion caused by the equine arteritis virus. Cornell Vet 47:69–75. [PubMed] [Google Scholar]
- 7.Plagemann PG, Moennig V. 1992. Lactate dehydrogenase-elevating virus, equine arteritis virus, and simian hemorrhagic fever virus: a new group of positive-strand RNA viruses. Adv Virus Res 41:99–192. doi: 10.1016/S0065-3527(08)60036-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Balasuriya UB. 2014. Equine viral arteritis. Vet Clin North Am Equine Pract 30:543–560. doi: 10.1016/j.cveq.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 9.Timoney PJ, McCollum WH, Roberts AW, Murphy TW. 1986. Demonstration of the carrier state in naturally acquired equine arteritis virus infection in the stallion. Res Vet Sci 41:279–280. [PubMed] [Google Scholar]
- 10.Timoney PJ, McCollum WH. 1993. Equine viral arteritis. Vet Clin North Am Equine Pract 9:295–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Go YY, Bailey E, Cook DG, Coleman SJ, Macleod JN, Chen KC, Timoney PJ, Balasuriya UB. 2011. Genome-wide association study among four horse breeds identifies a common haplotype associated with in vitro CD3+ T cell susceptibility/resistance to equine arteritis virus infection. J Virol 85:13174–13184. doi: 10.1128/JVI.06068-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Go YY, Zhang J, Timoney PJ, Cook RF, Horohov DW, Balasuriya UB. 2010. Complex interactions between the major and minor envelope proteins of equine arteritis virus determine its tropism for equine CD3+ T lymphocytes and CD14+ monocytes. J Virol 84:4898–4911. doi: 10.1128/JVI.02743-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Balasuriya UB, Go YY, MacLachlan NJ. 2013. Equine arteritis virus. Vet Microbiol 167:93–122. doi: 10.1016/j.vetmic.2013.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Calvert JG, Slade DE, Shields SL, Jolie R, Mannan RM, Ankenbauer RG, Welch SK. 2007. CD163 expression confers susceptibility to porcine reproductive and respiratory syndrome viruses. J Virol 81:7371–7379. doi: 10.1128/JVI.00513-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howitt J, Anderson CW, Freimuth P. 2003. Adenovirus interaction with its cellular receptor CAR. Curr Top Microbiol Immunol 272:331–364. [DOI] [PubMed] [Google Scholar]
- 16.Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. 2003. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crocker PR, Mucklow S, Bouckson V, McWilliam A, Willis AC, Gordon S, Milon G, Kelm S, Bradfield P. 1994. Sialoadhesin, a macrophage sialic acid binding receptor for haemopoietic cells with 17 immunoglobulin-like domains. EMBO J 13:4490–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramos-Castaneda J, Imbert JL, Barron BL, Ramos C. 1997. A 65-kDa trypsin-sensible membrane cell protein as a possible receptor for dengue virus in cultured neuroblastoma cells. J Neurovirol 3:435–440. doi: 10.3109/13550289709031189. [DOI] [PubMed] [Google Scholar]
- 19.Asagoe T, Inaba Y, Jusa ER, Kouno M, Uwatoko K, Fukunaga Y. 1997. Effect of heparin on infection of cells by equine arteritis virus. J Vet Med Sci 59:727–728. doi: 10.1292/jvms.59.727. [DOI] [PubMed] [Google Scholar]
- 20.Lu Z, Sarkar S, Zhang J, Balasuriya UB. 6 January 2016. Conserved arginine residues in the carboxyl terminus of the equine arteritis virus E protein may play a role in heparin binding but may not affect viral infectivity in equine endothelial cells. Arch Virol. doi: 10.1007/s00705-015-2733-3. [DOI] [PubMed] [Google Scholar]
- 21.Mi H, Muruganujan A, Thomas PD. 2013. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res 41:D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mi H, Thomas P. 2009. PANTHER pathway: an ontology-based pathway database coupled with data analysis tools. Methods Mol Biol 563:123–140. doi: 10.1007/978-1-60761-175-2_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prabhudas M, Bowdish D, Drickamer K, Febbraio M, Herz J, Kobzik L, Krieger M, Loike J, Means TK, Moestrup SK, Post S, Sawamura T, Silverstein S, Wang XY, El Khoury J. 2014. Standardizing scavenger receptor nomenclature. J Immunol 192:1997–2006. doi: 10.4049/jimmunol.1490003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petit SJ, Chayen NE, Pease JE. 2008. Site-directed mutagenesis of the chemokine receptor CXCR6 suggests a novel paradigm for interactions with the ligand CXCL16. Eur J Immunol 38:2337–2350. doi: 10.1002/eji.200838269. [DOI] [PubMed] [Google Scholar]
- 25.Jung Y, Kim JK, Shiozawa Y, Wang J, Mishra A, Joseph J, Berry JE, McGee S, Lee E, Sun H, Wang J, Jin T, Zhang H, Dai J, Krebsbach PH, Keller ET, Pienta KJ, Taichman RS. 2013. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun 4:1795. doi: 10.1038/ncomms2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Landro L, Damas JK, Halvorsen B, Fevang B, Ueland T, Otterdal K, Heggelund L, Froland SS, Aukrust P. 2009. CXCL16 in HIV infection—a link between inflammation and viral replication. Eur J Clin Invest 39:1017–1024. doi: 10.1111/j.1365-2362.2009.02207.x. [DOI] [PubMed] [Google Scholar]
- 27.Ludwig A, Weber C. 2007. Transmembrane chemokines: versatile ‘special agents' in vascular inflammation. Thromb Haemost 97:694–703. [PubMed] [Google Scholar]
- 28.Abel S, Hundhausen C, Mentlein R, Schulte A, Berkhout TA, Broadway N, Hartmann D, Sedlacek R, Dietrich S, Muetze B, Schuster B, Kallen KJ, Saftig P, Rose-John S, Ludwig A. 2004. The transmembrane CXC-chemokine ligand 16 is induced by IFN-gamma and TNF-alpha and shed by the activity of the disintegrin-like metalloproteinase ADAM10. J Immunol 172:6362–6372. doi: 10.4049/jimmunol.172.10.6362. [DOI] [PubMed] [Google Scholar]
- 29.Matloubian M, David A, Engel S, Ryan JE, Cyster JG. 2000. A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat Immunol 1:298–304. doi: 10.1038/79738. [DOI] [PubMed] [Google Scholar]
- 30.Ruth JH, Haas CS, Park CC, Amin MA, Martinez RJ, Haines GK III, Shahrara S, Campbell PL, Koch AE. 2006. CXCL16-mediated cell recruitment to rheumatoid arthritis synovial tissue and murine lymph nodes is dependent upon the MAPK pathway. Arthritis Rheum 54:765–778. doi: 10.1002/art.21662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lehrke M, Millington SC, Lefterova M, Cumaranatunge RG, Szapary P, Wilensky R, Rader DJ, Lazar MA, Reilly MP. 2007. CXCL16 is a marker of inflammation, atherosclerosis, and acute coronary syndromes in humans. J Am Coll Cardiol 49:442–449. doi: 10.1016/j.jacc.2006.09.034. [DOI] [PubMed] [Google Scholar]
- 32.Balasuriya UB, Snijder EJ, Heidner HW, Zhang J, Zevenhoven-Dobbe JC, Boone JD, McCollum WH, Timoney PJ, MacLachlan NJ. 2007. Development and characterization of an infectious cDNA clone of the virulent Bucyrus strain of equine arteritis virus. J Gen Virol 88:918–924. doi: 10.1099/vir.0.82415-0. [DOI] [PubMed] [Google Scholar]
- 33.MacLachlan NJ, Balasuriya UB, Rossitto PV, Hullinger PA, Patton JF, Wilson WD. 1996. Fatal experimental equine arteritis virus infection of a pregnant mare: immunohistochemical staining of viral antigens. J Vet Diagn Invest 8:367–374. doi: 10.1177/104063879600800316. [DOI] [PubMed] [Google Scholar]
- 34.Balasuriya UBR, Heidner HW, Davis NL, Wagner HM, Hullinger PJ, Hedges JF, Williams JC, Johnston RE, Wilson WD, Liu IK, MacLachlan NJ. 2002. Alphavirus replicon particles expressing the two major envelope proteins of equine arteritis virus induce high level protection against challenge with virulent virus in vaccinated horses. Vaccine 20:1609–1617. doi: 10.1016/S0264-410X(01)00485-6. [DOI] [PubMed] [Google Scholar]
- 35.Mondal SP, Cook RF, Chelvarajan RL, Henney PJ, Timoney PJ, Balasuriya UB. 28 December 2015. Development and characterization of a synthetic infectious cDNA clone of the virulent Bucyrus strain of equine arteritis virus expressing mCherry (red fluorescent protein). Arch Virol. doi: 10.1007/s00705-015-2633-6. [DOI] [PubMed] [Google Scholar]
- 36.Hedges JF, Demaula CD, Moore BD, McLaughlin BE, Simon SI, MacLachlan NJ. 2001. Characterization of equine E-selectin. Immunology 103:498–504. doi: 10.1046/j.1365-2567.2001.01262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore BD, Balasuriya UB, Hedges JF, MacLachlan NJ. 2002. Growth characteristics of a highly virulent, a moderately virulent, and an avirulent strain of equine arteritis virus in primary equine endothelial cells are predictive of their virulence to horses. Virology 298:39–44. doi: 10.1006/viro.2002.1466. [DOI] [PubMed] [Google Scholar]
- 38.McCollum WH. 1970. Vaccination for equine viral arteritis, p 143–151. Proceedings of the 2nd International Conference on Equine Infectious Diseases Karger, New York, NY. [Google Scholar]
- 39.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 40.Sarkar S, Balasuriya UB, Horohov DW, Chambers TM. 2015. Equine herpesvirus-1 suppresses type-I interferon induction in equine endothelial cells. Vet Immunol Immunopathol 167:122–129. doi: 10.1016/j.vetimm.2015.07.015. [DOI] [PubMed] [Google Scholar]
- 41.Sarkar S, Balasuriya UB, Horohov DW, Chambers TM. 2016. The neuropathogenic T953 strain of equine herpesvirus-1 inhibits type-I IFN mediated antiviral activity in equine endothelial cells. Vet Microbiol 183:110–118. doi: 10.1016/j.vetmic.2015.12.011. [DOI] [PubMed] [Google Scholar]
- 42.Wu Y, Li Q, Chen XZ. 2007. Detecting protein-protein interactions by far Western blotting. Nat Protoc 2:3278–3284. doi: 10.1038/nprot.2007.459. [DOI] [PubMed] [Google Scholar]
- 43.Wagner HM, Balasuriya UB, MacLachlan NJ. 2003. The serologic response of horses to equine arteritis virus as determined by competitive enzyme-linked immunosorbent assays (c-ELISAs) to structural and non-structural viral proteins. Comp Immunol Microbiol Infect Dis 26:251–260. doi: 10.1016/S0147-9571(02)00054-1. [DOI] [PubMed] [Google Scholar]
- 44.Balasuriya UB, Rossitto PV, DeMaula CD, MacLachlan NJ. 1993. A 29K envelope glycoprotein of equine arteritis virus expresses neutralization determinants recognized by murine monoclonal antibodies. J Gen Virol 74(Pt 11):2525–2529. doi: 10.1099/0022-1317-74-11-2525. [DOI] [PubMed] [Google Scholar]
- 45.Kabithe E, Hillegas J, Stokol T, Moore J, Wagner B. 2010. Monoclonal antibodies to equine CD14. Vet Immunol Immunopathol 138:149–153. doi: 10.1016/j.vetimm.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 46.Smith AE, Helenius A. 2004. How viruses enter animal cells. Science 304:237–242. doi: 10.1126/science.1094823. [DOI] [PubMed] [Google Scholar]
- 47.Marsh M, Helenius A. 2006. Virus entry: open sesame. Cell 124:729–740. doi: 10.1016/j.cell.2006.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nitschke M, Korte T, Tielesch C, Ter-Avetisyan G, Tunnemann G, Cardoso MC, Veit M, Herrmann A. 2008. Equine arteritis virus is delivered to an acidic compartment of host cells via clathrin-dependent endocytosis. Virology 377:248–254. doi: 10.1016/j.virol.2008.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duan X, Nauwynck HJ, Favoreel HW, Pensaert MB. 1998. Identification of a putative receptor for porcine reproductive and respiratory syndrome virus on porcine alveolar macrophages. J Virol 72:4520–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang YW, Dryman BA, Li W, Meng XJ. 2009. Porcine DC-SIGN: molecular cloning, gene structure, tissue distribution and binding characteristics. Dev Comp Immunol 33:464–480. doi: 10.1016/j.dci.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Delputte PL, Van Breedam W, Delrue I, Oetke C, Crocker PR, Nauwynck HJ. 2007. Porcine arterivirus attachment to the macrophage-specific receptor sialoadhesin is dependent on the sialic acid-binding activity of the N-terminal immunoglobulin domain of sialoadhesin. J Virol 81:9546–9550. doi: 10.1128/JVI.00569-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jusa ER, Inaba Y, Kouno M, Hirose O. 1997. Effect of heparin on infection of cells by porcine reproductive and respiratory syndrome virus. Am J Vet Res 58:488–491. [PubMed] [Google Scholar]
- 53.Kim JK, Fahad AM, Shanmukhappa K, Kapil S. 2006. Defining the cellular target(s) of porcine reproductive and respiratory syndrome virus blocking monoclonal antibody 7G10. J Virol 80:689–696. doi: 10.1128/JVI.80.2.689-696.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang YP, Zhang RW, Chang WS, Wang YY. 2010. Cxcl16 interact with SARS-CoV N protein in and out cell. Virol Sin 25:369–374. doi: 10.1007/s12250-010-3129-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Van Gorp H, Van Breedam W, Delputte PL, Nauwynck HJ. 2008. Sialoadhesin and CD163 join forces during entry of the porcine reproductive and respiratory syndrome virus. J Gen Virol 89:2943–2953. doi: 10.1099/vir.0.2008/005009-0. [DOI] [PubMed] [Google Scholar]
- 56.Sanchez-Torres C, Gomez-Puertas P, Gomez-del-Moral M, Alonso F, Escribano JM, Ezquerra A, Dominguez J. 2003. Expression of porcine CD163 on monocytes/macrophages correlates with permissiveness to African swine fever infection. Arch Virol 148:2307–2323. doi: 10.1007/s00705-003-0188-4. [DOI] [PubMed] [Google Scholar]
- 57.Graversen JH, Svendsen P, Dagnaes-Hansen F, Dal J, Anton G, Etzerodt A, Petersen MD, Christensen PA, Moller HJ, Moestrup SK. 2012. Targeting the hemoglobin scavenger receptor CD163 in macrophages highly increases the anti-inflammatory potency of dexamethasone. Mol Ther 20:1550–1558. doi: 10.1038/mt.2012.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fukumoto N, Shimaoka T, Fujimura H, Sakoda S, Tanaka M, Kita T, Yonehara S. 2004. Critical roles of CXC chemokine ligand 16/scavenger receptor that binds phosphatidylserine and oxidized lipoprotein in the pathogenesis of both acute and adoptive transfer experimental autoimmune encephalomyelitis. J Immunol 173:1620–1627. doi: 10.4049/jimmunol.173.3.1620. [DOI] [PubMed] [Google Scholar]
- 59.Kneidl J, Loffler B, Erat MC, Kalinka J, Peters G, Roth J, Barczyk K. 2012. Soluble CD163 promotes recognition, phagocytosis and killing of Staphylococcus aureus via binding of specific fibronectin peptides. Cell Microbiol 14:914–936. doi: 10.1111/j.1462-5822.2012.01766.x. [DOI] [PubMed] [Google Scholar]
- 60.Scholz F, Schulte A, Adamski F, Hundhausen C, Mittag J, Schwarz A, Kruse ML, Proksch E, Ludwig A. 2007. Constitutive expression and regulated release of the transmembrane chemokine CXCL16 in human and murine skin. J Investig Dermatol 127:1444–1455. doi: 10.1038/sj.jid.5700751. [DOI] [PubMed] [Google Scholar]