

ABSTRACT
In the oral epithelium, peripheral stores of Epstein-Barr virus (EBV) are transmitted from infiltrating B cells to epithelial cells. Once the virus is transmitted to epithelial cells, the highly permissive nature of this cell type for lytic replication allows virus amplification and exchange to other hosts. Since the initial transfer of EBV from B cells to epithelial cells requires transitioning of the B-cell to a state that induces virus reactivation, we hypothesized that there might be epithelium-specific signals that allow the infiltrating B cells to sense the appropriate environment to initiate reactivation and begin this exchange process. We previously found that the epithelium-specific miR-200 family of microRNAs promotes EBV lytic replication. Here we show that there are high levels of miR-200 family members in oral and tonsillar epithelia and in saliva. Analysis of cultured oral epithelial cells (OKF6) showed that they actively secrete membrane vesicles (exosomes) that are enriched with miR-200 family members. Coculturing of EBV-positive B cells with OKF6 cells induced viral reactivation. Further, treatment of EBV-positive B cells with OKF6 cell-derived membrane vesicles promoted reactivation. Using a cell system that does not naturally express miR-200 family members, we found that enforced expression of a miR-200 family member produced membrane vesicles that were able to induce the lytic cascade in EBV-positive B cells. We propose that membrane vesicles secreted by oral and tonsillar epithelial cells may serve as a tissue-specific environmental cue that initiates reactivation in B cells, promoting the transfer of virus from peripheral B-cell stores to the oral epithelium to facilitate virus amplification and exchange to other hosts.
IMPORTANCE Epstein-Barr virus (EBV) is an important human pathogen that is causally associated with several lymphomas and carcinomas. The switch from latency to the lytic cycle is critical for successful host infection and for EBV pathogenesis. Although the EBV lytic cycle can be triggered by certain agents in vitro, the mechanisms that signal reactivation in vivo are poorly understood. We previously reported that endogenously expressed miR-200 family members likely play a role in facilitating the lytic tendencies of EBV in epithelial cells. Here we show that membrane vesicles secreted from oral epithelial cells contain miR-200 family members and that they can be transmitted to proximal EBV-positive B cells, where they trigger reactivation. We propose that this intercellular communication pathway may serve as a sensor mechanism for infiltrating B cells to recognize an appropriate environment to initiate reactivation, thereby allowing the exchange of virus to the oral epithelium.
INTRODUCTION
Membrane vesicles (MVs), such as exosomes and microvesicles, can be actively released by cells into the extracellular environment, where they can facilitate intercellular communication. Exosomes are a class of small membrane vesicles (30 to 150 nm in diameter) of endocytic origin that are secreted from most cell types, including epithelial cells and lymphocytes, under both physiological and pathological conditions (1–8). Exosomes are composed of proteins, lipids, and nucleic acids that are derived from their cells of origin. Through the delivery of biologically active components from the cells of origin to neighboring and/or distant cells, exosomes are able to modulate many biological activities, such as tumorigenesis, immunosurveillance, cell proliferation, and angiogenesis (1, 9–12). MicroRNAs (miRNAs) are important exosomal cargo that facilitate signaling pathway alterations in recipient cells (2, 13–17). EBV-encoded BART miRNAs are selectively enriched in EBV-positive B-cell-derived exosomes (13, 14) and have been shown to inhibit NLRP3 inflammasome-mediated interleukin 1β (IL-1β) production (14) and to suppress the expression of the CXCL11 gene (13), an immunoregulatory gene involved in antiviral activity and lymphomagenesis.
Epstein-Barr virus (EBV) causes a lifelong infection, with more than 90% of the adult population worldwide being persistent carriers (18). EBV primarily utilizes B cells and epithelial cells in its infection cascade (18, 19). As a bona fide human tumor virus, EBV plays an etiological role in a number of lymphoid and epithelial malignancies, including non-Hodgkin's and Hodgkin's lymphoma, nasopharyngeal carcinoma, and gastric carcinoma (18). Similar to other herpesviruses, EBV has a biphasic infection cycle that includes a replicative phase (lytic cycle) and a latency phase (19, 20). Following initial infection, EBV preferentially exists in host B cells in a state of latency in which no viral production occurs. Nevertheless, under certain circumstances, latency in B cells can be disrupted and the virus can enter into a productive viral replication phase (19).
The switch from latency to the lytic cycle in B cells is a fundamental component of the virus infection cycle that is critical for virus persistence and pathogenesis. Chemicals that alter certain intracellular regulatory pathways, such as phorbol ester, calcium ionophores, histone deacetylase inhibitors (e.g., butyrate), and DNA-demethylating agents (e.g., 5-aza-cytidine), can be used to artificially induce reactivation in latently infected cell lines (21–24). Possibly more physiologically relevant than these chemical inducers, ectopic transforming growth factor β (TGF-β) or B-cell receptor (BCR) engagement can induce reactivation in tissue culture (25–27). This information gives clues about some of the regulatory pathways that facilitate reactivation, but even the significance of TGF-β or BCR engagement in triggering virus production in vivo has been a subject of debate (19, 28).
Probably the most significant site of virus production is the oral cavity, where salivary virions are produced and spread from host to host through deep kissing or other salivary exchange mechanisms (29). In contrast to the case with B cells, infection of epithelial tissues tends to lead to productive lytic replication rather than latency, and we (30) and Ellis-Connell et al. (31) have shown that this may result in part through a mechanism involving the epithelium-specific microRNA-200 family of microRNAs which inhibit expression of a key immediate early gene repressor, ZEB1 (32, 33). The propensity of epithelial cells to support lytic replication makes good sense from the standpoint of host-to-host transmission because it presumably facilitates the production of high viral titers in the oral epithelium that can be shed to the saliva.
The Hutt-Fletcher group (34, 35) has shown a cross-tissue infection bias for virus produced in B cells and epithelial cells, with virus produced in B cells preferentially infecting epithelial cells and vice versa. This speaks to the likely importance of cross-tissue virus exchange in the EBV infection cycle. The storage site of EBV in the persistently infected host is considered to be latently infected B cells (18). The transfer of virus from this storage vehicle to the oral epithelium likely occurs through reactivating B cells that infiltrate the oral epithelium or otherwise in proximity to the oral epithelium. The higher propensity of virus produced in B cells to infect epithelial tissue presumably helps facilitate this process. Nevertheless, we posited that the rarity of “random” reactivation in B cells would present a substantial inefficiency in this B-cell-to-epithelial-cell exchange process. We hypothesized that epithelium-derived transmissible agents capable of inducing reactivation in B cells might favor this B-cell-to-epithelial-cell exchange. Such an agent would act to trigger B-cell reactivation when the appropriate epithelial milieu is detected. Since we knew that the epithelium-specific miR-200 family could induce reactivation in B cells (30, 31) and since microRNAs are commonly found in shed membrane vesicles, we hypothesized that epithelium-derived membrane vesicles might provide such a sensor mechanism. Here we provide evidence that epithelium-derived membrane vesicles can induce reactivation in latently infected B cells and that this is likely at least partly due to the presence of miR-200 family members.
MATERIALS AND METHODS
Collection of clinical specimens.
Buccal epithelium and saliva samples were collected from two healthy participants in the department of psychiatry and behavioral sciences at the Tulane medical school in accordance with a protocol approved by the Institutional Review Board (IRB) of Tulane University. For the saliva samples, participants were asked to rinse their mouths well with distilled drinking water for 1 min. Five minutes after the oral rinse, participants were asked to spit ∼5 ml of saliva into a 50-ml sterile tube placed on ice. Following collection, the saliva samples were briefly vortexed and centrifuged at 2,600 × g for 15 min at 4°C. The cell-free supernatant was then collected and stored in a −80°C freezer for RNA extraction. Buccal swabs were collected by gently touching the oral mucosa with a cotton swab and were diluted in 1× phosphate-buffered saline (PBS). The samples were then centrifuged at 2,600 × g for 15 min at 4°C. Cell pellets were kept and stored in a −80°C freezer for RNA extraction. Tonsils were obtained from two 4-year-old patients scheduled for routine adenotonsillectomy at the Ochsner Clinic Foundation in accordance with a protocol approved by the IRB of the Ochsner Clinic Foundation. The tonsil surface epithelial tissues were carefully removed and homogenized using a homogenizing device (OMNI International) according to the manufacturer's instructions. The resulting samples were immediately subjected to total RNA isolation.
Cell culture.
OKF6/TERT2 (OKF6) is a telomerase-immortalized human oral epithelial cell line which retains its original oral epithelial cell morphology and was obtained from its creator, James Rheinwald (Dana-Farber Cancer Institute/Brigham and Women's Hospital). OKF6 cells were grown in keratinocyte serum-free medium (K-SFM) supplemented with 0.2 ng/ml of human recombinant epidermal growth factor (rEGF), 25 μg/ml of bovine pituitary extract (BPE), and 0.5% penicillin-streptomycin (pen-strep; Invitrogen). Both Mutu I and Akata are EBV-positive Burkitt's lymphoma cell lines (type I latency) and were grown in RPMI 1640 medium (ThermoFisher Scientific; catalog number SH30027) plus 10% fetal bovine serum (FBS; Invitrogen-Gibco; catalog number 16000-069) with 0.5% pen-strep (Invitrogen-Gibco; catalog number 15070-063). 293 cells are human kidney embryonic fibroblasts and were grown in a chemically defined serum-free medium (Pro293a-CDM; Lonza; catalog number 12-764Q). All cells used in this study were grown at 37°C in a humidified 5% CO2 incubator.
Plasmid constructions.
The pMSCV-puro-GFP-miR-429 and pMSCV-puro-GFP-CNTL retroviral expression vectors were generated as previously described (30). The pCEP4-BMRF1p-GFP vector was established by cloning a 1-kb region of the BMRF1 promoter upstream from the green fluorescent protein (GFP) reading frame in the pCEP4 vector (ThermoFisher Scientific).
Generation of stable cell lines.
Retrovirus preparations were generated through transient cotransfection of HEK293 cells with retroviral expression vectors plus packaging vectors. Transient transfections were performed using a modified version of the calcium phosphate precipitation procedure (36). Forty-eight hours later, viral supernatants were collected and subjected to one round of centrifugation followed by filtration through a 0.45-μm surfactant-free cellulose acetate filter. Infections were carried out in six-well plates with 1 ml of virus plus parental 293 cells. Cells were spun in six-well plates at 1,000 × g for 1 h at room temperature, followed by a 4-h incubation at 37°C with 5% CO2. Cells were then cultured in fresh medium for 2 days and subsequently selected in medium with 1 μg/ml of puromycin. After selection, 293 stable cells were maintained in complete Pro293a-CDM medium plus 1 μg/ml of puromycin. To generate membrane vesicles and exosomes, 293 stable cells were temporarily cultured in complete Pro293a-CDM without puromycin.
To create the Mutu-BMRF1p-GFP stable cell line, the pCEP4-BMRF1p-GFP vector was transfected into Mutu I cells, using the Lonza Nucleofector kit (Lonza). Stable cell lines were then selected in medium supplemented with 50 μg/ml of hygromycin. After selection, the generated stable cells were maintained in RPMI 1640 medium supplemented with 10% FBS plus hygromycin (50 μg/ml) and 0.5% pen-strep.
Membrane vesicle (exosome) isolation.
Membrane vesicles, including exosomes, were purified using the differential centrifugation method (37). Briefly, ∼100 ml of medium was collected from cell cultures (∼3.2 × 107 cells) and centrifuged at 3,500 × g for 20 min at 4°C to remove cell debris. Supernatant fractions were then collected and subjected to ultrafiltration using Amicon stirred cell (model number 8400; Millipore) with a molecular mass cutoff membrane of 100 kDa (Millipore) on ice. After ultrafiltration, the concentrates were centrifuged at 100,000 × g for 90 min at 4°C to generate vesicle pellets. The pellets were then washed with 12.5 ml of 1× PBS and centrifuged at 100,000 × g for 90 min at 4°C again. Finally, the pellets were resuspended in 100 μl of 1× PBS for downstream analysis.
NTA.
The quantity (concentration) and size distribution of collected OKF6 cell MVs (exosomes) were analyzed by the nanoparticle tracking analysis (NTA) assay using NanoSight LM20 equipment (Malvern Instruments, United Kingdom). All measurements were performed at room temperature according to the manufacturer's instructions. Samples were diluted 1:5,000 in 1× PBS, and triplicate samples were used to ensure measurement consistency.
RNA extraction.
Total cellular or saliva RNA and MV (exosome) RNA were isolated using the miRNeasy minikit (Qiagen; catalog number 217004) or TRIzol reagent (Life Technologies; catalog number 15596-018) according to the vendors' protocols and treated with RNase-free DNase (Life Technologies; catalog number AM1906) according to the vendor's protocol. The quantity and quality of the isolated RNA were further analyzed using a NanoDrop 2000 spectrophotometer (Thermo Fisher) and an Agilent 2100 bioanalyzer with an RNA 6000 pico kit. Intact MV (exosome) preparations were treated with RNase before total RNA isolation to ensure that the purified MV (exosome) RNAs were from the lumens of MVs and exosomes.
Coculture analysis.
One day before coculture, Mutu I cells were split to a concentration of 1 × 106 cells/ml and OKF6 cells were seeded in a 6-well plate with 1 × 106 cells per well. The next day, 6 × 105 Mutu I cells were collected, resuspended in 2 ml of K-SFM medium, and added to the confluent OKF6 culture. Seventy-two hours after coculture, the medium (containing Mutu I cells) was harvested and spun at 1,000 × g and room temperature for 2 min to collect the Mutu I cells. The cells were immediately lysed in 1× Laemmli buffer (Sigma), heated at 95°C for 15 min, and saved for Western blot analysis.
Membrane vesicle (exosome) treatment.
To monitor the uptake of OKF6 MVs (exosomes) by Mutu I cells, isolated OKF6 cell-derived MVs (exosomes) were first labeled with Vybrant dye (Invitrogen) according to the vendor's protocol. Labeled purified MVs (exosomes) were then washed in 12.5 ml of 1× PBS, collected by ultracentrifugation at 100,000 × g and 4°C for 90 min, and resuspended in 100 μl of 1× PBS. Fifty microliters of labeled MV (exosome) preparation (equivalent to ∼ 6.5 × 1010 particles) was added to a culture of 6 × 105 Mutu I cells in 0.5 ml of medium in a 24-well plate. Twenty-four hours postincubation, cells were harvested and mounted on glass slides with Vectashield with 4′,6-diamidino-2-phenylindole (DAPI) mounting solution (Vector Laboratories) and visualized by fluorescence microscopy analysis.
To evaluate the functionality of purified MVs (exosomes), the isolated MVs (exosomes) from OKF6, 293-miR-429, or 293-cntl cells (∼6.5 ×1010 particles each) were added to a culture of 6 × 105 Mutu I or Akata cells in 0.5 ml of medium in a 24-well plate. Seventy-two hours postincubation, cell samples were harvested, lysed in 1× Laemmli buffer, and heated at 95°C for 15 min. The resulting samples were then saved for Western blot analysis.
Induction of EBV lytic cycle.
Mutu-BMRF1p-GFP cells were spun down and resuspended at a concentration of 1 × 106 cells/ml in fresh RPMI 1640 medium (10% FBS, 0.5% pen-strep). Anti-human IgM (Sigma-Aldrich; catalog number I0759) was added to the Mutu-BMRF1p-GFP cell suspensions to a final concentration of 100 μg/ml. Cells were harvested 72 h later for fluorescence analysis.
Transmission electron microscopy and deconvolution fluorescence microscopy.
For transmission electron microscopy, purified MVs (exosomes) were washed with 1× PBS and collected by ultracentrifugation at 100,000 × g and 4°C for 90 min. The vesicle pellets were then fixed in 2% paraformaldehyde in Sorensen's buffer. Five microliters of the fixed vesicles was then dropped onto a Formvar-carbon-coated grid and dried at room temperature for 20 min. The grid was then carefully washed three times with Sorenson's wash buffer (0.1 M sodium phosphate, pH 7.4). Next, the grid was fixed in 1% glutaraldehyde in Sorensen's buffer for 5 min. The grid was then washed in water and stained with saturated aqueous uranyl acetate for 10 min. Next, the grid was embedded in 0.4% uranyl acetate–1.8% methylcellulose on ice and dried at room temperature. The MVs (exosomes) were subsequently analyzed with a JEOL 2010 scanning transmission electron microscope. For deconvolution fluorescence microscopy, slides were examined with a Leica DMRXA2 deconvolution upright microscope as described previously (38).
Quantification of miR-200 family miRNAs.
The mature forms of miR-200 family miRNAs, including miR-429, miR-200b, and miR-200c, were quantified using the TaqMan microRNA assay (Applied Biosystems) as previously described (30). The expression of miR-200 family miRNAs in tonsil, buccal, and saliva samples was calculated by the comparative threshold cycle (CT) method (2−ΔΔCT) versus RNU6B. The expression of MV (exosome) miR-200 family miRNAs was calculated by directly comparing to the average CT value for the respective miR-200 family member in cells using the following formula: 2[−(CTMV − CTcell)].
Small RNA sequencing (small RNA-seq) data sets from human serum and saliva samples were downloaded from the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under accession numbers SRR2136525, SRR2136526, SRR2136528, SRR2136529, SRR2136530, and SRR2136531 for serum samples (39) and DRR006759 for a saliva sample (40). Raw sequencing reads were then analyzed with miRDeep2 (41) for the expression of mature forms of miR-200 family miRNAs, including miR-429, miR-200b, and miR-200c.
Real-time RT-PCR analysis.
Total RNA was reverse transcribed using the SuperScript III first-strand synthesis system for reverse transcription-PCR (RT-PCR) (Invitrogen, Carlsbad, CA). Random hexamers were used along with 1 μg of RNA in a 20-μl reaction volume according to the manufacturer's instructions. For the incubation steps (25°C for 10 min followed by 50°C for 50 min), a Mastercycler ep (Eppendorf, Hamburg, Germany) was used. The resulting cDNA was subjected to quantitative (real-time) PCR using sequence-specific forward and reverse primers (Integrated DNA Technologies). For real-time PCR, 1 μl of the resulting cDNA was used in a 10-μl reaction volume that included 5 μl of Sybr green and a 500 nM concentration each of forward and reverse primers. Amplification was carried out using the following conditions: 95°C for 3 min followed by 40 cycles of 95°C for 15 s and 60°C for 60 s. Melt curve analysis was performed at the end of every qRT-PCR run. Samples were tested in triplicates. No-template controls were included in each PCR run. PCRs were performed on a Bio-Rad CFX96 real-time system, and data analysis was performed using CFX Manager 3.0 software. Relative detection levels were calculated by normalizing with the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene as a reference gene. Primer sequences for Zta, Rta, and GAPDH have been previously described (38, 42).
Quantification of EBV genomes in culture medium.
Seventy-two hours after incubation of Mutu I and Akata cells with OKF6 cell-derived MVs (exosomes), cells were spun down and the media were filtered through 0.45-μm-pore-size surfactant-free cellulose acetate filters. Proteinase K was added to the media to a final concentration of 1 mg/ml and the media were incubated at 37°C for 1 h, followed by incubation at 95°C for 10 min. The media were then subjected to phenol-chloroform extraction and diluted 10-fold in H2O. Two microliters of each diluted sample was used for real-time PCR analysis of viral DNA, and PCRs were carried out using two different sets of primers designed to amplify EBV genomic regions as previously described (43). Real-time PCR was conducted using an iQ5 PCR machine (Bio-Rad) with the following conditions: 95°C for 3 min followed by 40 cycles of 95°C for 30 s, 62°C for 40 s, and 72°C for 40 s using Eva green Supermix (Bio-Rad).
Western blot analysis.
Cell and/or MV (exosome) lysates were suspended in 1× Laemmli buffer (Sigma) and heated at 95°C for 15 min to shear genomic DNA. The resulting samples were measured with the Bio-Rad protein assay kit (catalog number 5000002) according to the manufacturer's instructions. An equal weight of lysates was subjected to SDS-polyacrylamide gel electrophoresis using Bio-Rad Criterion gel system and transferred to nitrocellulose membranes (Millipore). The blots were blocked for 30 min in Tris-buffered saline containing 5% low-fat powdered milk and 1% FBS and then incubated with the primary antibody (in blocking buffer) overnight at 4°C. The blots were washed three times with 1× TBST (140 mM NaCl, 3 mM KCl, 25 mM Tris-HCl [pH 7.4], 0.1% Tween 20) (each wash was carried out for approximately 10 min). The blots were then incubated with horseradish peroxidase-conjugated secondary antibody (Bio-Rad) in blocking buffer for 1 h at room temperature. Blots were washed as described above and analyzed with an enhanced chemiluminescence detection system (Perkin-Elmer) according to the manufacturer's recommendations, and filters were exposed to Fuji Super RX films which were then processed using a Kodak X-ray processor. The following primary antibodies were used for Western blot analysis: anti-Zta (Argene; catalog number 11-007), anti-Rta (Argene; catalog number 11-008), anti-BMRF1 (Capricorn; catalog number EBV-018-48180), anti-actin (Santa Cruz; catalog number sc-1615), anti-cytochrome c (Abcam, Epitomics), and anti-CD63 (Santa Cruz; catalog number sc-15363).
RESULTS
The oral microenvironment is enriched for miR-200 family members.
MicroRNA-200 family members are typically expressed in epithelial cells, where they help maintain the epithelial phenotype. To assess a possible role of miR-200 family members in signaling in the oral epithelial environment, we evaluated the levels of the miR-200 family members in the oral cavity by collecting both buccal (oral mucosal) epithelium and saliva samples from 2 healthy individuals. Total RNAs were extracted and the levels of representative miR-200 family members from two miR-200 gene loci, miR-200b plus miR-429 from one locus and miR-200c from the other, were examined using a TaqMan miRNA assay. The levels of miR-200b, miR-200c, and miR-429 in saliva were found to be similar to the levels observed in in vitro-cultured cells of OKF6, a telomerase-immortalized human oral epithelial cell line that retains its original oral epithelial cell morphology (Fig. 1A), while substantially higher miR-200b, miR-200c, and miR-429 levels were detected in the buccal epithelial samples (Fig. 1A). Consistent with these findings, our analysis of small RNA-seq data sets from normal saliva (40) and serum (39) found substantially higher levels of mature miR-200b, miR-200c, and miR-429 in saliva than in serum (Fig. 1B). Thus, these results indicate that mature miR-200 family members are expressed in the oral epithelium and that they are present in its surrounding environment (e.g., saliva).
FIG 1.

High levels of the miR-200 family of microRNAs are present in the oral and tonsil epithelia and in saliva. (A and C) Buccal epithelium and saliva samples were collected from two healthy individuals. OKF6 is a human oral epithelial cell system. Akata is an EBV-positive Burkitt's lymphoma cell line. Total RNA was extracted and subjected to the TaqMan microRNA assay for mature miR-200 family miRNAs. RNU6B was analyzed as a reference. The expression of miR-200 family miRNAs, including miR-200b, miR-200c, and miR-429, was determined by the comparative CT method (2−ΔΔCT). (B) MiRDeep2 analysis of serum and saliva small RNA-seq data sets for miR-200 family member expression.
We next examined the levels of miR-200 family members in tonsillar epithelium. Fresh tonsil samples were obtained from patients undergoing routine adenotonsillectomies, and the tonsillar epithelial tissues were excised to make total RNA. Both miR-200c and miR-429 were found to be expressed in tonsil epithelium (Fig. 1C) but not in the B-cell line Akata. Together, these results demonstrate the presence of miR-200 family members in the oral and tonsillar epithelia and the surrounding environments.
Oral epithelial cells produce a microenvironment that favors EBV lytic replication in B cells.
Since miR-200 family members were readily detected in saliva and the oral and tonsillar epithelia and because subepithelial and mucosal plasmacytoid B cells are major virus producers in the oral cavity (28, 29, 44–50), we hypothesized that the oral epithelial microenvironment may promote lytic replication in latently infected B cells. To examine this possibility, cells of the telomerase-immortalized human oral epithelial cell line OKF6 were seeded onto culture dishes, and 1 day later, latently infected Mutu I cells were added to the OKF6 cell monolayer. After 3 days of coculture, the Mutu I B cells were taken off, leaving an intact OKF6 cell monolayer that displayed no notable morphological changes. The Mutu I cells were spun down and RNA and protein were isolated and subjected to RT-PCR and Western blot analyses, respectively. Higher expression of the immediate early and early Zta, Rta, and BMRF1 genes was observed for the biological replicates of OKF6 cocultured cells than for cells cultured in OKF6 cell growth medium alone (Fig. 2). These results suggest that latency in EBV-infected B cells can be disrupted by this simulated oral epithelial microenvironment.
FIG 2.
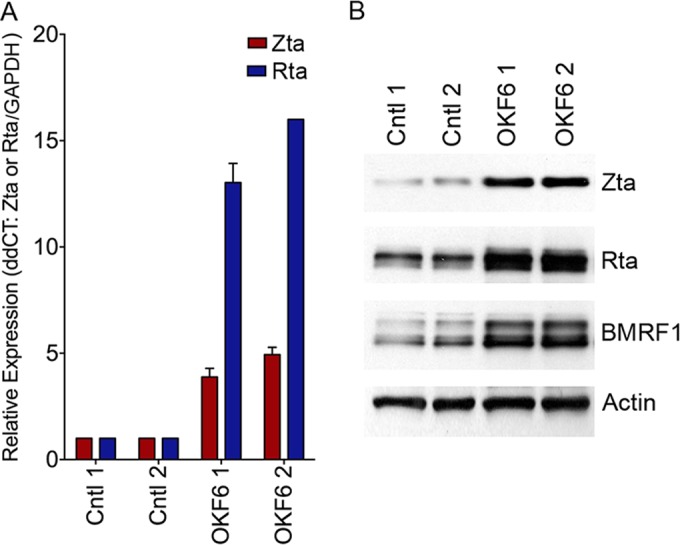
An oral epithelial cell microenvironment favors EBV lytic replication. EBV-positive Mutu I cells were cocultured with human oral epithelial OKF6 cells for 72 h, and Mutu I cells were then harvested for qRT-PCR analysis (A) and Western blot analysis (B). For qRT-PCR, the expression of Zta and Rta was determined by the comparative CT method (2−ΔΔCT) using GAPDH as a reference. For Western blot analysis, the levels of the EBV lytic antigens Zta (immediate early), Rta (immediate early), and BMRF1 (early) were measured. Actin was detected as a loading control and reference. For the control group, Mutu I cells were cultured in OKF6 cell culture medium for 72 h without OKF6 cells. Two biological replicates were included in each group.
Exosome-enriched membrane vesicles (eMVs) derived from oral OKF6 cells harbor miR-200 family members.
The induction of reactivation in Mutu I cells cocultured with OKF6 cells indicates that cell-cell contact and/or the presence of transmissible agents can signal reactivation in these latently infected B cells. Since membrane vesicles such as exosomes are important extracellular carriers of microRNAs and since we detected miR-200 family members in saliva, we reasoned that secreted MVs carrying miR-200 family members may function as one such signal. We therefore assessed whether exosome-enriched MVs (eMVs) secreted from OKF6 cells contained miR-200 family members.
Secreted membrane vesicles were collected and purified from OKF6 cell culture media using the differential centrifugation method (37) and were visualized with transmission electron microscopy. The isolated vesicles exhibited a typical cup-shaped exosomal morphology (Fig. 3A) with sizes ranging from approximately 30 to 100 nm, consistent with the size of exosome vesicles (2). Further, subsequent nanoparticle tracking analysis (NTA) showed a mode size distribution of ∼86 nm, indicating that the MV preparations were enriched with exosome-sized vesicles (Fig. 3B). To further evaluate the isolated vesicles, Western blot analysis was performed to assess the presence of exosome- and cell-specific markers. The exosome-specific marker CD63 was readily detected in the vesicle lysate (Fig. 3C). In contrast, cytochrome c, which is present in intact cells, apoptotic bodies, cell debris, and other types of vesicles, was undetectable in the vesicle lysate (Fig. 3C). These results indicated that our OKF6 cell vesicle preparations were enriched for exosomes.
FIG 3.

Characterization of membrane vesicles (exosomes) purified from OKF6 cell cultures. (A) Secretory membrane vesicles (exosomes) were isolated from the culture media of human oral epithelial OKF6 cells and analyzed by transmission electron microscopy. Representative images show that the majority of the vesicles were within the size range of exosomes (30 to 150 nm). The scale bar indicates 100 nm. (B) Nanoparticle tracking analysis (NTA) of an OKF6 cell-derived MV (exosome) preparation. (C) Equal amounts of the OKF6 whole-cell lysate and the lysates of two independent OKF6 cell-derived MV (exosome) preparations were analyzed for the indicated proteins by Western blotting. CD63 is an exosome-specific marker, and cytochrome c (Cyto C) is a mitochondrial protein marker. (D) Equal amounts of OKF6 cellular RNA and RNA isolated from an OKF6 cell-derived MV (exosome) preparation were analyzed using an Agilent bioanalyzer. (E) A TaqMan microRNA assay was used to analyze mature miR-200 family miRNAs in OKF6 cells and OKF6 cell-derived MVs (exosomes).
As a first step to investigate the possible incorporation of miR-200 family members in OKF6 cell-derived exosome-enriched membrane vesicles (eMVs), total RNA was extracted from the vesicle pellets and the RNA size distribution profile was analyzed using an Agilent bioanalyzer. The OKF6 cell-derived eMVs were found to be highly enriched with small RNA species (<200 nucleotides long) (Fig. 3D). To specifically assess the presence of miR-200 family members in these eMVs, equal amounts of RNAs from the vesicle preparations and from cells were analyzed using the TaqMan miRNA assay to quantify the levels of mature miR-200b, miR-200c, and miR-429. All three of these family members were readily detected in the OKF6 cell-derived eMVs, whereas another small RNA, RNU6B, was not (Fig. 3E). This finding showed active loading of these microRNAs into OKF6 cell-derived eMVs relative to RNU6B.
OKF6 cell-derived eMVs promote EBV reactivation in B cells.
The presence of miR-200 family members in OKF6 cell-derived eMVs raised the possibility that these MVs might facilitate at least some of the signaling between OKF6 cells and Mutu I B cells in our coculture experiments. To test this possibility, we first examined whether Mutu I cells can bind and internalize OKF6 cell-derived eMVs. The green fluorescent lipid dye Vybrant was used to label the purified OKF6 cell-derived eMVs, and the labeled eMVs were incubated with Mutu I cells for 24 h. The Mutu I cells were then collected and examined by fluorescence microscopy. Visualization of exposed Mutu I cells showed binding and internalization of labeled eMVs (Fig. 4A).
FIG 4.

OKF6 cell-derived eMVs break EBV latency in Mutu I cells. (A) OKF6 cell-derived eMVs were labeled with the green fluorescent dye Vybrant and incubated with Mutu I cells. Twenty-four hours postincubation, cells were examined by fluorescence microscopy. Nuclei were visualized by DAPI staining. (B and C) Mutu-BMRF1p-GFP cells were treated with OKF6 cell-derived eMVs, the control vehicle (1× PBS), or anti-IgM. Seventy-two hours posttreatment, cells were fixed and examined using fluorescence microscopy. Blue, Hoechst; green, GFP. Induction rates were determined by manually counting the GFP-positive cells in a total of 10 randomly selected fields from duplicate treatments for each condition. (D) Mutu I cells were incubated with purified OKF6 cell-derived eMVs or the control vehicle (1× PBS). Seventy-two hours postincubation, cell pellets were harvested for Western blot analysis of the EBV lytic antigens Zta (immediate early), Rta (immediate early), and BMRF1 (early). Actin was detected as a loading control and reference. Two biological replicates were included in each group. (E) The production of new virions was determined by the real-time PCR analysis of EBV genomic DNA in the cell culture media. Two biological replicates were included in each group. Two primer sets were utilized that amplify two different regions in the EBV genome (43). The amounts of EBV genomic DNA were calculated by the comparative CT method (2−ΔΔCT) versus GAPDH DNA.
To investigate whether uptake of OKF6 cell-derived eMVs can break EBV latency in recipient B cells, we created a lytic reporter system that expresses GFP in cells entering the lytic cascade. Initial iterations of this reporter contained a GFP cassette driven by a minimal promoter with multiple copies of Zta response elements. Although these reporters were responsive to ectopic Zta, they were poorly responsive to endogenously produced immediate early genes (data not shown). In contrast, a reporter containing a GFP reading frame driven by the EBV BMRF1 promoter was found to be highly responsive to the expression of both ectopic and endogenous EBV immediately early gene expression (data not shown). By introducing this plasmid into Mutu I cells, we were able to monitor initiation of the EBV lytic cycle through the detection of increased green fluorescence signals. To assess whether epithelium-derived eMVs can facilitate reactivation, eMVs were isolated from OKF6 cell culture medium and incubated with Mutu-BMRF1p-GFP cells. After 3 days of incubation, Mutu-BMRF1p-GFP cells were collected and analyzed by fluorescence microscopy. As shown in Fig. 4B and C, GFP signals were detected in nearly 20% of eMV-treated cells but rarely detected in cells treated with the control vehicle (1× PBS). Further, OKF6 cell-derived eMVs exhibited lytic cycle-inducing potency similar to that of the anti-IgM-mediated BCR engagement (Fig. 4C).
To further investigate the eMV-mediated reactivation, we incubated Mutu I cells with freshly prepared OKF6 cell-derived eMVs. Three days later, Mutu I cell pellets were collected and examined for viral lytic gene expression. As shown in Fig. 4D, expression of the viral immediate early and early Zta and Rta, and BMRF1 genes was increased in Mutu I cells exposed to OKF6 cell-derived eMVs, indicating disruption of latency. Further, real-time PCR analysis showed an increased level of viral genomic DNA in the supernatant of Mutu I cells incubated with OKF6 cell-derived eMVs (Fig. 4E). These results further establish that exposure of EBV-positive B cells to OKF6 cell-derived eMVs promotes entry into the lytic replication cycle and the production of secreted virions.
To ensure that the induction of reactivation by OKF6 cell-derived eMVs is not specific to a single cell line, we incubated OKF6 cell-derived eMVs with a second EBV-positive B-cell line, Akata. Similar to the results obtained with Mutu I cells, expression of the immediate early and early Zta, Rta, and BMRF1 genes and secreted virus were induced by OKF6 cell-derived eMVs (Fig. 5).
FIG 5.
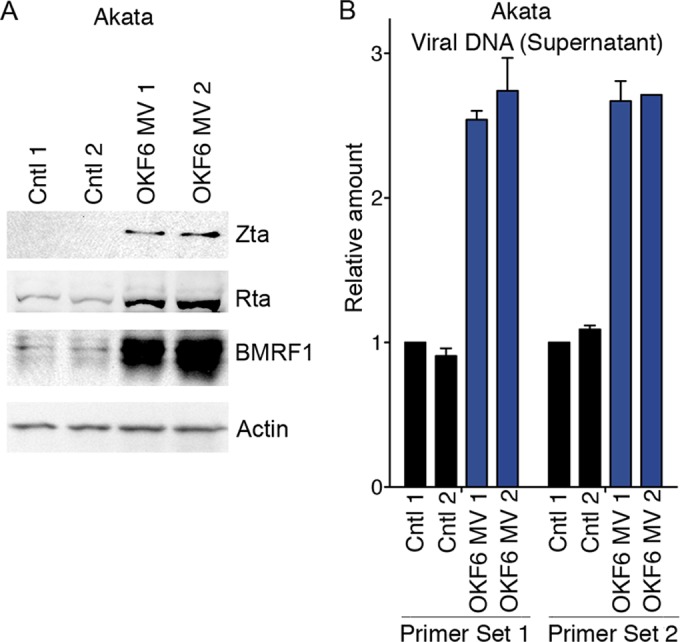
OKF6 cell-derived eMVs break EBV latency in Akata cells. Akata cells were incubated with purified OKF6 cell-derived eMVs or the control vehicle (1× PBS). Seventy-two hours postincubation, (A) cell pellets were harvested for Western blot analysis of the EBV lytic antigens Zta (immediate early), Rta (immediate early), and BMRF1 (early). Actin was detected as a loading control and reference. Two biological replicates were included in each group. (B) The production of new virions was determined by real-time PCR analysis of EBV genomic DNA in the cell culture media. Two biological replicates were included in each group. Two primers sets were utilized that amplify two different regions in the EBV genome (43). The amounts of EBV genomic DNA were calculated by the comparative CT method (2−ΔΔCT) versus GAPDH DNA.
miR429-containing eMVs promote EBV reactivation in B cells.
The data presented above show that OKF6 cell-derived eMV contain miR-200 family members and that these eMVs can induce reactivation in EBV-positive B-cell lines. To determine whether miR-200 family members per se play a role in eMV-mediated reactivation, we set out to compare the efficacies of miR-200 family-positive and miR-200 family-negative eMVs in facilitating reactivation. Most nonepithelial cells, such as 293 fibroblasts, do not express miR-200 family miRNAs (30). Since 293 cells have been previously shown to actively produce and release eMVs (51), we created 293 cell lines transduced in duplicate with either an empty vector control (293-cntl) or a miR-429 expressing retrovirus (293-miR-429).
Mature miR-429 but not other miR-200 family members were detected in eMVs derived from 293-miR-429 cells (Fig. 6A). To examine whether these miR-429-positive eMVs can disrupt viral latency, we incubated either the 293-miR-429- or 293-cntl-derived eMVs with Mutu I cells. After 3 days of incubation, cell pellets were collected and extracted proteins were analyzed by Western blotting for viral lytic gene expression. Exposure of Mutu I cells to miR-429-positive eMVs led to increased expression of the immediate early and early Zta, Rta, and BMRF1 genes (Fig. 6B). Mutu I cells incubated with eMVs generated from empty control retrovirus-transduced 293 cells exhibited levels of viral lytic gene expression similar to those observed with Mutu I cells incubated with the control vehicle (1× PBS) (Fig. 6B). Together, these results show that miR-429-containing eMVs promote viral lytic reactivation in recipient B cells.
FIG 6.
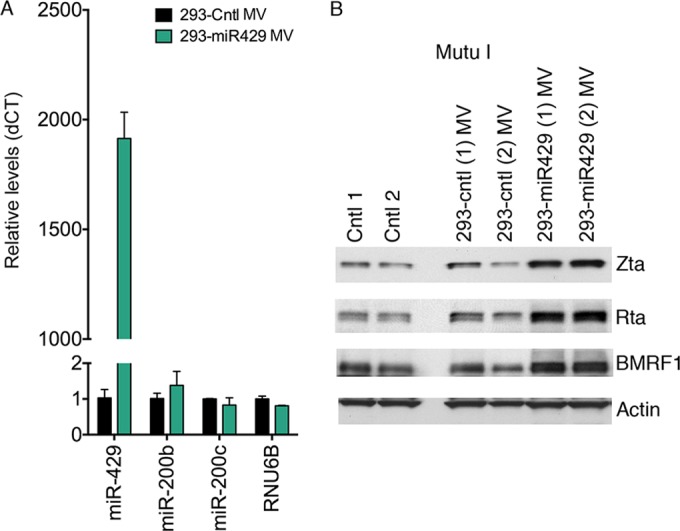
miR-429-containing eMVs promote lytic reactivation in Mutu I cells. (A) eMVs were purified from 293 cells stably expressing either miR-429 (293-miR429) or its control (293-cntl). RNAs were then isolated from the purified eMVs and subjected to TaqMan microRNA analysis of mature miR-200 family miRNAs. (B) Mutu I cells were incubated with purified 293-miR429 eMVs, 293-cntl eMVs, or the control vehicle (1× PBS). Seventy-two hours postincubation, cell pellets were harvested for Western blot analysis of the EBV lytic antigens Zta (immediate early), Rta (immediate early), and BMRF1 (early). Actin was detected as a loading control and reference. Two biological replicates were included in each group.
DISCUSSION
Based on the results presented here, we propose that the oral epithelium-associated MV (exosome) pathway plays a role in orchestrating EBV's lytic replication in B cells in vivo. In this model, oral epithelial cells actively load miR-200 family miRNAs into exosomes (and other possible membrane vesicles such as microvesicles) (Fig. 7) and secret these vesicles into the epithelial microenvironment to create conditions that favor EBV lytic replication. The transfer of eMVs containing miR-200 family members to infiltrating and/or proximal EBV-positive B cells leads to the disruption of latency and production of new viruses. This facilitates the exchange of virus from the peripheral B-cell stores to the oral epithelium, where virus is amplified and shed into the saliva.
FIG 7.

Model of eMV miR-200-mediated communication between oral epithelium and virally infected B cells.
Emerging evidence indicates that the EBV infection cascade relies on both the oral epithelium and B cells. One current opinion is that the exchange of virus generated from one tissue type to the other is an integral part of EBV's infection cycle, which is supported by a reciprocal infection preference for virus produced in epithelial cells versus B cells (34, 35). In agreement with this view, our proposed microenvironment sensing mechanism can potentially contribute further to facilitating the virus exchange between B cells and epithelial cells. This sensing system would also help enable such a limited number of virally infected B cells (approximately only one in one million circulating B cells) to maintain constant viral replication foci at oral epithelial sites.
Previously, our laboratory and others discovered an intimate relationship between the miR-200 signaling pathways and the EBV life cycle (30, 31). Specifically, we found that the miR-200 family of microRNAs functions as a cellular switch that regulates the transition from latency to the lytic cascade. The miR-200 family of microRNAs forms a double negative-feedback loop with the ZEB1 and ZEB2 transcriptional repressors, forming a regulatory axis that facilitates the committed transition of cells from either a mesenchymal to epithelial phenotype or an epithelial to mesenchymal phenotype. Specifically, the 3′ untranslated regions (UTRs) of both ZEB1 and ZEB2 contain multiple seed sites for miR-200 family members, and their expression is inhibited by high levels of miR-200s (52). Conversely, both miR-200 family member clusters are transcriptionally repressed by ZEB1 and/or ZEB2 (53). This forms a self-reinforcing loop that helps establish commitment to either high miR-200 family expression with low ZEB1 and ZEB2 expression or high ZEB1 and ZEB2 expression with low miR-200 family member expression. For example, once miR-200 family member expression increases, this suppresses the expression of their repressors, ZEB1 and ZEB2, which, in turn, leads to a further increase in miR-200 family expression and so on. Conversely, an increase in ZEB1 or ZEB2 is self-reinforced by repression of miR-200 family members, causing relief from inhibition by these microRNAs.
During B-cell latency, the low levels of endogenous miR-200s allow the expression of the transcriptional suppressors ZEB1 and ZEB2, which inhibit the viral immediate early Zta gene (32, 33), thereby promoting viral latency. In response to lytic induction signals, an upregulation of miR-200 family miRNAs will relieve the inhibitory effect of ZEB1 and ZEB2 on miR-200 promoters, further enhancing the expression of miR-200 family miRNAs and leading to further suppression of ZEB1 and ZEB2. This causes a committed transition from high ZEB1 and ZEB2 and low miR-200 family expression to a low ZEB1 and ZEB2 and high miR-200 expression, enforcing a commitment to the loss of ZEB1 and ZEB2 suppression of the Zta promoter.
In the context of intercellular communication between epithelial cells and B cells, we propose that the internalized miR-200s in recipient B cells will likely activate lytic replication by relieving ZEB1- and ZEB2-mediated suppression of the viral Zta gene. We think that miR-200s are ideal eMV signaling molecules for facilitating sustained pathway alterations. By taking advantage of this self-reinforcing feedback mechanism, a single dose of eMV miR-200s not only will directly inhibit ZEB1 and ZEB2 expression but also should be able to enforce elevated cellular expression of miR-200 family expression, thereby enforcing a commitment to break viral latency in recipient cells.
In our coculture system, a monolayer of OKF6 cells was shown to disrupt viral latency in B cells. Others have argued that both the concentration and transfer efficiency of MVs (exosomes) in in vivo environments are substantially higher than in in vitro simulated systems because of the high concentration of cells and the low intercellular volume in the in vivo settings (13, 54). This raises the possibility that reactivation efficiencies in vivo may, in fact, be greater than those observed in our tissue culture model.
Using a well-established differential centrifugation method, we were able to isolate eMVs from the supernatant of OKF6 cell culture media. The presence of exosomes in the OKF6 cell-derived vesicle preparation was confirmed by the presence of CD63 exosomal marker and the absence of cytochrome c, a key indicator for the contamination of apoptotic bodies and cell debris. The additional morphological analysis and particle size analysis further indicated that the majority of the isolated vesicles were exosomes. Nevertheless, we did detect a fraction of microvesicle-like particles (150 to 230 nm in diameter) in our vesicle preparations. Generally, microvesicles (or shedding vesicles) belong to another subpopulation of membrane vesicles and are more heterogeneous in size (100 to 1,000 nm in diameter) (2, 55, 56). Microvesicles are formed by outward budding of the plasma membrane and, like exosomes, contain proteins, lipids, and miRNAs (2). Since many studies have shown that miRNAs and other genetic materials can be carried by both exosomes and microvesicles and functionally delivered to recipient cells (16, 56), it is possible that this fraction of microvesicles may also contribute to the creation of a miR-200s-rich epithelial microenvironment and cooperate with exosomes to regulate the EBV life cycle.
Here, we have shown that eMV miR-200s can break viral latency in recipient B cells, but we cannot rule out the possibility that other epithelial signaling molecules carried by exosomes and other membrane vesicles are also involved in this regulatory process. Further analysis may uncover additional components within oral epithelial secretory vesicles that also help facilitate EBV reactivation.
In this study, we focused only on communication from oral epithelial cells to virally infected B cells. It is also interesting to consider, however, whether the virally infected B cells can simultaneously “talk to” oral epithelial cells, particularly through a paracrine-like manner in the microenvironment. Previous studies have shown that EBV-infected lymphoblastoid B cells can actively pack both viral and cellular nucleic acids (including miRNAs) and proteins into the exosomes, which can influence the recipient cells (12, 13, 54). When reaching the oral epithelial microenvironment, the virally infected B cells may similarly signal the epithelial cells, thereby modulating signaling pathways that may play a role in facilitating the needs of the virus. For instance, uptake of such vesicles could enhance permissiveness of recipient epithelial cells to EBV infection. This and other possible roles for B-cell-derived exosomes in the process of transfer of virus from B cells to epithelial cells require further investigation, however.
ACKNOWLEDGMENTS
This work was supported by a Louisiana Clinical & Translational Science Center Pilot Grant (1 U54 GM104940 from the National Institute of General Medical Sciences of the National Institutes of Health, which funds the Louisiana Clinical and Translational Science Center) to Z.L., Ruth L. Kirschstein National Research Service Award F30CA177267 from the NCI to M.J.S., and National Institutes of Health grants R01AI101046, R01CA124311, R01CA138268, and R01AI106676 to E.K.F. (and P20GM103518 to Prescott Deininger, Tulane Cancer Center).
Funding Statement
This work was also supported through core resources from a grant awarded to a nonauthor, Prescott Deininger (Tulane Cancer Center), under award P20GM103518.
REFERENCES
- 1.Théry C, Zitvogel L, Amigorena S. 2002. Exosomes: composition, biogenesis and function. Nat Rev Immunol 2:569–579. [DOI] [PubMed] [Google Scholar]
- 2.Théry C, Ostrowski M, Segura E. 2009. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 3.Schageman J, Zeringer E, Li M, Barta T, Lea K, Gu J, Magdaleno S, Setterquist R, Vlassov AV. 2013. The complete exosome workflow solution: from isolation to characterization of RNA cargo. Biomed Res Int 2013:253957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conde-Vancells J, Rodriguez-Suarez E, Embade N, Gil D, Matthiesen R, Valle M, Elortza F, Lu SC, Mato JM, Falcon-Perez JM. 2008. Characterization and comprehensive proteome profiling of exosomes secreted by hepatocytes. J Proteome Res 7:5157–5166. doi: 10.1021/pr8004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brugger B, Simons M. 2008. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 319:1244–1247. doi: 10.1126/science.1153124. [DOI] [PubMed] [Google Scholar]
- 6.Johnstone RM. 2005. Revisiting the road to the discovery of exosomes. Blood Cells Mol Dis 34:214–219. doi: 10.1016/j.bcmd.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 7.Fevrier B, Raposo G. 2004. Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr Opin Cell Biol 16:415–421. doi: 10.1016/j.ceb.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 8.Kourembanas S. 2015. Exosomes: vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu Rev Physiol 77:13–27. doi: 10.1146/annurev-physiol-021014-071641. [DOI] [PubMed] [Google Scholar]
- 9.Meckes DG Jr, Raab-Traub N. 2011. Microvesicles and viral infection. J Virol 85:12844–12854. doi: 10.1128/JVI.05853-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schorey JS, Bhatnagar S. 2008. Exosome function: from tumor immunology to pathogen biology. Traffic 9:871–881. doi: 10.1111/j.1600-0854.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Niel G, Porto-Carreiro I, Simoes S, Raposo G. 2006. Exosomes: a common pathway for a specialized function. J Biochem 140:13–21. doi: 10.1093/jb/mvj128. [DOI] [PubMed] [Google Scholar]
- 12.Meckes DG Jr, Gunawardena HP, Dekroon RM, Heaton PR, Edwards RH, Ozgur S, Griffith JD, Damania B, Raab-Traub N. 2013. Modulation of B-cell exosome proteins by gamma herpesvirus infection. Proc Natl Acad Sci U S A 110:E2925–E2933. doi: 10.1073/pnas.1303906110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MA, Hopmans ES, Lindenberg JL, de Gruijl TD, Wurdinger T, Middeldorp JM. 2010. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A 107:6328–6333. doi: 10.1073/pnas.0914843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haneklaus M, Gerlic M, Kurowska-Stolarska M, Rainey AA, Pich D, McInnes IB, Hammerschmidt W, O'Neill LA, Masters SL. 2012. Cutting edge: miR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1beta production. J Immunol 189:3795–3799. doi: 10.4049/jimmunol.1200312. [DOI] [PubMed] [Google Scholar]
- 15.Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. 2007. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 16.Mittelbrunn M, Sanchez-Madrid F. 2012. Intercellular communication: diverse structures for exchange of genetic information. Nat Rev Mol Cell Biol 13:328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, Li S, Li L, Li M, Guo C, Yao J, Mi S. 2015. Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteomics Bioinformatics 13:17–24. doi: 10.1016/j.gpb.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rickinson AB, Kieff E. 2007. Epstein-Barr virus, p 2655–2700. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 19.Lin Z, Flemington EK. 2010. Regulation of EBV latency by viral lytic proteins, p 167–192. In Robertson ES. (ed), Epstein-Barr virus: latency and transformation. Caister Academic Press, Wymondham, Norfolk, United Kingdom. [Google Scholar]
- 20.Kieff E, Rickinson AB. 2007. Epstein-Barr virus and its replication, p 2603–2654. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 21.Luka J, Kallin B, Klein G. 1979. Induction of the Epstein-Barr virus (EBV) cycle in latently infected cells by n-butyrate. Virology 94:228–231. doi: 10.1016/0042-6822(79)90455-0. [DOI] [PubMed] [Google Scholar]
- 22.zur Hausen H, Bornkamm GW, Schmidt R, Hecker E. 1979. Tumor initiators and promoters in the induction of Epstein-Barr virus. Proc Natl Acad Sci U S A 76:782–785. doi: 10.1073/pnas.76.2.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ben-Sasson SA, Klein G. 1981. Activation of the Epstein-Barr virus genome by 5-aza-cytidine in latently infected human lymphoid lines. Int J Cancer 28:131–135. doi: 10.1002/ijc.2910280204. [DOI] [PubMed] [Google Scholar]
- 24.Faggioni A, Zompetta C, Grimaldi S, Barile G, Frati L, Lazdins J. 1986. Calcium modulation activates Epstein-Barr virus genome in latently infected cells. Science 232:1554–1556. doi: 10.1126/science.3012779. [DOI] [PubMed] [Google Scholar]
- 25.Takada K. 1984. Cross-linking of cell surface immunoglobulins induces Epstein-Barr virus in Burkitt lymphoma lines. Int J Cancer 33:27–32. doi: 10.1002/ijc.2910330106. [DOI] [PubMed] [Google Scholar]
- 26.Takada K, Ono Y. 1989. Synchronous and sequential activation of latently infected Epstein-Barr virus genomes. J Virol 63:445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chasserot-Golaz S, Schuster C, Dietrich JB, Beck G, Lawrence DA. 1988. Antagonistic action of RU38486 on the activity of transforming growth factor-beta in fibroblasts and lymphoma cells. J Steroid Biochem 30:381–385. doi: 10.1016/0022-4731(88)90127-6. [DOI] [PubMed] [Google Scholar]
- 28.Laichalk LL, Thorley-Lawson DA. 2005. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J Virol 79:1296–1307. doi: 10.1128/JVI.79.2.1296-1307.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hadinoto V, Shapiro M, Sun CC, Thorley-Lawson DA. 2009. The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog 5:e1000496. doi: 10.1371/journal.ppat.1000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin Z, Wang X, Fewell C, Cameron J, Yin Q, Flemington EK. 2010. Differential expression of the miR-200 family microRNAs in epithelial and B cells and regulation of Epstein-Barr virus reactivation by the miR-200 family member miR-429. J Virol 84:7892–7897. doi: 10.1128/JVI.00379-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ellis-Connell AL, Iempridee T, Xu I, Mertz JE. 2010. Cellular microRNAs 200b and 429 regulate the Epstein-Barr virus switch between latency and lytic replication. J Virol 84:10329–10343. doi: 10.1128/JVI.00923-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu X, Wang Z, Mertz JE. 2007. ZEB1 regulates the latent-lytic switch in infection by Epstein-Barr virus. PLoS Pathog 3:e194. doi: 10.1371/journal.ppat.0030194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kraus RJ, Perrigoue JG, Mertz JE. 2003. ZEB negatively regulates the lytic-switch BZLF1 gene promoter of Epstein-Barr virus. J Virol 77:199–207. doi: 10.1128/JVI.77.1.199-207.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Borza CM, Hutt-Fletcher LM. 2002. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat Med 8:594–599. doi: 10.1038/nm0602-594. [DOI] [PubMed] [Google Scholar]
- 35.Jiang R, Scott RS, Hutt-Fletcher LM. 2006. Epstein-Barr virus shed in saliva is high in B-cell-tropic glycoprotein gp42. J Virol 80:7281–7283. doi: 10.1128/JVI.00497-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin Z, Yin Q, Flemington E. 2004. Identification of a negative regulatory element in the Epstein-Barr virus Zta transactivation domain that is regulated by the cell cycle control factors c-Myc and E2F1. J Virol 78:11962–11971. doi: 10.1128/JVI.78.21.11962-11971.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zeringer E, Barta T, Li M, Vlassov AV. 2015. Strategies for isolation of exosomes. Cold Spring Harb Protoc 2015:pdb.top074476. [DOI] [PubMed] [Google Scholar]
- 38.Cao S, Moss W, O'Grady T, Concha M, Strong MJ, Wang X, Yu Y, Baddoo M, Zhang K, Fewell C, Lin Z, Dong Y, Flemington EK. 2015. New noncoding lytic transcripts derived from the Epstein-Barr virus latency origin of replication, oriP, are hyperedited, bind the paraspeckle protein, NONO/p54nrb, and support viral lytic transcription. J Virol 89:7120–7132. doi: 10.1128/JVI.00608-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ignacio C, Hicks SD, Burke P, Lewis L, Szombathyne-Meszaros Z, Middleton FA. 2015. Alterations in serum microRNA in humans with alcohol use disorders impact cell proliferation and cell death pathways and predict structural and functional changes in brain. BMC Neurosci 16:55. doi: 10.1186/s12868-015-0195-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ogawa Y, Taketomi Y, Murakami M, Tsujimoto M, Yanoshita R. 2013. Small RNA transcriptomes of two types of exosomes in human whole saliva determined by next generation sequencing. Biol Pharm Bull 36:66–75. [DOI] [PubMed] [Google Scholar]
- 41.Friedländer MR, Mackowiak SD, Li N, Chen W, Rajewsky N. 2012. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40:37–52. doi: 10.1093/nar/gkr688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strong MJ, Baddoo M, Nanbo A, Xu M, Puetter A, Lin Z. 2014. Comprehensive high-throughput RNA sequencing analysis reveals contamination of multiple nasopharyngeal carcinoma cell lines with HeLa cell genomes. J Virol 88:10696–10704. doi: 10.1128/JVI.01457-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin Q, Wang X, Fewell C, Cameron J, Zhu H, Baddoo M, Lin Z, Flemington EK. 2010. MicroRNA miR-155 inhibits bone morphogenetic protein (BMP) signaling and BMP-mediated Epstein-Barr virus reactivation. J Virol 84:6318–6327. doi: 10.1128/JVI.00635-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tao Q, Srivastava G, Chan AC, Chung LP, Loke SL, Ho FC. 1995. Evidence for lytic infection by Epstein-Barr virus in mucosal lymphocytes instead of nasopharyngeal epithelial cells in normal individuals. J Med Virol 45:71–77. doi: 10.1002/jmv.1890450114. [DOI] [PubMed] [Google Scholar]
- 45.Niedobitek G, Agathanggelou A, Steven N, Young LS. 2000. Epstein-Barr virus (EBV) in infectious mononucleosis: detection of the virus in tonsillar B lymphocytes but not in desquamated oropharyngeal epithelial cells. Mol Pathol 53:37–42. doi: 10.1136/mp.53.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Niedobitek G, Herbst H, Young LS, Brooks L, Masucci MG, Crocker J, Rickinson AB, Stein H. 1992. Patterns of Epstein-Barr virus infection in non-neoplastic lymphoid tissue. Blood 79:2520–2526. [PubMed] [Google Scholar]
- 47.Anagnostopoulos I, Hummel M, Kreschel C, Stein H. 1995. Morphology, immunophenotype, and distribution of latently and/or productively Epstein-Barr virus-infected cells in acute infectious mononucleosis: implications for the interindividual infection route of Epstein-Barr virus. Blood 85:744–750. [PubMed] [Google Scholar]
- 48.Niedobitek G, Agathanggelou A, Herbst H, Whitehead L, Wright DH, Young LS. 1997. Epstein-Barr virus (EBV) infection in infectious mononucleosis: virus latency, replication and phenotype of EBV-infected cells. J Pathol 182:151–159. [DOI] [PubMed] [Google Scholar]
- 49.Karajannis MA, Hummel M, Anagnostopoulos I, Stein H. 1997. Strict lymphotropism of Epstein-Barr virus during acute infectious mononucleosis in nonimmunocompromised individuals. Blood 89:2856–2862. [PubMed] [Google Scholar]
- 50.Crawford DH, Ando I. 1986. EB virus induction is associated with B-cell maturation. Immunology 59:405–409. [PMC free article] [PubMed] [Google Scholar]
- 51.Chairoungdua A, Smith DL, Pochard P, Hull M, Caplan MJ. 2010. Exosome release of beta-catenin: a novel mechanism that antagonizes Wnt signaling. J Cell Biol 190:1079–1091. doi: 10.1083/jcb.201002049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park SM, Gaur AB, Lengyel E, Peter ME. 2008. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, Brabletz T. 2008. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep 9:582–589. doi: 10.1038/embor.2008.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Meckes DG Jr, Shair KH, Marquitz AR, Kung CP, Edwards RH, Raab-Traub N. 2010. Human tumor virus utilizes exosomes for intercellular communication. Proc Natl Acad Sci U S A 107:20370–20375. doi: 10.1073/pnas.1014194107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. 1999. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 94:3791–3799. [PubMed] [Google Scholar]
- 56.Cocucci E, Racchetti G, Meldolesi J. 2009. Shedding microvesicles: artefacts no more. Trends Cell Biol 19:43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]