

ABSTRACT
Hepatitis B virus (HBV) is a causative agent for chronic liver diseases such as hepatitis, cirrhosis, and hepatocellular carcinoma (HCC). HBx protein encoded by the HBV genome plays crucial roles not only in pathogenesis but also in replication of HBV. Although HBx has been shown to bind to a number of host proteins, the molecular mechanisms by which HBx regulates HBV replication are largely unknown. In this study, we identified jumonji C-domain-containing 5 (JMJD5) as a novel binding partner of HBx interacting in the cytoplasm. DNA microarray analysis revealed that JMJD5-knockout (JMJD5KO) Huh7 cells exhibited a significant reduction in the expression of transcriptional factors involved in hepatocyte differentiation, such as HNF4A, CEBPA, and FOXA3. We found that hydroxylase activity of JMJD5 participates in the regulation of these transcriptional factors. Moreover, JMJD5KO Huh7 cells exhibited a severe reduction in HBV replication, and complementation of HBx expression failed to rescue replication of a mutant HBV deficient in HBx, suggesting that JMJD5 participates in HBV replication through an interaction with HBx. We also found that replacing Gly135 with Glu in JMJD5 abrogates binding with HBx and replication of HBV. Moreover, the hydroxylase activity of JMJD5 was crucial for HBV replication. Collectively, these results suggest that direct interaction of JMJD5 with HBx facilitates HBV replication through the hydroxylase activity of JMJD5.
IMPORTANCE HBx protein encoded by hepatitis B virus (HBV) plays important roles in pathogenesis and replication of HBV. We identified jumonji C-domain-containing 5 (JMJD5) as a novel binding partner to HBx. JMJD5 was shown to regulate several transcriptional factors to maintain hepatocyte function. Although HBx had been shown to support HBV replication, deficiency of JMJD5 abolished contribution of HBx in HBV replication, suggesting that HBx-mediated HBV replication is largely dependent on JMJD5. We showed that hydroxylase activity of JMJD5 in the C terminus region is crucial for expression of HNF4A and replication of HBV. Furthermore, a mutant JMJD5 with Gly135 replaced by Glu failed to interact with HBx and to rescue the replication of HBV in JMJD5-knockout cells. Taken together, our data suggest that interaction of JMJD5 with HBx facilitates HBV replication through the hydroxylase activity of JMJD5.
INTRODUCTION
Hepatitis B virus (HBV) is an enveloped virus belonging to the Hepadnaviridae family (1) and possessing a partially double-stranded circular DNA genome. HBV is transmitted by blood via perinatal and sexual routes and infects more than 300 million people worldwide. HBV infection leads to chronic infection in 90% of perinatal individuals, 20 to 30% of children, and less than 1% of adults (2). Chronic infection often results in development of cirrhosis and hepatocellular carcinoma (HCC). Although reverse transcriptase inhibitors, including lamivudine and entecavir, are currently available for the treatment of patients infected with HBV, patients must take these drugs for life, and emergence of drug-resistant breakthrough viruses is a matter of concern.
HBx protein consists of 154 amino acids and is encoded by the viral genome as a nonstructural phosphoprotein involved in viral replication and pathogenesis, such as in the development of HCC (3). HBx has been shown to stimulate several signaling pathways, including AP-1 (3), NF-κΒ (4), CREB (5), and AP-2 (5), and to enhance transcription of SREBP-1a through the interaction with DNA-binding sites (6). HBx also modulates the cell cycle and apoptosis through the activation of RAS (7), cyclin D1 (8), and cyclin A (9) and the interaction with damage-specific DNA-binding protein 1 (DDB1) (10) and Bcl-2 family proteins (11–14). In addition, HBx in some genotypes participates in the apoptotic response through phosphorylation at Ser31 by AKT1 (15) and is degraded in a ubiquitin-independent proteasome (16), suggesting that some HBx functions may be regulated by posttranslational modifications.
Recent investigations on HCC in HBx transgenic mice generated in several laboratories have suggested that HBx participates in the pathogenesis of HBV (17–20). In addition, HBx has been shown to be involved in HBV replication in vitro and in vivo by using a recombinant HBV plasmid, pHBVΔX, possessing a stop codon in the coding region of HBx (21–24). However, the molecular mechanisms of HBx in HBV replication remain unclear.
A number of host proteins have been identified as binding partners for HBx, including HBx-interacting protein (22), p53 (25), COP9 signalosome (4), apolipoprotein A1 (26), Bc-2/Bcl-x (11), nuclear receptor coactivator 3 (27), protein arginine methyltransferase 1 (28), peptidylprolyl cis/trans isomerase, NIMA-interacting 1 (29), IPS-1 (30), and S-phase kinase-associated protein 2 (31). However, the biological significance of the interaction of HBx with these host factors in the life cycle of HBV is still unclear.
In addition, HBx has been shown to interact with DDB1 (32), leading to enhancement of the stability of HBx (33) and competing with the interaction between DDB1 and CUL4-associated factor-1 (DCAF1). Intervention in the interaction between DDB1 and DCAF1 by HBx results in hijacking of the cellular E3 ubiquitin ligase complex of DDB1/CUL4 (34) and participates in HBV replication and HBx-induced cell death (35). These findings suggest that interaction of HBx and DDB1 is required for HBV replication; however, precise mechanisms underlying the interaction of the proteins on HBV replication remain unknown.
In this study, we identified jumonji C-domain (JmjC)-containing 5 (JMJD5) as a novel binding partner for HBx and revealed that JMJD5 participates in HBV replication. Structural analyses have shown that JMJD5 exhibits hydroxylase activity rather than demethylase activity (36, 37). The structure of the jumonji C domain of JMJD5 was conserved with the C-terminal transactivation domains of the factor inhibiting hypoxia-inducible 1 alpha (FIH-1) and JMJD6 (36, 38). The C-terminal transactivation domain of FIH-1 exhibits an asparaginyl hydroxylase activity to catalyze hydroxylation of Asp803 of hypoxia-inducible factor (39). The hydroxylase activity of FIH-1 is dependent on Fe(II), and mutation of His199 to Ala abolished both the interaction with Fe(II) and the hydroxylase activity (39). Although a mutation of His321 to Ala in JMJD5, which corresponds to a mutation of His199 to Ala in FIH-1 (37), abrogates the enzymatic activity of JMJD5 in cultured cells (40), the biological functions and substrates of JMJD5 in hepatocytes remain unknown.
By using the CRISPR/Cas9 system, we established a JMJD5-knockout (JMJD5KO) human hepatoma (High) cell line and reveled that JMJD5 is essential for HBx-mediated HBV replication. Moreover, the deficiency of JMJD5 suppressed the expression of several transcriptional factors involved in hepatocyte differentiation, such as HNF4A, CEBPA, and FOXA3. Expression of a mutant JMJD5 in which His321 was replaced with Ala failed to rescue the expression of HNF4A in JMJD5-knockout Huh7 cells, suggesting that JMJD5 is involved in the maintenance of hepatocyte homeostasis. Further, a random mutagenesis experiment identified Gly135 in JMJD5 as a critical amino acid residue for the interaction with HBx. Replacing Gly135 with Glu in JMJD5 abrogated the interaction with HBx and failed to rescue the replication of HBV in JMJD5-knockout Huh7 cells. We also demonstrated that mutation of His321 to Ala in JMJD5 failed to rescue the replication of HBV. Collectively, these data suggest that direct interaction of JMJD5 with HBx through its Gly135 and hydroxylase activity of JMJD5 facilitates HBV replication.
MATERIALS AND METHODS
Cell lines and viruses.
HEK293T, Huh7, HepG2, HepG2.2.15-7 (41), and Vero cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. Hepatitis C virus (HCV) derived from the genotype 2a JFH-1 strain mutated in E2, p7, and NS2, as shown previously (42), was prepared by serial passages in Huh7.5.1 cells. Japanese encephalitis virus (JEV) (AT31 strain) (43) was prepared in C6/36 cells. Herpes simplex virus 1 (HSV-1) (KOS strain) was prepared in Vero cells. Infectious titers of HCV and JEV were determined by focus-forming assay. Mouse monoclonal antibodies against HCV NS5A (5A27) (44) and JEV NS3 (1791) (45) were used to visualize their foci. The titers of HSV-1 were determined by plaque assay. HBV was prepared from the culture supernatant of HepAD38.7 cells (41). HepAD38.7 cells were cultured in DMEM/F-12 medium supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 18 μg/ml hydrocortisone (Sigma), 5 μg/ml insulin (Sigma), 400 μg/ml G418 (Nacalai Tesque), and 400 ng/ml tetracycline (Nacalai Tesque). To obtain the HBV-containing culture supernatants, HepAD38.7 cells (1 × 106 cells) were seeded on a 10-cm dish (Greiner) coated with collagen type 1 (Nitta Gelatin, Inc.) and cultured with tetracycline-free medium. Culture supernatants were replaced with fresh medium every 3 days postpassage and collected. HBV was concentrated by 15% polyethylene glycol (PEG) solution (molecular weight [MW], 8,000; 0.75 M NaCl and 60 mM EDTA) at 3,000 rpm for 20 min. After PEG precipitation, the pellets were dissolved in primary maintenance medium consisting of Williams E medium supplemented with 5 μg/ml transferrin, 10 ng/ml epidermal growth factor (EGF), 3 μg/ml insulin, 2 mM l-glutamine, 18 μg/ml hydrocortisone, 40 ng/ml dexamethasone, 5 ng/ml sodium selenite, 2% dimethyl sulfoxide (DMSO), 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% FBS (46). For HBV infection, HepG2-hNTCP-C4 cells (2 × 105) (47) were seeded on 24-well plates (Greiner) coated with collagen type 1 and incubated overnight. For HBV infection, 1,000 genome equivalents (GEq)/ml of HBV in the primary maintenance medium containing 4% PEG 8000 (Nacalai Tesque) was inoculated, and the culture medium was replaced every 2 days.
Plasmids.
HBV expression plasmids, pUC19 HBV Ae_US, pUC19 HBV Bj_JPN35, and pUC19 HBV_JPNAT (48), were used for amplification of the regions of HBx. PCR products were cloned into pEF FLAG pGK puro (49), pGEX6P-1 (GE Healthcare), and pGBKT7 (Clontech) vectors. cDNA of JMJD5 was cloned into pEF HA pGK hygro (where HA is hemagglutinin and hygro is hygromycin) and pGADT7 (Clontech) vectors. pEF OSF-HBx was generated by insertion of a One-Strep-FLAG (OSF) tag and HBx cDNA into pEF pGK puro (where puro is puromycin) vector. cDNAs of DDB1, STUB1, SMC4, PSME3, and TIF1B were amplified by using cDNA derived from Huh7 cells and cloned into pGADT7 vector. A cDNA of HNF4A was amplified by using cDNA derived from Huh7 cells and cloned into pEF FLAG pGK puro vector. To express HBV pregenomic RNA (pgRNA), full-length HBV DNA (C_JPNAT strain) was amplified by PCR and cloned into pcDNA3.1(+), with the resulting construct named pCMV pgRNA (where CMV is cytomegalovirus). A lentiviral vector that expresses a short hairpin RNA (shRNA) was generated by using pFTRE3G_pGK_GFP (where GFP is green fluorescent protein) (50). Briefly, pFTRE3G_pGK_GFP was cut by PacI/AscI and cloned into the U6 promoter and pGK_puro cassette, with the resulting product designated FU6_pGKpuro. The shRNA target sequence of JMJD5 was 5′-GCCTGACATGCAGACAGCATT-3′ and was located in the 5′ noncoding region. The shRNA sequence of LacZ was 5′-CTACACAAATCAGCGATTTC-3′. The reporter plasmids of the nucleocapsid promoter of HBV were generated by insertion of a DNA fragment of the HBV nucleocapsid promoter (Ae_US strain). DNA fragments of the core upstream regulatory sequence (CURS) and basic core promoter (BCP) were cloned into pGL4.18 (Promega). The mutations of JMJD5, HBx, and pHBVΔX (C_JPNAT strain) were generated by the method of splicing by overlap extension. For the CRISPR/Cas9 system, the plasmids of pX330 (catalog number 42230) and pCAG EGxxFP (catalog number 50716) were obtained from Addgene. The target sequence of the JMJD5 gene was 5′-CCACTGAGCTCTTCTACGAGGGC-3′. Genomic DNAs of Huh7 were amplified by using the primer set 5′-CAACCACTGAGGATCCCTGCCCAACAGAGAAGACTGC-3′ and 5′-TGCCGATATCGAATTCCACTACTGCACTCCAGCCTG-3′ and cloned into pCAG EGxxFP by using an In-Fusion HD cloning kit (TaKaRa). For lentivirus production, pCMV δR8.2 and pCMV-VSV-G (where VSV-G is vesicular stomatitis virus G protein) were obtained from Addgene. The plasmids used in this study were confirmed by sequencing with an ABI Prism 3130 genetic analyzer (Applied Biosystems).
Immunoprecipitation and immunoblotting.
Cells were lysed with lysis buffer consisting of 20 mM Tris-HCl (pH 7.4), 135 mM NaCl, 1% Triton X-100, 1% glycerol, and protease inhibitor cocktail tablets (Roche), incubated for 30 min at 4°C, and subjected to centrifugation at 14,000 × g for 15 min at 4°C. The supernatants were boiled at 95°C for 5 min and then incubated with anti-FLAG antibody at 4°C for 90 min. After incubation with protein G-Sepharose 4B (GE Healthcare) at 4°C for 90 min, the beads were washed three times by lysis buffer and boiled at 95°C for 5 min. The proteins were resolved by SDS-PAGE (Novex gels; Invitrogen), transferred onto nitrocellulose membranes (iBlot; Life Technologies), blocked with phosphate-buffered saline (PBS) containing 0.05% Tween 20 and 5% skim milk, incubated with primary antibody at room temperature for 12 h, and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody at room temperature for 1 h. The immune complexes were visualized with Super Signal West Femto substrate (Pierce) and detected by an LAS-3000 image analyzer system (Fujifilm).
Mass spectrometry analysis.
293T cells transfected with pEF OSF-HBx or empty vector were incubated for 48 h and lysed by lysis buffer. The cell lysates were incubated with Strep-Tactin Sepharose (IBM GmbH) for 90 min and washed with lysis buffer three times, and then the precipitants were boiled in sample buffer at 95°C for 5 min. Copurified proteins were subjected to SDS-PAGE, stained with Coomassie brilliant blue, and digested by trypsin. The extracted peptides from the SDS-PAGE gel were directly analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using an L-column (C18, 0.1 by 150 mm; CERI) on a Nanoflow LC system (Advance UHPLC; Bruker Daltonics) coupled to an linear trap quadrupole (LTQ) Orbitrap Velos mass spectrometer (Thermo Scientific). Tandem mass spectra were acquired automatically and then searched against a human database from Swiss-Prot with the Mascot server (Matrix Science).
Antibodies.
pGEX6P-1 JMJD5 was transformed into Escherichia coli [E. coli BL21(DE3)/pLysS; Novagen] and glutathione S-transferase (GST) fused with JMJD5 was purified by glutathione-Sepharose 4B (GE Healthcare). Recombinant JMJD5 proteins were immunized into BALB/c mice (4-week-old females; SLC Japan), and hybridomas were generated by fusion with the myeloma cell line PAI. We successfully obtained a clone, named AI6, as an anti-JMJD5 monoclonal antibody. Antibodies to the following proteins were purchased from the indicated manufacturers: anti-FLAG mouse monoclonal antibody (clone M2; Sigma), anti-HA rat monoclonal antibody (clone 3F10; Roche), horseradish peroxidase (HRP)-conjugated anti-FLAG mouse monoclonal antibody (clone M2; Sigma), and anti-β-actin mouse monoclonal antibody (A2228; Sigma).
Yeast two-hybrid assays.
The interaction of HBx and host proteins was analyzed by using a Matchmaker 3 two-hybrid system (Clontech) according to the manufacturer's protocol. Saccharomyces cerevisiae (AH109) cells were transformed with pGBKT7 HBx and pGADT7 host proteins and plated onto yeast medium (synthetic dextrose [SD]) depleted of Leu and Trp (SD/−LW) or depleted of Leu, Trp, Ade, His (SD/−LWAH).
Production of lentiviruses.
HEK293T cells (2 × 106) were seeded on a 10-cm dish and incubated at 37°C for 1 day. A lentiviral transfer vector (FU6_pGKpuro; 1.5 μg), 2.5 μg of pCMV-dR8.2dvpr, and 1 μg of pCMV-VSV-G were mixed with 500 μl of Opti-MEM and 40 μl of polyethylenimine ([PEI], 1 mg/ml; MW, 25,000) (Polysciences, Inc.) and incubated for 15 min. DNA complex was inoculated into HEK293T cells, and the culture medium was changed at 4 h posttransfection. Culture supernatants at 3 days posttransfection were passed through a 0.45-μm-pore-size filter. For the infection of lentivirus, 2 × 105 HepG2.2.15-7 cells seeded on a six-well plate were incubated for 1 day, and the virus-containing culture supernatants (2 ml) and hexadimethrine bromide (polybrene; 4 mg/ml; Sigma) were inoculated onto cells and centrifuged at 2,500 rpm for 45 min at 32°C. Puromycin was added at 2 days postinfection to select stable cell lines.
Purification of intracellular and supernatant HBV relaxed circular DNA (rcDNA) and cellular RNA.
Extracellular and intracellular HBV DNA was extracted as reported previously (48). Briefly, the cell pellets were lysed by using lysis buffer (50 mM Tris-HCl [pH 7.4], 1 mM EDTA, 1% NP-40) at 4°C for 15 min. After centrifugation at 15,000 rpm for 5 min, supernatants were incubated with 7 mM magnesium acetate (MgOAc), 0.2 mg/ml of DNase I (Roche), and 0.1 mg/ml of RNase A (Sigma) at 37°C for 3 h. After the addition of 10 mM EDTA, the lysates were digested by proteinase K (0.3 mg/ml; Life Technologies) and 2% SDS at 37°C for 12 h. Extracted HBV DNA was purified by phenol-chloroform-isoamyl alcohol, precipitated with ethanol, and resolved in pure water. Total RNA was extracted by using IsogenII (Nippon Gene) according to the manufacturer's protocol.
qPCR.
Quantitative PCR (qPCR) for HBV DNA was performed by using Fast SYBR green master mix (Applies Biosystems). The following primers were used for detection of HBV DNA: 5′-GGAGGGATACATAGAGGTTCCTTGA-3′ and 5′-GTTGCCCGTTTGTCCTCTAATTC-3′. Total RNA was used for quantification of mRNA expression by using a Power SYBR green RNA-to-Ct 1-Step kit (Applied Biosystems). The PCR was performed by using a ViiA7 real-time PCR system (Life Technologies). The following primers were used: for HNF4A, 5′-GCAGTGCGTGGTGGACAAAG-3′ and 5′-CTTCGAGTGCTGATCCGGTC-3′; for FOXA3, 5′-GTGAAGATGGAGGCCCATGAC-3′ and 5′-GAGGATTCAGGGTCATGTAGGAGTT-3′; for CEBPA, 5′-GAAGTCGGTGGACAAGAACAG-3′ and 5′-GTCATTGTCACTGGTCAGCTC-3′; for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-TGTAGTTGAGGTCAATGAAGGG-3′ and 5′-ACATCGCTCAGACACCATG-3′. The expression level of each gene was determined by the ΔΔCT (where CTis threshold cycle) method using GAPDH as an internal control.
HBcAg measurement.
The supernatants from HBV-infected HepG2-hNTCP-C4 cells were collected at 10 days postinfection, and the amounts of HBV core antigen (HBcAg) were determined by Lumipulse assay (Fujirebio, Inc.) according to the manufacturer's protocol.
Immunofluorescence microscopy.
HepG2 cells cultured on glass slides for 1 day were transfected with plasmids for the expression of HBx and JMJD5 and fixed with 4% paraformaldehyde in PBS for 2 h at 1 day posttransfection; they were then washed with PBS three times and permeabilized by incubation with 0.2% Triton X-100 in PBS for 15 min. After cells were washed with PBS three times, they were incubated with anti-HA and anti-FLAG antibodies (1/1,000) diluted by 2% FBS in PBS at room temperature for 1 h. The cells were again washed with PBS three times and then incubated with Alexa Fluor 488 (AF488)-conjugated anti-mouse antibody (1/2,000) and AF488-conjugated anti-rat antibody (1/2,000) diluted by 2% FBS in PBS at room temperature for 1 h. The stained cells were covered with Prolong Gold AntiFade reagent with 4′,6′-diamidino-2-phenylindole (DAPI; Life Technologies) and observed under a FluoView FV1000 confocal microscope (Olympus).
Generation of JMJD5-knockout Huh7 cells.
The gene knockout Huh7 cell line was constructed by using CRISPR/Cas9 as described previously (51, 52). Huh7 cells were transfected with pX330 and pCAG EGxxFP and incubated for 1 week. GFP-positive Huh7 cells were sorted by fluorescence-activated cell sorting (FACS) and formed single colonies. Gene deficiency was confirmed by sequencing and Western blotting.
Reporter assay.
Huh7 cells were transfected with either CURS BCP-Luc or BCP-Luc together with pRL-SV40 (where SV40 is simian virus 40), and luciferase activity in the cell lysate was assayed by using a Dual-Luciferase Reporter Assay system (Promega) according to the manufacturer's protocol at 24 h posttransfection.
Southern blotting.
HBV core-incorporated DNA was electrophoresed in 1% agarose with Tris-acetate EDTA (TAE) buffer and transferred onto a nylon membrane (Hybond-N+; GE Healthcare). The pUC19 HBV (C_JPNAT) was digested by AatII and AclI, and the DNA fragment was extracted and purified by using a QIAquick gel extraction kit (Qiagen). Labeling probes, hybridization, and detection were performed by using a Gene Images AlkPhos Direct Labeling and Detection system (Amersham), according to the manufacturer's protocol.
Northern blotting.
Total RNA (5 μg) was subjected to electrophoresis in 1.2% agarose-formaldehyde gel and morpholinepropanesulfonic acid (MOPS) buffer. rRNA was visualized by ethidium bromide staining, and electrophoresed RNA was transferred onto a positively charged nylon membrane (Roche). A pBluscriptII (SK−) HBV-chloramphenicol acetyltransferase (CAT) construct (53) was linearized by HindIII, an RNA probe was synthesized by a digoxigenin (DIG) RNA labeling kit (Roche), and hybridization and detection were performed by using a DIG Northern Starter kit (Roche) according to the manufacturer's protocols. The signals were detected by an LAS-3000 image analyzer system (Fujifilm).
Microarray analyses of Huh7 and JMJD5KO cells.
Total RNA was extracted from Huh7 cells and from JMJD5KO Huh7 cells and purified with an miRNeasy kit (Qiagen) according to the manufacturer's instructions. Microarray analysis was performed as described previously (54). Briefly, 200 ng of total RNA was reverse transcribed, and the resulting cDNAs were used for in vitro transcription. Each cRNA sample was hybridized using a SurePrint G3 human gene expression (8 by 60,000) microarray kit, version 2.0 (model 039494; Agilent Technologies). Raw data were imported into a Subio platform, version 1.18 (Subio), for database management and quality control. Gene expression profiling was compared between Huh7 and JMJD5KO cells.
Random mutagenesis assay.
Random mutagenesis against JMJD5 was introduced by using a GeneMorph II random mutagenesis kit (Agilent Technologies) according to the manufacturer's protocol. The primers used were 5′-ATCGATACGGGATCCATATGGCTGGAGACACCCACTG-3′ and 5′-TCATCTGCAGCTCGAGCTACGACCACCAGAAGCTGA-3′. PCR products were electrophoresed, purified by using a QIAquick gel extraction kit (Qiagen), and cloned into pGADT7 vector by using an In-Fusion HD cloning kit (TaKaRa). After transformation, the bacterial colonies were picked up, and their plasmids were extracted and used to transform yeasts into yeasts expressing HBx, which were then plated on SD/−LW or SD/−LWAH medium.
JMJD5 structural modeling.
The JMJD5 secondary structure and the disordered regions were predicted from the amino acid sequence using the programs PSIPRED (55) and DISOPRED (56), respectively. The putative structure of the N-terminal region (residues 1 to 150) of JMJD5 was modeled using I-TASSER (57). The electrostatic potential was calculated by the APBS tool, and figures were rendered by PyMol (The PyMol Molecular Graphics System, version 1.7.6, Schrödinger, LLC).
Microarray data accession number.
The microarray data have been deposited in the NCBI Gene Expression Omnibus (GEO) database under accession number GSE73491.
RESULTS
Identification of JMJD5 as a binding partner for HBx.
To identify a novel HBx-interacting protein, we performed coimmunoprecipitation and MS analyses. 293T cells transfected with an empty vector or a One-Strep-FLAG (OSF) tag HBx expression vector and HBx and HBx-binding proteins were purified using Strep-Tactin beads and subjected to SDS-PAGE, and the resolving gel was visualized by Coomassie brilliant blue solution (Fig. 1A, left). Proteins that had entered the running gel were identified by using LC-MS/MS (Fig. 1A, right). We identified the DDB1/CUL4 complex, which was previously reported as HBx-binding proteins (33). CSN5, a component of the COP9 signalosome, was also reported to be an interacting protein with HBx by using yeast two-hybrid screening (4). In addition, all of the COP9 signalosome components, CSN1 to CSN7, were coimmunoprecipitated with HBx in this assay. HBx has been reported to be involved in the ubiquitin proteasome system (58) and in the regulation of gene expression (59). Therefore, we selected STUB1, TIF4, and PSME3, which are known to be involved in the induction of proteasome degradation, and nuclear proteins such as SMC4 and JMJD5. We next determined whether these candidate proteins directly bind to HBx in yeasts. Yeast cells were transformed by using a GAL4-activated domain fused with DDB1, STUB1, SMC4, PSME3, TIF1B, or JMJD5 and a GAL4-binding domain fused with HBx. The transformed yeast cells were plated on yeast medium depleted of Leu and Trp (SD/−LW) or depleted of Leu, Trp, Ade, and His (SD/−LWAH). DDB1 was originally identified as an HBx-binding protein by using yeast two-hybrid screening (60), and in this study we used DDB1 as a positive control. Interestingly, only JMJD5 and DDB1, but not the other candidates, exhibited binding to HBx in yeasts (Fig. 1B). Therefore, we focused on JMJD5 for further experiments. Because HBV has eight different genotypes (A to H), we next examined whether JMJD5 is capable of binding to HBx derived from different genotypes. Although FLAG-HBx of genotypes A, B, and C were coprecipitated with HA-JMJD5 in 293T cells expressing these proteins, the interaction between JMJD5 and HBx of genotype B was weaker than that of other genotypes (Fig. 1C). To further determine the cellular localization of HBx and JMJD5, FLAG-HBx and/or HA-JMJD5 was expressed in Huh7 cells. Although HBx and JMJD5 were localized in the cytoplasm and nucleus, respectively, some fractions of JMJD5 were translocated to the cytoplasm in cells coexpressing HBx (Fig. 1D), suggesting that HBx interacts with JMJD5 in the cytoplasm. Collectively, these results suggest that JMJD5 is a novel binding partner of HBx.
FIG 1.

Identification of JMJD5 as a binding partner for HBx. (A) Empty vector or OSF-HBx was expressed in 293T cells, and cell lysates were purified by Strep-Tactin beads. Samples were subjected to SDS-PAGE and stained by Coomassie brilliant blue. Gels were cut, and proteins were identified by LC-MS/MS. The identified proteins are listed. (B) Yeasts were transformed by the indicated cDNAs and HBx and cultured on SD/−LWAH or SD/−LW plates. AD, GAL4-activated domain; BD, GAL4 DNA binding domain. (C) 293T cells were transfected with HA-JMJD5 and FLAG-HBx of genotype A, B, or C. Proteins immunoprecipitated (IP) with anti-FLAG antibody were subjected to Western blotting. (D) mCherry-JMJD5 and/or Aequorea coerulescens GFP (AcGFP)-HBx were expressed in Huh7 cells and stained with DAPI at 48 h posttransfection after fixation.
JMJD5 participates in HBV replication.
To examine the roles of JMJD5 in HBV replication, shRNA against JMJD5 was lentivirally transduced in HepG2.2.15-7 cells, which produce HBV particles (41, 61), resulting in JMJD5-knockdown (JMJD5KD) HepG2.2.15-7 cells. We also prepared a monoclonal antibody against JMJD5 (AI6) after immunization of mice with recombinant JMJD5 proteins. Endogenous expression of JMJD5 detected by AI6 antibody was reduced in JMJD5KD HepG2.2.15-7 cells (Fig. 2A). No significant difference in cell growth was observed between control and JMJD5KD HepG2.2.15-7 cells by MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay (Fig. 2B). To examine the roles of JMJD5 on HBV replication, JMJD5KD HepG2.2.15-7 cells were incubated for 6 days. The levels of both intracellular and extracellular HBV DNAs in JMJD5KD HepG2.2.15-7 cells were lower than those of cells transfected with control shRNA (Fig. 2C). Southern blot analysis revealed a reduction of HBV DNA in JMJD5KD HepG2.2.15-7 cells (Fig. 2D). Northern blot analysis showed no significant difference in transcription levels of HBV RNA between control and JMJD5KD HepG2.2.15-7 cells (Fig. 2E). In addition, the reductions of HBV DNA by reverse transcriptase inhibitor treatment were comparable between control and JMJD5KD HepG2.2.15-7 cells (Fig. 2F), suggesting that susceptibility to treatment with the reverse transcriptase inhibitor is not affected by suppression of JMJD5 expression. To further confirm the roles of JMJD5 in HBV replication, we established JMJD5KD HepG2.2.15-7 cells expressing HA-tagged RNA interference (RNAi)-resistant JMJD5. The amounts of intracellular HBV DNA were significantly enhanced by the expression of RNAi-resistant JMJD5 in JMJD5KD HepG2.2.15-7 cells (Fig. 2G). In addition, the reduction of HBcAg expression in JMJD5KD HepG2.2.15-7 cells was rescued by the exogenous expression of RNAi-resistant JMJD5 (Fig. 2H). Recently, HepG2 cells expressing sodium taurocholate cotransporting polypeptide (NTCP) were shown to be susceptible to HBV infection (46). Next, therefore, we established the JMJD5KD cell line upon transfection with shRNA into HepG2-hNTCP-C4 cells stably expressing NTCP (47) (Fig. 2I) and inoculated with HBV derived from the supernatants of HepAD38.7 cells (41). Although the amounts of HBV DNA were comparable between the HepG2-hNTCP-C4 cell lines established by transfection with either an shRNA targeting LacZ (shLacZ) or shJMJD5 at 2 days postinfection, HBV DNA in JMJD5KD HepG2-hNTCP-C4 cells was significantly decreased at 10 days postinfection (Fig. 2J). The amounts of HBcAg in the culture supernatants were also decreased in the JMJD5KD HepG2-hNTCP-C4 cells (Fig. 2K). Finally, Northern blot analysis revealed that HBV RNA was significantly reduced in JMJD5KD HepG2-hNTCP-C4 dells (Fig. 2L). Taken together, these results suggest that JMJD5 participates in HBV replication.
FIG 2.

JMJD5 participates in HBV replication. (A) The control and JMJD5KD HepG2.2.15-7 cell lines were lysed and subjected to Western blotting. (B) Control and JMJD5KD HepG2.2.15-7 cells were seeded onto 24-well plates and incubated for 3 days. The cell growth of each cell line was analyzed by MTT assay. Data represent the means ± standard deviations of two independent experiments. (C) Control and JMJD5KD HepG2.2.15-7 cells (2 × 105) were treated with DNase and RNase, and core-incorporated HBV DNA resistant to the treatment was precipitated by ethanol after proteinase K digestion and quantified by qPCR (left). Culture supernatants of the control and JMJD5KD HepG2.2.15-7 cells were collected after 3 days of incubation, and the core-incorporated HBV DNA was extracted and quantified by qPCR (right). Data represent the means ± standard deviations of two independent experiments. (D) Core-incorporated HBV DNA was extracted from the control and JMJD5KD HepG2.2.15-7 (2 × 106) cells and subjected to Southern blotting. ssDNA, single-stranded DNA. (E) The total RNA was extracted from the control and JMJD5KD HepG2.2.15-7 cells and subjected to Northern blotting. (F) The control and JMJD5KD HepG2.2.15-7 cells were treated with 10 μM lamivudine for 6 days, and intracellular HBV DNA was quantified by qPCR. Data represent the means ± standard deviations of two independent experiments. NTC, no-treatment control. (G) Core-incorporated HBV DNA was extracted from control HepG2.2.15-7 cells, JMJD5KD HepG2.2.15-7 cells, and JMJD5KD HepG2.2.15-7 cells stably expressing shRNA-resistant HA-JMJD5 and quantified by qPCR. Data represent the means ± standard deviations of two independent experiments. (H) Western blotting of the control, JMJD5KD, and JMJD5KD stably expressing shRNA-resistant HA-JMJD5 HepG2.2.15-7 cells. (I) Western blotting of the control and JMJD5KD HepG2-hNTCP-C4 cells. (J) The viral DNA levels of control and JMJD5KD HepG2-hNTCP-C4 cells infected with HBV (1000 GEq/cell) were determined by Southern blotting at 2 days (left) and 10 days (right) postinfection. (K) HBcAg in the culture supernatants of the control and JMJD5KD HepG2-hNTCP-C4 cells infected with HBV (1,000 GEq/cell) was quantified at 10 days postinfection. Data represent the means ± standard deviations of two independent experiments. (L) The viral RNA levels of control and JMJD5KD HepG2-hNTCP-C4 cells infected with HBV (1,000 GEq/cell) were determined by Northern blotting at 10 days postinfection.
Establishment of JMJD5-knockout (KO) Huh7 cells by the CRISPR/Cas9 system.
Next, to examine the roles of JMJD5 on viral replication in a human hepatoma cell line, Huh7, we established two JMJD5-deficient Huh7 cell lines (JMJD5KO Huh7 lines 45 and 47) by the CRISPR/Cas9 system (Fig. 3A) and JMJD5-deficient Huh7 cell lines complemented with HA-JMJD5 (Fig. 3B). The growth of JMJD5KO Huh7 cells was similar to that of parental Huh7 cells (Fig. 3C). Although complementation with HA-JMJD5 in JMJD5-deficient Huh7 cells exhibited a slight enhancement of HSV-1 propagation, there was no effect on the propagation of JEV and HCV (Fig. 3D), suggesting that deficiency of JMJD5 in Huh7 cells has only a moderate or no effect on the propagation of HSV-1, JEV and HCV.
FIG 3.

Establishment of JMJD5-knockout Huh7 cells by a CRISPR/Cas9 system. (A) Capitalized letters indicate the sequences of the CRISPR/Cas9 system targeting JMJD5. Gene knockout by sequence modification in the JMJD5 gene is shown. The dotted lines and the characters in red indicate deletion and insertion of the sequences, respectively. WT, wild type. (B) Western blotting of Huh7 cells, JMJD5-knockout Huh7 cell lines (JMJD5KO Huh7 lines 45 and 47) and JMJD5KO Huh7 cells complemented with HA-JMJD5. (C) Growth kinetics of Huh7 cells, and JMJD5KO Huh7 lines 45 and 47 cells. Cells seeded onto 24-well plates were cultured for 3 days, and cell growth was analyzed by MTT assay. Data represent the means ± standard deviations of two independent experiments. (D) Huh7 cells, JMJD5KO Huh7 (line 45) cells, and JMJD5KO Huh7 cells complemented with HA-JMJD5 were infected with either HSV-1 (multiplicity of infection of 0.1), JEV (multiplicity of infection of 1), or HCV (multiplicity of infection of 3), and infectious titers in the culture supernatants were determined at 2 to 3 days postinfection. Data represent the means ± standard deviations of two independent experiments. FFU, focus-forming units.
Deficiency of JMJD5 downregulates several host factors involved in liver homeostasis.
Although JMJD5 has been shown to be a tumor suppressor against lymphoma (62), JMJD5 has also been suggested to function as an oncogene by regulating hypoxia-inducible factor 1α (HIF-1α) through its interaction with pyruvate kinase muscle isozyme (PKM2) (40) or cyclin A1 (63) and to be involved in colon carcinogenesis (64). Therefore, the function of JMJD5, especially in hepatocytes, remains unclear. To examine the roles of JMJD5 in hepatocytes, we performed DNA microarray analysis by using parental and JMJD5KO Huh7 cells and showed that numbers of transcription factors involved in the hepatocyte development and differentiation listed by Qiagen's Ingenuity Pathway Analysis (IPA) were downregulated in JMJD5KO Huh7 cells (see Table S1 in the supplemental material). Among them, HNF4A, FOXA1, FOXA2, and FOXA3 have been shown to convert primary mouse embryonic fibroblasts into hepatocyte-like cells (65). Therefore, we next examined the roles of JMJD5 on the expression of HNF4A, CEBPA, and FOXA3 by qPCR. Deficiency of JMJD5 significantly reduced the expression of HNF4A, and exogenous expression of JMJD5 in JMJD5KO Huh7 cells recovered the expression (Fig. 4A). Similar to HNF4A expression, the expression levels of CEBPA and FOXA3 were downregulated in JMJD5KO Huh7 cells and then rescued by exogenous expression of JMJD5 (Fig. 4B and C). The jumonji C (JmjC) domain of JMJD5 has been predicted to possess activity of hydroxylase rather than demethylase (37, 38). To determine the enzymatic activity of JMJD5 on HNF4A expression, an enzymatically deficient mutant, JMJD5 H321A, that possessed a substitution of Ala for His321 in the JmjC domain, was expressed in JMJD5KO Huh7 cells, but no significant enhancement of HNF4A expression was observed by the expression of this mutant (Fig. 4D), suggesting that JMJD5 regulates the expression of several transcriptional factors involved in the maintenance of hepatocyte function through its enzymatic activity. The HBV nucleocapsid promoter region contains binding sites for HNF4 and CEBPA (Fig. 4E, top) (66), and HNF4A has been suggested to be involved in the expression of the HBV nucleocapsid (67–69). Therefore, we considered that downregulation of HNF4A in JMJD5KO Huh7 cells likely suppresses the promoter activity of HBV nucleocapsid. To test this, we constructed a luciferase reporter plasmid to express firefly luciferase under the control of the HBV nucleocapsid promoter. The region of the basic core promoter (BCP) contains an HNF4-binding site. The region of the core upstream regulatory sequence (CURS) contains one HNF4A- and two CEBP-binding sites (Fig. 4E, top). The luciferase assay revealed that there was no significant difference in the promoter activities of the HBV nucleocapsid between JMJD5KO Huh7 cells and Huh7 cells with restored JMJD5 expression (Fig. 4E, bottom). Previous studies showed that the activity of the HBV nucleocapsid promoter was enhanced by the overexpression of HNF4A. Therefore, we examined the effect of overexpression of HNF4A on the nucleocapsid promoter. In spite of an 80-fold increase in HNF4A expression by transfection (Fig. 4F), only marginal and 2-fold increases in luciferase activity were observed in the BCP and CURS promoters, respectively (Fig. 4G). These results suggest that a 50% reduction of HNF4A expression by the knockdown of JMJD5 has no effect on the promoter activity of the HBV nucleocapsid, but JMJD5 might play some roles in the maintenance of hepatocytes. Further studies are needed to clarify the biological significance of JMJD5 in liver homeostasis.
FIG 4.

Deficiency of JMJD5 downregulates several host factors involved in liver homeostasis. (A to C) The expression levels of HNF4A, CEBPA, and FOXA1 in Huh7 cells, JMJD5KO Huh7 cells (lines 45 and 47), and JMJD5KO Huh7 cells complemented with HA-JMJD5 were determined by qPCR and expressed as the relative expression after normalization with expression of GAPDH. Data represent the means ± standard deviations of two independent analyses. Significant differences are indicated by asterisks (*, P < 0.05). (D) Expression levels of HNF4A in Huh7 cells, JMJD5KO Huh7 cells (line 45), and JMJD5KO Huh7 cells complemented with either the wild-type or the H321A mutant of HA-JMJD5 were determined by qPCR and expressed as the relative expression after normalization with expression of GAPDH. Data represent the means ± standard deviations of two independent analyses. Significant differences are indicated by asterisks (*, P < 0.05). (E) Reporter plasmids carrying a firefly luciferase gene under either the basic core promoter (BCP-Luc) or the core upstream regulatory sequence and BCP (CURS BCP-Luc) of the HBV nucleocapsid were transfected with pRL-SV40 into JMJD5KO Huh7 cells (line 45) and JMJD5KO Huh7 cells complemented with HA-JMJD5. Relative luciferase activities were determined by using a dual-luciferase kit (Promega) at 24 h posttransfection. Data represent the means ± standard deviations of two independent analyses. (F) Levels of mRNA of HNF4A were determined by qPCR upon overexpression of HNF4A. Data are shown as relative expression levels normalized by the expression of GAPDH. (G) The reporter plasmids (BCP-Luc and CURS BCP-Luc) were transfected with pRL-SV40 into Huh7 cells and those overexpressing HNF4A. Relative luciferase activities were determined by using a dual-luciferase kit (Promega) at 24 h posttransfection. Data represent the means ± standard deviations of two independent analyses. Significant differences are indicated by asterisks (*, P < 0.05).
JMJD5 participates in HBx-mediated HBV replication.
Next, to determine the roles of JMJD5 in HBV replication, Huh7 cells, JMJD5KO Huh7 cells, and Huh7 cells with restored JMJD5 expression were transfected with an HBV expression plasmid (pHBV C_JPNAT), and the levels of intracellular HBV DNA were determined by qPCR and Southern blotting at 3 days posttransfection (Fig. 5A and B). To examine the effects of interaction between JMJD5 and HBx on transcription of HBV pgRNA, we constructed the expression vector of pgRNA under the CMV promoter (pCMV pgRNA) and transfected it into parental, JMJD5KO, and JMJD5-restored JMJD5KO Huh7 cells. Intracellular core-incorporated HBV DNA was reduced in JMJD5KO Huh7 cells but was restored by the exogenous expression of JMJD5 (Fig. 5C). In addition, Southern blot analysis also confirmed the reduction of HBV DNA replication by the knockout of JMJD5 in Huh7 cells (Fig. 5D). These data suggest that interaction of JMJD5 and HBx has no effect on the transcription of HBV RNA from the viral genome but participates in the posttranscriptional steps. Intracellular HBV core-incorporated DNA was significantly reduced in JMJD5KO Huh7 cells but was restored by the exogenous expression of JMJD5, suggesting that JMJD5 regulates HBV replication. To examine the role of JMJD5 on HBx-mediated HBV replication, JMJD5KO Huh7 cells were transfected with a plasmid encoding an HBV genome lacking the HBx region (pHBVΔX) (21). Although exogenous expression of HBx enhanced the replication of HBVΔX in a dose-dependent manner in Huh7 cells (Fig. 5E), rescue of HBVΔX replication by the expression of HBx was not observed in JMJD5KO Huh7 cells, in contrast to Huh7 and JMJD5KO Huh7 cells complemented with JMJD5 (Fig. 5F), suggesting that the interaction between HBx and JMJD5 participates in HBx-mediated HBV replication.
FIG 5.
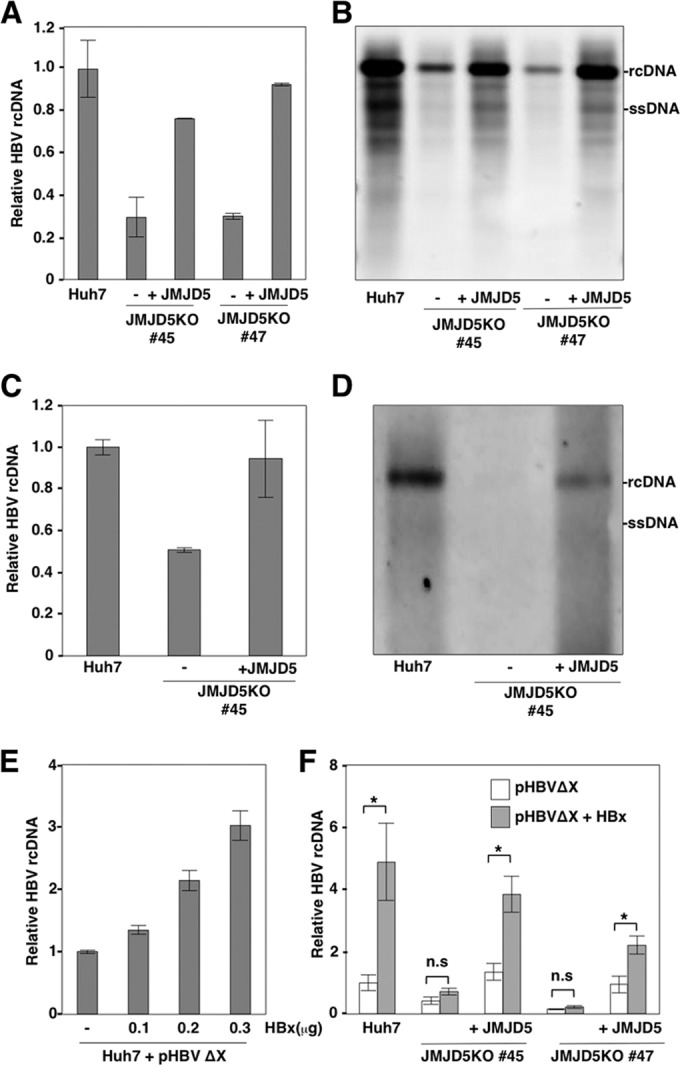
JMJD5 participates in HBx-mediated HBV replication. (A and B) JMJD5KO Huh7 cell lines (lines 45 and 47) and those complemented with HA-JMJD5 were transfected with pHBV plasmids (genotype C, C_JPNAT strain), and intracellular core-incorporated HBV DNA was quantified by qPCR (A) and Southern blotting (B) at 3 days posttransfection. Data of qPCR represent the means ± standard deviations of two independent experiments. (C) JMJD5KO Huh7 cells (line 45) and JMJD5KO Huh7 cells complemented with HA-JMJD5 were transfected with pCMV HBV pgRNA plasmids, and intracellular core-incorporated HBV DNA was quantified by qPCR. Data represent the means ± standard deviations of two independent analyses. (D) JMJD5KO Huh7 cells (line 45) and JMJD5KO Huh7 cells complemented with HA-JMJD5 were transfected with pCMV HBV pgRNA plasmids, and intracellular core-incorporated HBV DNA was examined by Southern blotting. (E) Huh7 cells were transfected with pHBVΔX together with various amounts of HBx expression vector (0, 0.1, 0.2, and 0.3 μg/well), and intracellular core-incorporated HBV DNA was quantified by qPCR at 3 days posttransfection. Data represent the means ± standard deviations of two independent experiments. (F) Huh7 cells, JMJD5KO Huh7 cells (lines 45 and 47), and JMJD5KO Huh7 cells complemented with HA-JMJD5 were transfected with pHBVΔX together with/without HBx expression vector (0.3 μg/well), and the level of intracellular core-incorporated HBV DNA was determined by qPCR at 3 days posttransfection. Data represent the means ± standard deviations of two independent experiments. Significant differences are indicated by asterisks (*, P < 0.05).
The hydroxylase activity of JMJD5 and the interaction of JMJD5 with HBx play crucial roles in HBV replication.
To further investigate the interaction between JMJD5 and HBx in more detail, random mutations were introduced in JMJD5 by using a commercial kit. PCR products were cloned into vectors for yeast two-hybrid screening, and the effects of JMJD5 mutations on HBx interaction were examined. Although most of the mutations introduced into JMJD5 were deletion and insertion of stop codons, we identified mutant 2 of JMJD5, which lacks growth in yeasts in the absence of Leu, Trp, Ade, and His (Fig. 6A). The cDNA of the JMJD5 mutant 2 was transferred to a mammalian expression vector and expressed in 293T cells, and subsequent immunoprecipitation analysis revealed that interaction of the JMJD5 mutant 2 with HBx was severely impaired (Fig. 6B). Sequence analysis identified six amino acid substitutions in JMJD5 mutant 2. To identify which amino acid residue in JMJD5 was crucial for the interaction with HBx, we established six mutant plasmids encoding JMJD5 possessing a single amino acid substitution in one of the six amino acids identified in mutant 2, and each of the resulting JMJD5 mutants was examined to determine its interaction with HBx by immunoprecipitation experiments. We found that mutation of Gly135 to Glu in JMJD5 (G135E) significantly reduced the interaction with HBx (Fig. 6C), suggesting that Gly135 is a critical amino acid residue for the interaction with HBx. To assess the impact of G135E mutation on HBV replication, JMJD5KO Huh7 cells were transfected with either the wild-type JMJD5 or G135E JMJD5 together with HBV expression plasmids. Although the expression of G135E JMJD5 restored the expression of HNF4A to the same level as in the wild-type JMJD5 (Fig. 6D), HBV replication was not rescued by the expression of G135E JMJD5 in JMJD5KO Huh7 cells (Fig. 6E), suggesting that the interaction between JMJD5 and HBx is crucial for HBV replication. In addition, to examine the role of the hydroxylase activity of JMJD5 in HBV replication, we complemented JMJD5KO Huh7 cells with either the wild-type JMJD5 or a hydroxylase-deficient mutant of JMJD5 H321A. H321A JMJD5 exhibited binding to HBx (Fig. 6F) but did not rescue the replication of HBV in JMJD5KO Huh7 cells (Fig. 6G), suggesting that hydroxylase activity is also essential for HBV replication. Taken together, our data suggest that the interaction of JMJD5 with HBx and the enzymatic activity of the JmjC domain in JMJD5 play crucial roles in HBV replication.
FIG 6.

The hydroxylase activity of JMJD5 and the interaction of JMJD5 with HBx play crucial roles in HBV replication. (A) JMJD5 cDNA was amplified by using random mutagenesis kit (Agilent Technologies). PCR products were cloned into pGADT7 vectors. Yeasts possessing BD-HBx were transformed with pGADT7 JMJD5 mutants. Representative yeast clones grown on SD/−LW or SD/−LWAH plates are shown. (B) A plasmid encoding FLAG-HBx was transfected into 293T cells together with a plasmid encoding either the wild type or mutants of HA-JMJD, and cell lysates were immunoprecipitated with anti-FLAG antibody and analyzed by Western blotting. (C) A schematic representation of JMJD5 mutant 2, with the shaded area indicating the JmjC domain (top). FLAG-HBx was transfected into 293T cells together with plasmids encoding HA-JMJD of either the wild type or substitution mutants of E42V, S68R, G135E, K209E, T244I, or E366G, and then the cell lysates were immunoprecipitated with anti-FLAG antibody and analyzed by Western blotting. (D and E) The levels of expression of HN4A (D) and intracellular core-incorporated HBV DNA (E) in Huh7 cells and JMJD5KO Huh7 cells (line 45) transfected with the HBV expression plasmid together with either the wild type or the G135E mutant of HA-JMJD5 were determined by qPCR at 3 days posttransfection. The expression of HN4A was normalized with that of GAPDH. Data represent the means ± standard deviations of two independent analyses. (F) A plasmid encoding FLAG-HBx was transfected into 293T cells together with a plasmid encoding either the wild type or the H321A mutant of HA-JMJD, and the cell lysates were immunoprecipitated with anti-FLAG antibody and analyzed by Western blotting. (G) The levels of intracellular core-incorporated HBV DNA in Huh7 cells and JMJD5KO Huh7 cells (line 45) transfected with pHBV plasmids (genotype C, C_JPNAT strain) together with either the wild type or the H321A mutant of JMJD5 were quantified by qPCR at 3 days posttransfection. Data represent the means ± standard deviations of two independent experiments. WT, wild type.
DISCUSSION
HBx is a multifunctional protein, and HBx transgenic mice generated by several groups develop HCC at around 1 year of age (17). HBx had been also shown to integrate into several host genes such as LINE1 and TERT, and these chimeric transcriptional products stimulate liver cancer incidents (20, 70), suggesting that HBx directly or indirectly participates in liver carcinogenesis. Numerous molecular mechanisms of HBx-induced HCC have already been reported. For example, HBx has been shown to directly interact with the tumor suppressor gene p53 and to inactivate p53-mediated transcription (25). Although HBx has been shown to activate RAS (7), the incidence of HBx-induced HCC in mice was accelerated by the knockdown of p53 (71), suggesting that inhibition of p53 by HBx is not sufficient to induce liver cancer in mice. In addition, cooperation with NRAS was not needed for HBx-induced liver cancer in mice (71). Further studies are needed to clarify the molecular mechanisms by which HBx promotes the development of HCC and the infection with HBV. The production of HBV particles in culture supernatants upon transfection with pHBVΔX was significantly lower than that with the wild-type HBV plasmid (21–23). Moreover, HBVΔX failed to induce viremia in chimeric mice transplanted with human hepatocytes (24), suggesting that HBx is required for HBV propagation.
In this study, we identified JMJD5 as a novel HBx-binding protein regulating HBV replication. Although HBx has been shown to localize in both the cytoplasm and nucleus (72), HBx was mainly detected in the cytoplasm in this study, while JMJD5 was clearly localized in the nucleus. Coexpression of JMJD5 with HBx resulted in the translocation of some fraction of JMJD5 from the nucleus to cytoplasm. The expression level of HBx is low in the liver of hepatitis B patients, and studies based on overexpression of HBx have not employed physiological conditions (3). Indeed, our data suggested that HBx specifically interacts with JMJD5 although further studies will be needed to investigate the interaction between JMJD5 and HBx under physiological conditions.
Knockdown of JMJD5 in HepG2.2.15-7 cells resulted in significant reductions of intracellular and extracellular HBV DNA, and expression of an RNAi-resistant JMJD5 in the knockdown cells restored the replication of viral DNA and expression of core protein, suggesting that the expression of JMJD5 protein participates in the replication of HBV. To further confirm the role of the interaction between JMJD5 and HBx in the replication of HBV, we generated JMJD5-knockout Huh7 cells and found that replication of HBV was severely impaired in the knockout cells. Although amounts of pgRNA were no different in control and JMJD5KD HepG2.2.15-7 cells (Fig. 2E), intracellular HBV DNA levels were lower in JMJD5KO Huh7 cells even with the transcription of pgRNA by the CMV promoter (Fig. 5C and D), suggesting that interaction of JMJD5 and HBx participates in posttranscriptional steps such as stability and multimerization of HBV capsids. In addition, rescue of the replication of HBVΔX in the knockout cells was required for exogenous expression of both HBx and JMJD5. Although our loss-of-function and gain-of-function data suggest that JMJD5 participates in the HBx-mediated HBV replication, further studies are needed to clarify the role of JMJD5 in the replication of HBV in more detail.
JMJD5 was originally identified as a tumor suppressor gene by gene trap screening (62); however, JMJD5 has also been reported as an oncogene by other groups (40, 63). DNA microarray analysis of JMJD5-knockout Huh7 cells revealed that deficiency of JMJD5 suppresses the expression of liver factors, as reported previously (65, 73, 74). Exogenous expression of JMJD5, particularly the JmjC domain of JMJD5, in the JMJD5-knockdown cells restored the expression of the liver factors, including HNF4A, CEBPA, and FOXA3. To confirm the role of JMJD5 on hepatocyte differentiation through the regulation of liver factors, investigation of the liver-specific JMJD5-knockout mice is needed.
Random mutagenesis and functional screening using yeasts identified Gly135 of JMJD5 as a critical amino acid residue for interaction with HBx, and replacing Gly135 with Glu abrogated the rescue of HBV replication by the expression of JMJD5 in the JMJD5-knockout cells. Furthermore, the expression of an enzymatically deficient JMJD5 mutant in which His321 was replaced with Ala in the JmjC domain was also incapable of restoring HBV replication in the JMJD5-knockout cells. These results suggest that both the association of JMJD5 with HBx and the hydroxylase activity in JMJD5 are required for efficient replication of HBV. JMJD6, which has a JmjC domain structurally similar to that of JMJD5 (36, 38), possesses lysyl-hydroxylase activity and catalyzes the U2 small nuclear ribonucleoprotein auxiliary factor 65-kDa subunit (U2AF65) to regulate RNA splicing (75) and hydroxylation of lysine residues in histone (76, 77). Further studies are needed to identify the molecular targets of JMJD5 in hepatocytes.
Structural modeling and disorder predictions in silico suggested the presence of an alpha helix domain in the N terminus of JMJD5 (Fig. 7A) and the possibility of direct interaction with partner proteins through the exposed Gly135 on the surface of JMJD5. In addition, the molecular surface around Gly135 in JMJD5 was deduced to be highly hydrophobic (Fig. 7B), and substitution of Glu for Gly135 drastically altered the electrostatic potential from positive to negative (Fig. 7C), consistent with a scenario in which substitution of Glu for Gly135 in JMJD5 leads to abrogation of the interaction with HBx.
FIG 7.

Structural modeling and disorder predictions in silico of the N-terminal domain of JMJD5. (A) Structural model of the N-terminal domain of JMJD5. White indicates Gly135. (B and C) Electrostatic potential models of the wild type (B) and G135E mutant (C) of the N-terminal domain of JMJD5. The orientation of the model was identical to that in shown in panel A. Red and blue indicate negatively and positively charged areas, respectively. Blue circles indicate the surface around Gly135.
In conclusion, we have identified JMJD5 as a novel HBx-binding partner and suggested that both the hydroxylase activity of JMJD5 and the direct interaction of JMJD5 with HBx through Gly135 facilitate HBV replication. Currently, reverse transcriptase inhibitors are available only for the treatment of hepatitis B patients. Intervention with the interaction between JMJD5 and HBx might be a novel target for the development of anti-HBV agents.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to M. Tomiyama for secretarial work and to M. Ishibashi and M. Nagao for their technical assistance. We also thank D. C. S. Huang, A. Ryo, and T. Suzuki for providing experimental materials. The Core Instrumentation Facility of Osaka University conducted the FACS sorting.
This work was supported by the Research Program on Hepatitis from the Japan Agency for Medical Research and Development, the Japanese Ministry of Education, Culture, Sports, Science, and Technology, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, and the Uehara Memorial Foundation.
T. Kouwaki, T. Okamoto, and Y. Matsuura designed the research; T. Kouwaki, T. Okamoto, A. Ito, Y. Sugiyama T. Suzuki, S. Kusakabe, J. Hirano, and T. Fukuhara performed most of the experiments; K. Saito performed the mass analysis; D. Okuzaki performed the DNA microarray analysis; K. Yamashita and D. M. Standley performed the prediction of the JMJD5 structure. A. Yamashita, K. Watashi, M. Sugiyama, S. Yoshio, T. Kanto, K. Moriishi, and M. Mizokami assisted with the experiments; and T. Kouwaki, T. Okamoto, and Y. Matsuura wrote the manuscript.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02776-15.
REFERENCES
- 1.Nassal M. 2008. Hepatitis B viruses: reverse transcription a different way. Virus Res 134:235–249. doi: 10.1016/j.virusres.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 2.Ott JJ, Stevens GA, Groeger J, Wiersma ST. 2012. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 30:2212–2219. doi: 10.1016/j.vaccine.2011.12.116. [DOI] [PubMed] [Google Scholar]
- 3.Benhenda S, Cougot D, Buendia M-A, Neuveut C. 2009. Hepatitis B virus X protein molecular functions and its role in virus life cycle and pathogenesis. Adv Cancer Res 103:75–109. doi: 10.1016/S0065-230X(09)03004-8. [DOI] [PubMed] [Google Scholar]
- 4.Tanaka Y, Kanai F, Ichimura T, Tateishi K, Asaoka Y, Guleng B, Jazag A, Ohta M, Imamura J, Ikenoue T, Ijichi H, Kawabe T, Isobe T, Omata M. 2006. The hepatitis B virus X protein enhances AP-1 activation through interaction with Jab1. Oncogene 25:633–642. [DOI] [PubMed] [Google Scholar]
- 5.Su F, Schneider RJ. 1996. Hepatitis B virus HBx protein activates transcription factor NF-κB by acting on multiple cytoplasmic inhibitors of rel-related proteins. J Virol 70:4558–4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maguire HF, Hoeffler JP, Siddiqui A. 1991. HBV X protein alters the DNA binding specificity of CREB and ATF-2 by protein-protein interactions. Science 252:842–844. doi: 10.1126/science.1827531. [DOI] [PubMed] [Google Scholar]
- 7.Benn J, Schneider RJ. 1995. Hepatitis B virus HBx protein deregulates cell cycle checkpoint controls. Proc Natl Acad Sci U S A 92:11215–11219. doi: 10.1073/pnas.92.24.11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park SG, Chung C, Kang H, Kim J-Y, Jung G. 2006. Up-regulation of cyclin D1 by HBx is mediated by NF-κB2/BCL3 complex through κB site of cyclin D1 promoter. J Biol Chem 281:31770–31777. doi: 10.1074/jbc.M603194200. [DOI] [PubMed] [Google Scholar]
- 9.Bouchard M, Giannakopoulos S, Wang EH, Tanese N, Schneider RJ. 2001. Hepatitis B virus HBx protein activation of cyclin A–cyclin-dependent kinase 2 complexes and G1 transit via a Src kinase pathway. J Virol 75:4247–4257. doi: 10.1128/JVI.75.9.4247-4257.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin-Lluesma S, Schaeffer C, Robert EI, van Breugel PC, Leupin O, Hantz O, Strubin M. 2008. Hepatitis B virus X protein affects S phase progression leading to chromosome segregation defects by binding to damaged DNA binding protein 1. Hepatology 48:1467–1476. doi: 10.1002/hep.22542. [DOI] [PubMed] [Google Scholar]
- 11.Geng X, Huang C, Qin Y, McCombs JE, Yuan Q, Harry BL, Palmer AE, Xia N-S, Xue D. 2012. Hepatitis B virus X protein targets Bcl-2 proteins to increase intracellular calcium, required for virus replication and cell death induction. Proc Natl Acad Sci U S A 109:18471–18476. doi: 10.1073/pnas.1204668109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geng X, Harry BL, Zhou Q, Skeen-Gaar RR, Ge X, Lee ES, Mitani S, Xue D. 2012. Hepatitis B virus X protein targets the Bcl-2 protein CED-9 to induce intracellular Ca2+ increase and cell death in Caenorhabditis elegans. Proc Natl Acad Sci U S A 109:18465–18470. doi: 10.1073/pnas.1204652109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. 1998. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol 72:8586–8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu L, Chen L, Li L, Sun H, Yang G, Chang Y, Tu Q, Wu M, Wang H. 2011. Hepatitis B virus X protein enhances cisplatin-induced hepatotoxicity via a mechanism involving degradation of Mcl-1. J Virol 85:3214–3228. doi: 10.1128/JVI.01841-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee W-P, Lan K-H, Li C-P, Chao Y, Lin H-C, Lee S-D. 2012. Pro-apoptotic or anti-apoptotic property of X protein of hepatitis B virus is determined by phosphorylation at Ser31 by Akt. Arch Biochem Biophys 528:156–162. doi: 10.1016/j.abb.2012.08.008. [DOI] [PubMed] [Google Scholar]
- 16.Kim J-H, Sohn S-Y, Benedict Yen TS, Ahn B-Y. 2008. Ubiquitin-dependent and -independent proteasomal degradation of hepatitis B virus X protein. Biochem Biophys Res Commun 366:1036–1042. doi: 10.1016/j.bbrc.2007.12.070. [DOI] [PubMed] [Google Scholar]
- 17.Kim CM, Koike K, Saito I, Miyamura T, Jay G. 1991. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature 351:317–320. doi: 10.1038/351317a0. [DOI] [PubMed] [Google Scholar]
- 18.Yu DY, Moon HB, Son JK, Jeong S, Yu SL, Yoon H, Han YM, Lee CS, Park JS, Lee CH, Hyun BH, Murakami S, Lee KK. 1999. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J Hepatol 31:123–132. doi: 10.1016/S0168-8278(99)80172-X. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Cui F, Lv Y, Li C, Xu X, Deng C, Wang D, Sun Y, Hu G, Lang Z, Huang C, Yang X. 2004. HBsAg and HBx knocked into the p21 locus causes hepatocellular carcinoma in mice. Hepatology 39:318–324. doi: 10.1002/hep.20076. [DOI] [PubMed] [Google Scholar]
- 20.Lau CC, Sun T, Ching AK, He M, Li JW, Wong AM, Co NN, Chan AW, Li PS, Lung RW, Tong JH, Lai PB, Chan HL, To KF, Chan TF, Wong N. 2014. Viral-human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell 25:335–349. doi: 10.1016/j.ccr.2014.01.030. [DOI] [PubMed] [Google Scholar]
- 21.Blum HE, Zhang ZS, Galun E, von Weizsäcker F, Garner B, Liang TJ, Wands JR. 1992. Hepatitis B virus X protein is not central to the viral life cycle in vitro. J Virol 66:1223–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melegari M, Scaglioni PP, Wands JR. 1998. Cloning and characterization of a novel hepatitis B virus X binding protein that inhibits viral replication. J Virol 72:1737–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Z, Torii N, Hu Z, Jacob J, Liang TJ. 2001. X-deficient woodchuck hepatitis virus mutants behave like attenuated viruses and induce protective immunity in vivo. J Clin Invest 108:1523–1531. doi: 10.1172/JCI200113787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsuge M, Hiraga N, Akiyama R, Tanaka S, Matsushita M, Mitsui F, Abe H, Kitamura S, Hatakeyama T, Kimura T, Miki D, Mori N, Imamura M, Takahashi S, Hayes CN, Chayama K. 2010. HBx protein is indispensable for development of viraemia in human hepatocyte chimeric mice. J Gen Virol 91:1854–1864. doi: 10.1099/vir.0.019224-0. [DOI] [PubMed] [Google Scholar]
- 25.Truant R, Antunovic J, Greenblatt J, Prives C, Cromlish JA. 1995. Direct interaction of the hepatitis B virus HBx protein with p53 leads to inhibition by HBx of p53 response element-directed transactivation. J Virol 69:1851–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang T, Xie N, He W, Liu R, Lei Y, Chen Y, Tang H, Liu B, Huang C, Wei Y. 2013. An integrated proteomics and bioinformatics analyses of hepatitis B virus X interacting proteins and identification of a novel interactor apoA-I. J Proteomics 84:92–105. doi: 10.1016/j.jprot.2013.03.028. [DOI] [PubMed] [Google Scholar]
- 27.Hong A, Han DD, Wright CJ, Burch T, Piper J, Osiowy C, Gao C, Chiang S, Magill T, Dick K, Booth TF, Li X, He R. 2012. The interaction between hepatitis B virus X protein and AIB1 oncogene is required for the activation of NFκB signal transduction. Biochem Biophys Res Commun 423:6–12. doi: 10.1016/j.bbrc.2012.05.021. [DOI] [PubMed] [Google Scholar]
- 28.Benhenda S, Ducroux A, Rivière L, Sobhian B, Ward MD, Dion S, Hantz O, Protzer U, Michel M-L, Benkirane M, Semmes OJ, Buendia M-A, Neuveut C. 2013. Methyltransferase PRMT1 is a binding partner of HBx and a negative regulator of hepatitis B virus transcription. J Virol 87:4360–4371. doi: 10.1128/JVI.02574-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pang R, Lee TK, Poon RT, Fan ST, Kwong YL, Tse E. 2007. PIN1 interacts with a specific serine-proline motif of hepatitis B virus X protein to enhance hepatocarcinogenesis. Gastroenterology 132:1088–1103. doi: 10.1053/j.gastro.2006.12.030. [DOI] [PubMed] [Google Scholar]
- 30.Kumar M, Jung SY, Hodgson AJ, Madden CR, Qin J, Slagle BL. 2011. Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of beta interferon. J Virol 85:987–995. doi: 10.1128/JVI.01825-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalra N, Kumar V. 2006. The X protein of hepatitis B virus binds to the F box protein Skp2 and inhibits the ubiquitination and proteasomal degradation of c-Myc. FEBS Lett 580:431–436. doi: 10.1016/j.febslet.2005.12.034. [DOI] [PubMed] [Google Scholar]
- 32.Becker SA, Lee TH, Butel JS, Slagle BL. 1998. Hepatitis B virus X protein interferes with cellular DNA repair. J Virol 72:266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bergametti F, Sitterlin D, Transy C. 2002. Turnover of hepatitis B virus X protein is regulated by damaged DNA-binding complex. J Virol 76:6495–6501. doi: 10.1128/JVI.76.13.6495-6501.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li T, Robert EI, van Breugel PC, Strubin M, Zheng N. 2010. A promiscuous alpha-helical motif anchors viral hijackers and substrate receptors to the CUL4-DDB1 ubiquitin ligase machinery. Nat Struct Mol Biol 17:105–111. doi: 10.1038/nsmb.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leupin O, Bontron S, Schaeffer C, Strubin M. 2005. Hepatitis B virus X protein stimulates viral genome replication via a DDB1-dependent pathway distinct from that leading to cell death. J Virol 79:4238–4245. doi: 10.1128/JVI.79.7.4238-4245.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Del Rizzo PA, Krishnan S, Trievel RC. 2012. Crystal structure and functional analysis of JMJD5 indicate an alternate specificity and function. Mol Cell Biol 32:4044–4052. doi: 10.1128/MCB.00513-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang H, Zhou X, Wu M, Wang C, Zhang X, Tao Y, Chen N, Zang J. 2013. Structure of the JmjC-domain-containing protein JMJD5. Acta Crystallogr D Biol Crystallogr 69:1911–1920. doi: 10.1107/S0907444913016600. [DOI] [PubMed] [Google Scholar]
- 38.Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, Ratcliffe PJ, Schofield CJ. 2003. Structure of factor-inhibiting hypoxia-inducible factor (HIF) reveals mechanism of oxidative modification of HIF-1 alpha. J Biol Chem 278:1802–1806. doi: 10.1074/jbc.C200644200. [DOI] [PubMed] [Google Scholar]
- 39.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. 2002. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev 16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang H-J, Hsieh Y-J, Cheng W-C, Lin C-P, Lin Y-S, Yang S-F, Chen C-C, Izumiya Y, Yu J-S, Kung H-J, Wang W-C. 2014. JMJD5 regulates PKM2 nuclear translocation and reprograms HIF-1α-mediated glucose metabolism. Proc Natl Acad Sci U S A 111:279–284. doi: 10.1073/pnas.1311249111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ogura N, Watashi K, Noguchi T, Wakita T. 2014. Formation of covalently closed circular DNA in Hep38.7-Tet cells, a tetracycline inducible hepatitis B virus expression cell line. Biochem Biophys Res Commun 452:315–321. doi: 10.1016/j.bbrc.2014.08.029. [DOI] [PubMed] [Google Scholar]
- 42.Russell RS, Meunier JC, Takikawa S, Faulk Engle RE, Bukh J, Purcell RH, Emerson SU. 2008. Advantages of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc Natl Acad Sci U S A 105:4370–4375. doi: 10.1073/pnas.0800422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao Z, Date T, Li Y, Kato T, Miyamoto M, Yasui K, Wakita T. 2005. Characterization of the E-138 (Glu/Lys) mutation in Japanese encephalitis virus by using a stable, full-length, infectious cDNA clone. J Gen Virol 86:2209–2220. doi: 10.1099/vir.0.80638-0. [DOI] [PubMed] [Google Scholar]
- 44.Okamoto K, Mori Y, Komoda Y, Okamoto T, Okochi M, Takeda M, Suzuki T, Moriishi K, Matsuura Y. 2008. Intramembrane processing by signal peptide peptidase regulates the membrane localization of hepatitis C virus core protein and viral propagation. J Virol 82:8349–8361. doi: 10.1128/JVI.00306-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Katoh H, Mori Y, Kambara H, Abe T, Fukuhara T, Morita E, Moriishi K, Kamitani W, Matsuura Y. 2011. Heterogeneous nuclear ribonucleoprotein A2 participates in the replication of Japanese encephalitis virus through an interaction with viral proteins and RNA. J Virol 85:10976–10988. doi: 10.1128/JVI.00846-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, Fu L, Song M, Chen P, Gao W, Bijie R, Yinyan S, Cai T, Feng X, Sui J, Li W. 2012. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iwamoto M, Watashi K, Tsukuda S, Aly HH, Fukasawa M, Fujimoto A, Suzuki R, Aizaki H, Ito T, Koiwai O, Kusuhara H, Wakita T. 2014. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem Biophys Res Commun 443:808–813. doi: 10.1016/j.bbrc.2013.12.052. [DOI] [PubMed] [Google Scholar]
- 48.Sugiyama M, Tanaka Y, Kato T, Orito E, Ito K, Acharya SK, Gish RG, Kramvis A, Shimada T, Izumi N, Kaito M, Miyakawa Y, Mizokami M. 2006. Influence of hepatitis B virus genotypes on the intra- and extracellular expression of viral DNA and antigens. Hepatology 44:915–924. doi: 10.1002/hep.21345. [DOI] [PubMed] [Google Scholar]
- 49.Huang DC, Cory S, Strasser A. 1997. Bcl-2, Bcl-XL and adenovirus protein E1B19kD are functionally equivalent in their ability to inhibit cell death. Oncogene 14:405–414. doi: 10.1038/sj.onc.1200848. [DOI] [PubMed] [Google Scholar]
- 50.Kelly GL, Grabow S, Glaser SP, Fitzsimmons L, Aubrey BJ, Okamoto T, Valente LJ, Robati M, Tai L, Fairlie WD, Lee EF, Lindstrom MS, Wiman KG, Huang DCS, Bouillet P, Rowe M, Rickinson AB, Herold MJ, Strasser A. 2014. Targeting of MCL-1 kills MYC-driven mouse and human lymphomas even when they bear mutations in p53. Genes Dev 28:58–70. doi: 10.1101/gad.232009.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujihara Y, Ikawa M. 2014. CRISPR/Cas9-based genome editing in mice by single plasmid injection. Methods Enzymol 546:319–336. doi: 10.1016/B978-0-12-801185-0.00015-5. [DOI] [PubMed] [Google Scholar]
- 52.Mashiko D, Fujihara Y, Satouh Y, Miyata H, Isotani A, Ikawa M. 2013. Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Sci Rep 3:3355. doi: 10.1038/srep03355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Okuyama-Dobashi K, Kasai H, Tanaka T, Yamashita A, Yasumoto J, Chen W, Okamoto T, Maekawa S, Watashi K, Wakita T, Ryo A, Suzuki T, Matsuura Y, Enomoto N, Moriishi K. 2015. Hepatitis B virus efficiently infects non-adherent hepatoma cells via human sodium taurocholate cotransporting polypeptide. Sci Rep 5:17047. doi: 10.1038/srep17047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fukuhara T, Kambara H, Shiokawa M, Ono C, Katoh H, Morita E, Okuzaki D, Maehara Y, Koike K, Matsuura Y. 2012. Expression of microRNA miR-122 facilitates an efficient replication in nonhepatic cells upon infection with hepatitis C virus. J Virol 86:7918–7933. doi: 10.1128/JVI.00567-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones DT. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 56.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. 2004. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol 337:635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 57.Yang J, Yan R, Roy A, Xu D, Zhang Y. 2015. The I-TASSER suite: protein structure and function prediction. Nat Methods 12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu Z, Zhang Z, Doo E, Coux O, Goldberg AL, Liang TJ. 1999. Hepatitis B virus X protein is both a substrate and a potential inhibitor of the proteasome complex. J Virol 73:7231–7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Belloni L, Pollicino T, De Nicola F, Guerrieri F, Raffa G, Fanciulli M, Raimondo G, Levrero M. 2009. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci U S A 106:19975–19979. doi: 10.1073/pnas.0908365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee TH, Elledge SJ, Butel JS. 1995. Hepatitis B virus X protein interacts with a probable cellular DNA repair protein. J Virol 69:1107–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sells MA, Chen ML, Acs G. 1987. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci U S A 84:1005–1009. doi: 10.1073/pnas.84.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Suzuki T, Minehata K-I, Akagi K, Jenkins NA, Copeland NG. 2006. Tumor suppressor gene identification using retroviral insertional mutagenesis in Blm-deficient mice. EMBO J 25:3422–3431. doi: 10.1038/sj.emboj.7601215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hsia DA, Tepper CG, Pochampalli MR, Hsia EYC, Izumiya C, Huerta SB, Wright ME, Chen H-W, Kung H-J, Izumiya Y. 2010. KDM8, a H3K36me2 histone demethylase that acts in the cyclin A1 coding region to regulate cancer cell proliferation. Proc Natl Acad Sci U S A 107:9671–9676. doi: 10.1073/pnas.1000401107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang R, Huang Q, Li Y, Song Y, Li Y. 2015. JMJD5 is a potential oncogene for colon carcinogenesis. Int J Clin Exp Pathol 8:6482–6489. [PMC free article] [PubMed] [Google Scholar]
- 65.Sekiya S, Suzuki A. 2011. Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature 475:390–393. doi: 10.1038/nature10263. [DOI] [PubMed] [Google Scholar]
- 66.Kramvis A, Kew MC. 1999. The core promoter of hepatitis B virus. J Viral Hepat 6:415–427. doi: 10.1046/j.1365-2893.1999.00189.x. [DOI] [PubMed] [Google Scholar]
- 67.Zheng Y, Li J, Ou JH. 2004. Regulation of hepatitis B virus core promoter by transcription factors HNF1 and HNF4 and the viral X protein. J Virol 78:6908–6914. doi: 10.1128/JVI.78.13.6908-6914.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bock CT, Malek NP, Tillmann HL, Manns MP, Trautwein C. 2000. The enhancer I core region contributes to the replication level of hepatitis B virus in vivo and in vitro. J Virol 74:2193–2202. doi: 10.1128/JVI.74.5.2193-2202.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Raney AK, Johnson JL, Palmer CN. 1997. Members of the nuclear receptor superfamily regulate transcription from the hepatitis B virus nucleocapsid promoter. J Virol 71:1058–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Keng VW, Tschida BR, Bell JB, Largaespada DA. 2011. Modeling hepatitis B virus X-induced hepatocellular carcinoma in mice with the sleeping beauty transposon system. Hepatology 53:781–790. doi: 10.1002/hep.24091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jhunjhunwala S, Jiang Z, Stawiski EW, Gnad F, Liu J, Mayba O, Du P, Diao J, Johnson S, Wong KF, Gao Z, Li Y, Wu TD, Kapadia SB, Modrusan Z, French DM, Luk JM, Seshagiri S, Zhang Z. 2014. Diverse modes of genomic alteration in hepatocellular carcinoma. Genome Biol 15:436. doi: 10.1186/s13059-014-0436-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Henkler F, Hoare J, Waseem N, Goldin RD, McGarvey MJ, Koshy R, King IA. 2001. Intracellular localization of the hepatitis B virus HBx protein. J Gen Virol 82:871–882. doi: 10.1099/0022-1317-82-4-871. [DOI] [PubMed] [Google Scholar]
- 73.Zaret KS. 2002. Regulatory phases of early liver development: paradigms of organogenesis. Nat Rev Genet 3:499–512. doi: 10.1038/nrg837. [DOI] [PubMed] [Google Scholar]
- 74.Zaret KS, Grompe M. 2008. Generation and regeneration of cells of the liver and pancreas. Science 322:1490–1494. doi: 10.1126/science.1161431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Webby CJ, Wolf A, Gromak N, Dreger M, Kramer H, Kessler B, Nielsen ML, Schmitz C, Butler DS, Yates JR, Delahunty CM, Hahn P, Lengeling A, Mann M, Proudfoot NJ, Schofield CJ, Böttger A. 2009. Jmjd6 catalyses lysyl-hydroxylation of U2AF65, a protein associated with RNA splicing. Science 325:90–93. doi: 10.1126/science.1175865. [DOI] [PubMed] [Google Scholar]
- 76.Han G, Li J, Wang Y, Li X, Mao H, Liu Y, Chen CD. 2012. The hydroxylation activity of Jmjd6 is required for its homo-oligomerization. J Cell Biochem 113:1663–1670. doi: 10.1002/jcb.24035. [DOI] [PubMed] [Google Scholar]
- 77.Unoki M, Masuda A, Dohmae N, Arita K, Yoshimatsu M, Iwai Y, Fukui Y, Ueda K, Hamamoto R, Shirakawa M, Sasaki H, Nakamura Y. 2013. Lysyl 5-hydroxylation, a novel histone modification, by Jumonji domain containing 6 (JMJD6). J Biol Chem 288:6053–6062. doi: 10.1074/jbc.M112.433284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qiao L, Wu Q, Lu X, Zhou Y, Fernández-Alvarez A, Ye L, Zhang X, Han J, Casado M, Liu Q. 2013. SREBP-1a activation by HBs and the effect on hepatitis B virus enhancer II/core promoter. Biochem Biophys Res Commun 432:643–649. doi: 10.1016/j.bbrc.2013.02.030. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.