

ABSTRACT
In addition to infectious viral particles, hepatitis B virus-replicating cells secrete large amounts of subviral particles assembled by the surface proteins, but lacking any capsid and genome. Subviral particles form spheres (22-nm particles) and filaments. Filaments contain a much larger amount of the large surface protein (LHBs) compared to spheres. Spheres are released via the constitutive secretory pathway, while viral particles are ESCRT-dependently released via multivesicular bodies (MVBs). The interaction of virions with the ESCRT machinery is mediated by α-taxilin that connects the viral surface protein LHBs with the ESCRT component tsg101. Since filaments in contrast to spheres contain a significant amount of LHBs, it is unclear whether filaments are released like spheres or like virions. To study the release of subviral particles in the absence of virion formation, a core-deficient HBV mutant was generated. Confocal microscopy, immune electron microscopy of ultrathin sections and isolation of MVBs revealed that filaments enter MVBs. Inhibition of MVB biogenesis by the small-molecule inhibitor U18666A or inhibition of ESCRT functionality by coexpression of transdominant negative mutants (Vps4A, Vps4B, and CHMP3) abolishes the release of filaments while the secretion of spheres is not affected. These data indicate that in contrast to spheres which are secreted via the secretory pathway, filaments are released via ESCRT/MVB pathway like infectious viral particles.
IMPORTANCE This study revises the current model describing the release of subviral particles by showing that in contrast to spheres, which are secreted via the secretory pathway, filaments are released via the ESCRT/MVB pathway like infectious viral particles. These data significantly contribute to a better understanding of the viral morphogenesis and might be helpful for the design of novel antiviral strategies.
INTRODUCTION
The human hepatitis B virus (HBV) is a spherical particle, 42 nm in diameter, consisting of an outer envelope and an inner icosahedral nucleocapsid. The nucleocapsid is formed by the core protein and harbors the viral genomic DNA. The HBV genome encodes at least four different open reading frames, coding for the viral polymerase, the core and the e antigen (HBcAg and HBeAg), the regulatory X protein (HBx), and the three different surface proteins (HBsAg): the large HBV surface protein (LHBs), the middle surface protein (MHBs) and the small surface protein (SHBs) (1). LHBs encompasses the PreS1 domain, the PreS2 domain, and the S domain, MHBs consists of the PreS2 and the S domain, and SHBs contains the S domain. These surface proteins are not only constitutive components of the envelope of viral particles but also assemble to capsid-free subviral particles lacking any viral genome having the shape of spheres and filaments (2) that are secreted in 1,000- to 100,000-fold excess relative to infectious viral particles. SHBs, the predominant part of these subviral particles, can assemble to 22-nm spherical particles. The incorporation of larger amounts of LHBs in these subviral particles results in the formation of filamentous structures with 22-nm diameters and variable lengths (3, 4). The relevance of subviral particles for the viral life cycle is not fully understood. It has been reported that the release of viral particles is not directly affected by interference with the secretion of subviral particles (5, 6), but they seem to enhance the infectivity of HBV (7). Apart from this, subviral particles are assumed to sequester HBV-specific antibodies.
Spheres self-assemble in the lumen of the endoplasmic reticulum (ER). They are transported to the ER-Golgi intermediate compartment (ERGIC) and released by the general secretory pathway (8, 9). They are efficiently secreted and do not accumulate within the hepatocytes. Recent work demonstrates that HBV particles are released by a different pathway. The release of virions occurs ESCRT (endosomal sorting complex required for transport)-dependently via multivesicular bodies (MVBs) (8, 10, 11). ESCRT-MVB complex is mainly composed of ESCRT-I, ESCRT-II, and ESCRT-III (12). ESCRT-III is the core component and formed by charged multivesicular body proteins (CHMPs), such as CHMP3 (13–15). ESCRT-III recruits the vacuolar protein sorting 4A and 4B (Vps4A/B) to constrict membranes and mediate fission (16, 17). It has been reported that by interaction with the HBV capsid and LHBs, the endosomal sorting and trafficking adaptor γ2-adaptin and endosomal ubiquitin ligase Nedd4 are involved in the egress of HBV (11, 18). Moreover, a recent study identified α-taxilin as an essential factor for the release of HBV. α-Taxilin mediates the interaction of the viral particle with the ESCRT machinery by binding, on the one hand, to the PreS1 domain of LHBs and, on the other hand, to the ESCRT-I component tsg101 (19). In addition, previous electron microscopy (EM) studies showed that HBV viral particles and filaments were formed by a tubular budding along the membrane of dilated intracellular cisternae in HBV stably expressing HepG2 cells (20, 21). In light of later observations that the release of HBV occurs MVB dependently, it was speculated that these structures could represent MVBs or early endosomes. Filaments are enriched in LHBs. LHBs was found to interact with γ2-adaptin. The LHBs/γ2-adaptin interaction was described to be relevant for the MVB-dependent release of HBV (11, 18). Therefore, it was speculated that filaments may assemble into MVB-related compartments by a conventional membrane-derived budding process (8).
However, it remained unclear whether filaments that are characterized by a significant larger amount of LHBs compared to spheres are released like viral particles or secreted as spheres. To address this question, we generated a core-deficient HBV mutant to study the release of filaments in the absence of virion formation. Moreover, the MVB-dependent release was blocked, and the effect on the amount of filaments in the supernatant was analyzed. The data obtained indicate that while spheres are released via the classic secretory pathway, filaments like viral particles are MVB-dependently released.
MATERIALS AND METHODS
Cells and transient transfection.
The human hepatocarcinoma cell lines Huh 7.5 and HepG2 were grown in Dulbecco modified Eagle medium (DMEM; Lonza, Belgium) and supplemented with 10% fetal calf serum, 0.1 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine (DMEM complete). Huh 7.5 and HepG2 cells were transfected using linear polyethyleneimine (Polysciences, Inc.) as described previously (22).
Antibodies and chemicals.
Rab7- and Rab 5B-specific antibodies were purchased from St. Cruz Biotech (St. Cruz, CA). The monoclonal MA18/7 anti-PreS1 domain of LHBs was kindly provided by D. Glebe (23), Giessen, Germany. The monoclonal anti-SHBs antibody HB01 which detects a linear epitope at amino acids 120 to 125 was kindly provided by Aurelia Zvirbliene (23), Department of Immunology and Cell Biology, Vilnius University, Vilnius, Lithuania. The polyclonal rabbit against denatured HBV core K46 (18) was a gift from Reinhild Prange, Department of Medical Microbiology and Hygiene, Johannes Gutenberg-Universität Mainz, Mainz, Germany. Polyclonal rabbit anti-HBV core B0586 was purchased from Dako, Denmark. Goat-derived polyclonal antibody ab17183 against hepatitis B virus surface antigen was ordered from Abcam. Anti-β-actin was ordered from Sigma-Aldrich (St. Louis, MO). Green fluorescent protein (GFP)-specific antibody A11122 was purchased from Invitrogen. Purified mouse anti-GM130 clone 35 as a marker for the Golgi apparatus and protein disulfide isomerase (PDI)-specific antibody were ordered from BD Transduction Laboratories. Alexa Fluor-FluoroNanogold conjugates and HQ silver enhancement kits were ordered from Nanoprobes, Inc., Yaphank, NY. For quantification of HBsAg, an Enzygnost HBsAg enzyme-linked immunosorbent assay (ELISA; Siemens, Germany) was used. For double immunofluorescence staining, a donkey-derived mouse-specific Alexa 488-labeled secondary antibody from Invitrogen (Darmstadt, Germany) and a donkey-derived rabbit-specific Cy3-labeled secondary antibody were used (Jackson Laboratories, Bar Harbor, ME). U18666A, which inhibits the transportation of cholesterol (24) and alters the trafficking of MVB-associated membrane proteins (25, 26), was ordered from Sigma-Aldrich (St. Louis, MO). PrestoBlue cell viability reagent purchased from Life Technologies was used to analyze the cytotoxic effect.
Plasmids.
The plasmid p1.2×HBV harbors a 1.2-fold HBV genome genotype D. Plasmid HBV/A carrying 1.5-fold autologous promoter-driven genomes of genotype A2 (pCEP-Pur-gtA1.5) (27) was used. The plasmid pSHBs/A was subcloned from pCEP-Pur-gtA1.5 and inserted into pCDNA.3 vector (Invitrogen, Carlsbad, CA). The core-deficient HBV mutant 1.2×HBVΔCore was generated by site-directed mutagenesis based on p1.2×HBV. The primers 5′-TGC CTT GGG TGG CTT TGG GGC TGA GAC ATC GAC CCT TAT AAA GAA and 5′-TTC TTT ATA AGG GTC GAT GTC TCA GCC CCA AAG CCA CCC AAG GCA target the start codon of the core protein. (The inserted stop codon is indicated in boldface type.) The mutations of core were verified by DNA sequencing. pGalT-eGFP encoding β-1,4-galactosyltransferase was kindly provided by R. Duden, Lübeck, Germany. peYFP-Rab7 was a gift from B. Munz, Tübingen. The plasmids peGFP-Vps4A.wt, peGFP-Vps4A.dn, pDsRed-Vps4B.wt, pDsRed-Vps4B.dn, and pCHMP3.YFP (11) were generously provided by Reinhild Prange, Department of Medical Microbiology and Hygiene, Johannes Gutenberg-Universität Mainz, Mainz, Germany.
Subcellular fractionation by sucrose and Percoll gradient ultracentrifugation.
The isolation of MVBs has recently been described in detail (28). For Percoll gradient ultracentrifugation, postnuclear supernatant (PNS) was centrifuged at 12,000 × g for 20 min. The supernatant was diluted with isotonic Percoll (pH 7.4; Pharmacia Fine Chemicals, Piscataway, NJ) to a density of 1.070 g/ml. This material was centrifuged in a Beckman ALT110 rotor for 45 min at 29,000 ×g. A total of 17 fractions were collected from the above and subjected to refractive index analysis and SDS-PAGE.
HBsAg-ELISA.
HBsAg was determined using a commercial ELISA system (Siemens). The results are expressed by the quotient of the absorbance signal of the sample (s) and the cutoff value (co).
SDS-PAGE and Western blot analysis.
SDS-PAGE and Western blot analysis were performed according to standard procedures (29). Detection of bound secondary antibody was performed by via enhanced chemiluminescence using Super-Signal West Pico chemiluminescent substrate (Thermo Scientific, Freiburg, Germany) and Immobilon Western chemiluminescent HRP substrate (Merck-Millipore, Germany).
Indirect immunofluorescence analysis.
Fixation and staining were performed as described recently (30). Immunofluorescence staining was analyzed using a confocal laser scanning microscope (CLSM 510; Carl Zeiss, Germany).
Electron microscopy.
HBV viral particles and subviral particles were produced by transient transfection and partially purified using sucrose gradient centrifugation as described previously (31). Freshly glow-discharged carbon-coated nickel grids were incubated with sucrose gradient-derived samples for 3 min and washed with distilled water four times. The grids were negatively stained with 2% aqueous solution of uranyl acetate for 10 s. For immunogold staining, sucrose gradient derived fractions were 1:5 diluted with phosphate-buffered saline. The samples were then incubated with polyclonal goat anti-HBsAg antibody for 3 h, followed by incubation with gold-conjugated rabbit anti-goat IgG for 1 h. The specimens were examined using a Zeiss EM-109 transmission electron microscope.
Correlative light and electron microscopy.
Huh 7.5 cells were transfected with p1.2×HBVΔCore. At 48 h after transfection, the cells were reseeded on Sarstedt Lumox multiwell plate. Then, 24 h later, the cells were fixed with 2% formaldehyde and stained with primary antibody MA18/7 specific to LHBs and secondary antibody coupled to Alexa Fluor-FluoroNanogold. To visualize bound FluoroNanogold in the ultrathin section by electron microscope, the gold particles were enlarged by using an HQ silver enhancement kit as recommended by the manual. Areas with LHBs-positive cells as evidenced by fluorescence microscopy were marked with the help of a diamond scratcher on the bottom of the culture dish and prepared for ultrathin sections according to standard procedures (32).
Statistical analysis.
The significance of results was analyzed by two-tailed t test using GraphPad Prism version 5.04. Error bars in the figures represent the standard deviations.
RESULTS
The synthesis and subcellular distribution of LHBs and SHBs are not affected by the lack of core protein.
To study the release of subviral particles in the absence of virion formation but without affecting the ratio between the viral surface proteins, The p1.2×HBVΔCore was generated by site-directed mutagenesis of the wild-type HBV (wtHBV) genome genotype D (p1.2×HBV) (33). The start codon of the core protein was mutated to stop codon (Fig. 1A). To clarify whether the lack of core protein affects the production of LHBs and SHBs, cellular lysates derived from Huh 7.5 cells transfected with p1.2×HBVΔCore and wtHBV (p1.2×HBV) were analyzed by HBsAg ELISA and Western blotting. Lysates from pUC18-transfected cells served as a negative control. Quantification of the intracellular HBsAg by ELISA (Fig. 1B) showed that comparable amounts of HBsAg were produced in cells expressing core-deficient HBV mutant compared to wtHBV-expressing cells. This was confirmed by Western blotting of these cellular lysates using the LHBs-specific monoclonal antibody MA18/7 (Fig. 1C). Comparable results were obtained for HepG2 cells expressing the core-deficient HBV mutant or wtHBV (Fig. 1C).
FIG 1.

Lack of virion production neither affects the synthesis nor the intracellular distribution of surface proteins. (A) Schematic illustration of the inserts encoding wtHBV (p1.2×HBV) and HBV core-deficient mutant (p1.2×HBVΔCore). The core-deficient mutant was generated based on p1.2×HBV harboring a 1.2-fold wtHBV genome (genotype D) by site-directed mutagenesis. (B) HBsAg ELISA of lysates derived from pUC18 (negative control)-, p1.2×HBVΔCore-, and p1.2×HBV-transfected Huh 7.5 cells, respectively. (C) Western blot analysis of lysates from Huh 7.5 and HepG2 cells transfected with the p1.2×HBVΔCore, p1.2×HBV, or pUC18. A mouse-derived LHBs-specific antibody (MA18/7) and a rabbit-derived HBV core-specific antibody K46 were used for detection. As indicated by arrows, LHBs was detected as double bands, the p39 and its glycosylated form gp42. The lower double bands marked with stars represent an unspecific binding of the antibody. When lysates from Huh 7.5 or HepG2 cells expressing HBV were analyzed by Western blotting with MA18/7, the p39-specific band overlaps the upper, nonspecific band. However, when the corresponding supernatants were analyzed by Western blotting with MA18/7, only p39 and gp42 could be detected. Detection of β-actin was used as a loading control. (D and E) p1.2×HBVΔCore- or p1.2×HBV-transfected Huh 7.5 cells were analyzed by confocal immunofluorescence microscopy staining with the LHBs-specific antibody MA18/7 (green) (D) and the Core-specific antibody B0586 (red) or the SHBs-specific monoclonal antibody HB01 (red) (E). Nuclei were stained with DAPI (4′,6′-diamidino-2-phenylindole; blue).
Moreover, confocal immunofluorescence microscopy, using MA18/7 and a core-specific polyclonal serum (Fig. 1D) or a SHBs-specific monoclonal antibody (HB01) (Fig. 1E), showed that comparable amounts of LHBs are present in Huh 7.5 cells expressing the core-deficient mutant or the wtHBV genome. Regarding the subcellular distribution of LHBs (Fig. 1D) and HBsAg (Fig. 1E), there was no difference detectable in Huh 7.5 cells expressing the core-deficient mutant compared to the wtHBV. The lack of any core-specific signal from Western blot (Fig. 1C) and immunofluorescence microscopy (Fig. 1D) in cells expressing the mutant confirms the deficiency of core protein in these cells. These data indicate that the deficiency in core protein neither impairs the synthesis of LHBs and SHBs nor affects their subcellular distribution.
Filaments are released from core-deficient HBV mutant.
To investigate whether HBsAg is released from cells expressing the core-deficient HBV mutant, HBsAg-specific ELISAs were performed. The result showed that the lack of core has no effect on the release of HBsAg (Fig. 2A). Western blot analysis of the supernatant using the LHBs-specific antibody MA18/7 showed that comparable amounts of LHBs are released from Huh 7.5 cells expressing wtHBV or the core-deficient mutant, respectively (Fig. 2B). Comparable results were obtained for HepG2 cells expressing wtHBV or the core-deficient mutant, respectively (Fig. 2B). Further analysis of the supernatants by density gradient centrifugation and subsequent Western blot analysis of the fractions showed for Huh 7.5 cells that in the case of supernatant derived from wtHBV, as well as in the case of supernatant from cells expressing the 1.2×HBVΔCore, LHBs was found in fractions 7 to 16, with a major peak in fractions 9 to 12. Comparable results were obtained for HepG2 cells expressing either wtHBV or the core-deficient mutant. Here, LHBs was detected in the fractions 8 to 15, with a major peak in fractions 10 to 12 (Fig. 2C). This was consistent with the HBsAg-specific ELISA of these fractions (Fig. 2D), demonstrating that particles with comparable density are released from wtHBV, as well as from 1.2×HBVΔCore-expressing cells. Furthermore, fractions 9 to 12 concentrated from 1.2×HBVΔCore-expressing Huh 7.5 cells were analyzed by electron microscopy. HBV filaments (Fig. 2E) were frequently observed with a length ranging from 200 nm to 1.2 μm. In addition, in these specimens a lot of spheres 22 nm in diameter were observed as well. To confirm that the filamentous particles are indeed HBV filaments, immunogold electron microscopy of the partially concentrated sample was performed with a goat-derived polyclonal anti-HBs antiserum. The gold particles specifically labeled the filaments (Fig. 2F). Taken together, these data indicate that the destruction of the core expression affects neither the synthesis nor the release of the filaments.
FIG 2.

HBsAg and subviral particles are released from core-deficient mutants. (A) Supernatants from pUC18-, p1.2×HBVΔCore-, and p1.2×HBV-transfected Huh 7.5 cells were analyzed by HBsAg-specific ELISA. (B) Supernatants from pUC18-, p1.2×HBVΔCore-, and p1.2×HBV-transfected Huh 7.5 or HepG2 cells were analyzed by Western blotting with the LHBs-specific antibody (MA18/7). (C) Supernatants from p1.2×HBVΔCore- or p1.2×HBV-transfected Huh 7.5 (upper panel) and HepG2 (lower panel) cells were concentrated with polyethylene glycol 6000 and then subjected to sucrose density gradient centrifugation. A total of 22 fractions were collected from top to bottom and analyzed by Western blotting with the LHBs-specific antibody (MA18/7). LHBs-specific bands were labeled with black arrows. (D) Fractions 7 to 15 of the sucrose density gradient centrifugation (see Fig. 2C) were 20-fold (Huh 7.5) or 50-fold (HepG2) diluted and quantified by the HBsAg-specific ELISA. (E) Electron microscopy of fractions 9 to 12 concentrated from p1.2×HBVΔCore-transfected Huh 7.5 supernatant (see Fig. 2C). A classic filament 22 nm in diameter (I), a python-shaped filament with a branch (II) or a long tail (III), and a horn-shaped filament (IV) were visualized by the negative staining. In addition, spherical particles of 22 nm were also observed. Arrows and arrowheads indicate HBV filaments and spheres, respectively. The black bars indicate the size. (F) Immunogold labeling of HBV filaments using a goat-derived anti-HBsAg serum and colloidal gold-conjugated rabbit-derived anti-goat IgG secondary antibody. The black bar indicates the size.
In cells expressing the core-deficient mutant, LHBs colocalizes with multivesicular bodies.
The data described above indicate that this system is suitable to analyze the release pathway of LHBs-containing subviral particles in the absence of any potential interference due to the formation of viral particles. So it could be investigated whether the LHBs-containing filaments are indeed released like spheres through the constitutive secretory pathway or like viral particles via the ESCRT/MVB pathway. To study this, Huh 7.5 cells were either cotransfected with p1.2×HBVΔCore or p1.2×HBV and with peYFP-Rab7 as marker for the endosomal/MVB pathway (34–36) or with pGalT-eGFP as a marker for the trans-Golgi apparatus (37, 38) and subsequently analyzed by confocal laser scanning microscopy (CLSM) using an LHBs-specific monoclonal antibody (MA18/7). The CLSM analysis of Huh 7.5 cells expressing the core-deficient mutant or wtHBV revealed that the eYFP-Rab7-positive subcellular compartments partially colocalize with a fraction of LHBs (Fig. 3A). The colocalization between LHBs and YFP-Rab7 could also be observed in HepG2 cells that were cotransfected with p1.2×HBVΔCore or p1.2×HBV and with peYFP-Rab7 (Fig. 3A). This was in detail analyzed for Huh 7.5 and HepG2 cells by the determination of the intensity profile for the YFP-Rab7-specific fluorescence and the LHBs-specific signal (Fig. 3B). The colocalization coefficient of the LHBs- and YFP-Rab7-specific signal was determined as almost 1.0 in Huh 7.5 and in HepG2 cells (Fig. 3C). In contrast to this, no significant colocalization between GalT-eGFP and LHBs could be found in cells expressing wtHBV or the core-deficient mutant (Fig. 3D). This was confirmed by the analysis of the intensity profile for the GalT-eGFP-specific fluorescence and the LHBs-specific signal (Fig. 3E/F), which shows no significant overlap between the GalT and LHBs-specific signals. Taken together, these data indicate that LHBs in the absence of virion production maintains the capacity to enter the MVB pathway.
FIG 3.
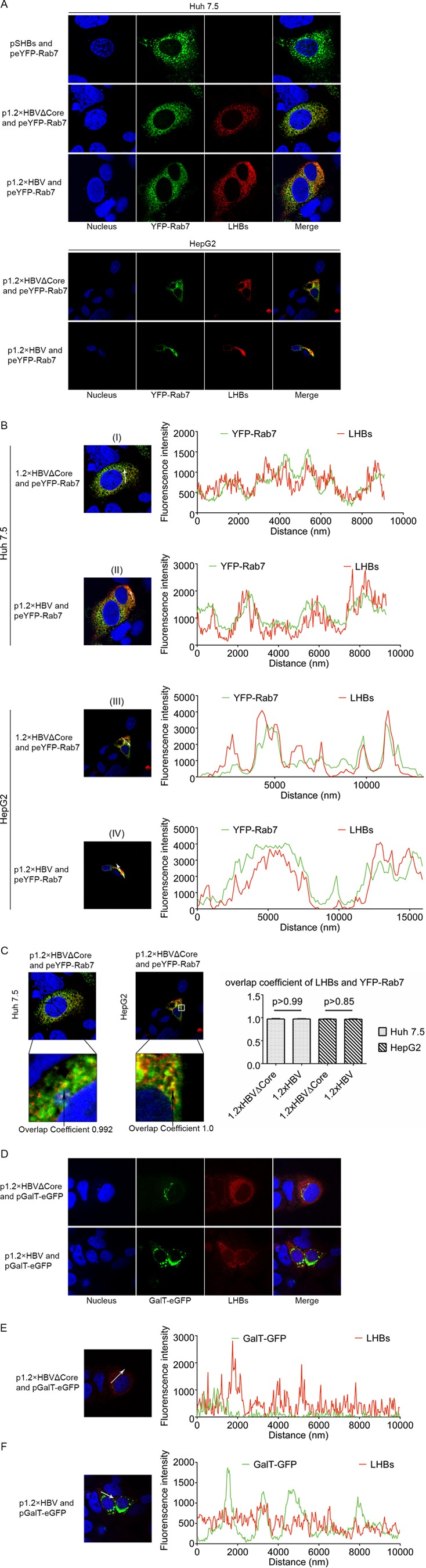
In cells expressing the core-deficient HBV genome LHBs is found in Rab7-positive structures as in wtHBV-expressing cells. (A) Huh 7.5 (upper field) and HepG2 (lower field) cells were cotransfected with YFP-Rab7 expression vector and the vectors pSHBs (negative control), p1.2×HBVΔCore, or p1.2×HBV and then analyzed by confocal immunofluorescence microscopy with the LHBs-specific antibody MA18/7 (red). (B) The colocalization of LHBs (red) and YFP-Rab7 (green) was investigated by analyzing the intensity profiles, which describes the distribution of YFP-Rab7- and LHBs-specific signals along the indicated white arrow. The colocalization was analyzed based on 15 LHBs- and YFP-Rab7-positive Huh 7.5 (I and II) and HepG2 cells (III and IV). Representative examples are shown here. (C) The colocalization of LHBs and of YFP-Rab7 was expressed by the colocalization coefficient, which describes the degree of overlap between YFP-Rab7- and LHBs-specific signals. The overlap coefficient was determined based on 15 Huh 7.5 or HepG2 cells. Representative examples are shown here. (D) Huh 7.5 cells were cotransfected with pGalT-eGFP (green) and the vector p1.2×HBVΔCore or p1.2×HBV and analyzed by confocal immunofluorescence microscopy with the LHBs-specific antibody MA18/7 (red). The figure showed a representative result; comparable data were obtained for the analysis of 10 different cells. (E and F) The intensity profile of the GalT-GFP-specific signals (green) and LHBs-specific signals (red), indicating the distribution of GalT-GFP and LHBs, was determined along the indicated white arrow. The figure shows a representative result; comparable data were obtained for the analysis of 10 different cells. Panels A to F only show a representative result; comparable data were obtained for analysis of different cells.
LHBs is found in isolated MVBs from cells expressing a core-deficient HBV genome.
To corroborate and extend the results of the colocalization studies, MVBs were isolated from p1.2×HBVΔCore-transfected or p1.2×HBVΔCore- and peYFP-Rab7-cotransfected Huh 7.5 cells by discontinuous sucrose gradient ultracentrifugation of the cellular homogenates. MVBs floated up to the interphase between the 8.6 and 35% layers of the gradient (39). The respective fractions were analyzed by Western blotting with LHBs and Rab7 (as MVB markers)-, YFP and Rab 5B (early endosomes/MVBs)-, PDI (ER)-, and GM-130 (Golgi)-specific antisera. The blot detects LHBs in the MVB fraction characterized by the presence of Rab7 and Rab5B) that contained only traces of PDI and GM130 (Fig. 4A). For deeper analysis, subcellular fractionation of homogenate derived from p1.2×HBVΔCore-/peYFP-Rab7-cotransfected Huh 7.5 cells was performed by Percoll-based linear gradient ultracentrifugation and Western blot analysis of the fractions using LHBs-, Rab7-, Rab 5B-, PDI-, and GM130-specific antisera. LHBs was detected in fractions 5 to 14, with a major peak in fractions 4 and 5. Rab7 could be detected in fractions 3 to 11, with a major peak in fractions 4 and 5. Rab 5B was found in fractions 4 to 7, with the same peak in fractions 4 and 5, as observed for Rab7. The Golgi marker GM130 was observed in fractions 4 to 14, with a major peak in fractions 6 and 7. PDI was distributed in fractions 4 to 8, with a major peak in fraction 6 and 7 (Fig. 4B). This demonstrates that over the gradient the peak of LHBs strongly overlaps with the peak for the MVB markers, which indicates that filaments enter MVBs in the absence of virion production. To demonstrate unequivocally that these structures indeed represent MVBs, we performed electron microscopy analysis of ultrathin sections derived from cells expressing the core-deficient HBV mutant (Fig. 4A). Unfortunately, using this approach makes it difficult to directly identify impressive filaments based on their shape, since they must be oriented in parallel to the section plane in the MVBs. Therefore, the ultrathin sections of Huh 7.5 cells expressing the core-deficient mutant were first stained LHBs with FluoroNanogold, which is a probe that contains fluorophores and nanogold particles. After silver enhancement, nanogold-silver polymer and the fluorescence could be observed in LHBs-positive cells by light and/or fluorescence microscopy (Fig. 4C). Such nanogold-silver compounds in the EM could mainly be found within large dilated compartments delimited by smooth membranes (Fig. 4D). This suggests that LHBs locates within such compartments. The lumena of these organelles were characterized by the presence of many small intraluminal vesicles with diameters ranging from 40 to 250 nm. This size and morphology corresponds to multivesicular bodies (MVBs). In case of pUC18-transfected Huh 7.5 cells, such large MVBs, reaching a diameter of up to 6 μm, were not observed (Fig. 4E). Taken together, these data indicate that LHBs even in the absence of virion production enters MVBs, indicating that filaments have the capacity to leave the cell like viral particles via MVBs.
FIG 4.

LHBs enters MVBs even in the absence of virion production. (A) Homogenates of p1.2×HBVΔCore-transfected or p1.2×HBVΔCore/peYFP-Rab7-cotransfected Huh 7.5 cells were subfractionated by a discontinuous sucrose density gradient. The isolated MVB fraction was analyzed by Western blotting with LHBs-, Rab7-, YFP-, Rab 5B-, PDI-, and GM130-specific antibodies. LHBs- and Rab7-specific bands are indicated by black arrows. (B) Homogenates of p1.2×HBVΔCore- and peYFP-Rab7-cotransfected Huh 7.5 cells were subfractionated using a Percoll-based linear density gradient. A total of 19 fractions were recovered from top to bottom and analyzed by Western blotting with LHBs-, Rab7-, Rab 5B-, PDI-, and GM130-specific antibodies. The LHBs, Rab7, and Golgi signals in the Western blot were determined by Image Studio Lite (LI-COR Biosciences), and the strongest signal was standardized as 1. LHBs-specific bands are labeled with black arrows, while the nonspecific bands are indicated with black stars. (C) Huh 7.5 cells transfected with p1.2×HBVΔCore were fixed 48 h after transfection. Intracellular LHBs was detected using an LHBs specific antibody (MA18/7) and a secondary antibody coupled to Alexa Fluor-FluoroNanogold. Areas with LHBs-positive cells were first visualized by use of a fluorescent channel (left) and marked with the help of a diamond scratcher on the bottom of the culture dish. After silver enhancement, the nanogold particles, representing the LHBs, were enlarged to be visualized by bright-field channel (middle). (D and E) Ultrathin sections were prepared from Fig. 4C and analyzed by electron microscopy. In the ultrathin sections of p1.2×HBVΔCore-transfected Huh 7.5 cells stained LHBs with FluoroNanogold, nanogold-silver dots with a diameter of about 20 nm, representing the LHBs, were predominantly found in the large dilated MVBs. Such dots and MVBs could not be observed in the pUC18-transfected cells (negative control) (E). The inlet shows a magnification to visualize the structure of MVBs (black arrows) and nanogold-silver dots after amplification (white arrows). Comparable data were obtained from the other experiments, and the most promising results are shown. The black bars indicate the size.
Inhibition of MVB biogenesis disrupts the release of filaments.
The data described above show that filaments enter into MVBs. To investigate the relevance of the MVB compartment for the release of filaments, we disrupted the MVB biogenesis and analyzed the effect on the release of LHBs. Huh 7.5 cells expressing the 1.2×HBVΔCore or 1.2×HBV/D or wtHBV genotype A (1.2×HBV/A) were treated with U18666A, an inhibitor of MVB biogenesis (24–26). At 48 h after treatment with U18666A, no cytotoxic effect could be observed by analyzing the cell viability (data not shown). Supernatants and cellular lysates were analyzed by Western blotting using the LHBs-specific antibody MA18/7 or the SHBs-specific antibody HB01. The data showed that U18666A significantly inhibited the release of LHBs from mutant HBV or wtHBV (genotypes A and D)-expressing cells in a dose-dependent manner, whereas the synthesis of LHBs was not significantly affected (Fig. 5). In contrast to LHBs, no significant inhibition of production and secretion of SHBs (HBV spheres) could be detected in cells expressing 1.2×HBV/A (Fig. 5C and D). 1.2×HBV/A was chosen since the monoclonal HB01 does not react with SHBs of 1.2×HBVΔCore and 1.2×HBV/D. This indicates that spheres and filaments are released via different routes: spheres via the ER-Golgi secretory pathway and filaments via the MVBs.
FIG 5.

Inhibition of MVB morphogenesis impairs the release of filaments but not the release of spheres. (A) p1.2×HBVΔCore-transfected Huh 7.5 cells were treated with different concentrations of MVBs inhibitor U18666A (U18). The lysates (LS) and corresponding supernatants (SPN) were analyzed by Western blotting with the LHBs-specific antibody (MA18/7). The signals in the Western blot were determined by using Image Studio Lite. Values are normalized to the untreated panel. The data represent the mean value from three independent experiments. (B) Huh 7.5 cells transfected with p1.2×HBV/D (genotype D) were treated with different concentrations of U18666A (U18). The lysates (LS) and corresponding supernatants (SPN) were analyzed by Western blotting with the LHBs-specific antibody (MA18/7). The signals on the blot were quantified by using Image Studio Lite. Values are normalized to the untreated panel. The data represent the mean value from three independent experiments. (C) Huh 7.5 cells transfected with p1.2×HBV/A (genotype A) were treated with different concentrations of U18666A (U18). The supernatants (sn) were analyzed by Western blotting using the SHBs-specific antibody (HB01). The signals on the blot were quantified by using Image Studio Lite. Values are normalized to the untreated panel. The data represent the mean value from three independent experiments. (D) Huh 7.5 cells transfected with p1.2×HBV/A (genotype A) were treated with different concentrations of U18666A (U18). The lysates (LS) were analyzed by Western blotting with the SHBs-specific antibody (HB01). The signals on blot were determined by Image studio lite from LI-COR Biosciences. Values are normalized to untreated panel. The data represent the mean value from three independent experiments.
Dominant-negative CHMP and Vps4 trap HBV filaments in class E-like compartments, leading to the inhibition of filaments secretion.
Previous studies showed that HBV exploits the functions of MVB for the egress of viral particle, and inhibitions of Vps4A, Vps4B, or ESCRT-III component CHMPs blocked the release of viral particles but not of spherical particles (10, 11). To investigate in more detail whether the release of HBV filaments is also dependent on the function of Vps4A, Vps4B, and CHMPs, Huh 7.5 cells were cotransfected with the HBV core-deficient mutant and with wt Vps4A/B, or dominant-negative (dn) mutants of Vps4A/B or dn CHMP3, respectively. After 72 h, the cells were analyzed by immunofluorescence microscopy using the LHBs-specific antibody MA18/7. As shown in Fig. 6A, GFP-tagged wt Vps4A and DsRed-tagged wtVps4B in cells coexpressing of the core-deficient HBV mutant exhibited diffuse staining allover the cell and partially colocalized with LHBs in the perinuclear region. In contrast to this, the coexpression of dn Vps4A/B and dn CHMP3 led to the formation of dysfunctional MVBs designated the “class E compartment” (16, 17, 40), accumulating in clustered foci, and dramatically redistributed LHBs into the punctate dots. To determine whether the dysfunction of MVB affects the release of HBV filaments, supernatants from Huh 7.5 cells expressing wt Vps4A/B or the dn mutants were analyzed by Western blotting using the LHBs-specific MA18/7. Significant reduction of LHBs could be found in case of the coexpression of dn mutants (Fig. 6B). A similar inhibition could also be observed when dn CHMP3 was coexpressed (Fig. 6B). Overexpression of SHBs in Huh 7.5 cells, which can mimic the assembly and secretion of HBV spheres (41), was used to investigate the effect of the inhibition on the secretion of spheres. Supernatants from Huh 7.5 cells coexpressing these dn mutants with SHBs were analyzed by Western blotting and HBsAg ELISA. Here, no significant reduction of the secretion of HBV spheres was caused by the dn mutants compared to the controls (Fig. 6C). To analyze the relevance of MVBs for the release of SHBs and LHBs in cells expressing wtHBV, Huh 7.5 cells expressing 1.2×HBV/A were cotransfected with the dn mutants Vps4A.dn, Vps4B.dn, or CHMP3.dn. The supernatants were analyzed by Western blotting using the SHBs-specific antibody HB01. Although the release of LHBs was significantly inhibited, the secretion of SHBs was slightly affected in the expression of dn mutants (Fig. 6D).
FIG 6.
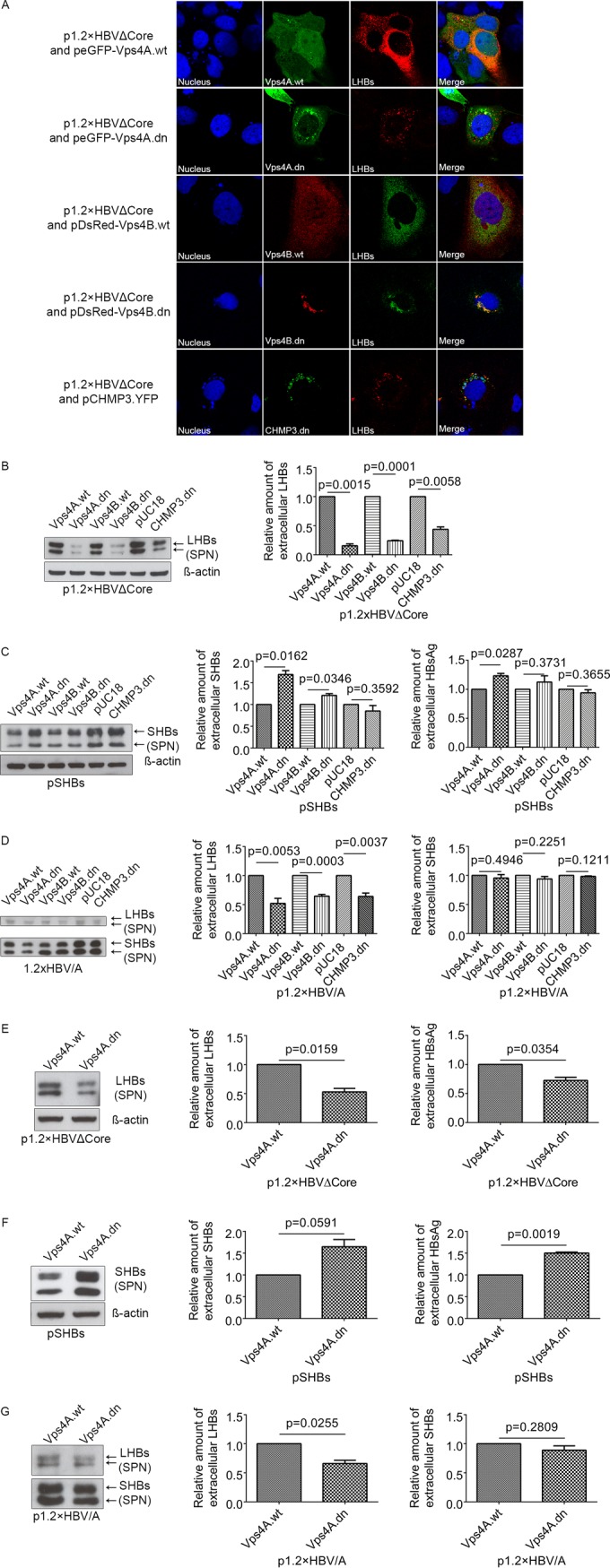
Interference with MVB functionality by coexpression of dominant-negative (dn) mutants of the ESCRT machinery blocks the release of filaments, whereas the secretion of spheres is not affected. (A) Huh 7.5 cells were cotransfected with p1.2×HBVΔCore and peGFP-Vps4A.wt (Vps4A.wt), peGFP-Vps4A.dn (Vps4A.dn), pDsRed-Vps4B.wt (Vps4B.wt), pDsRed-Vps4B.dn (Vps4B.dn), or pCHMP3.YFP (CHMP3.dn) and analyzed by confocal immunofluorescence microscopy using the LHBs-specific antibody MA18/7. Overlays of the fluorescence images are shown in the right column, with yellow indicating colocalization. Nuclei were stained with DAPI (blue). Analysis was based on at least 10 cells, and representative examples are presented. (B) Supernatants from Huh 7.5 cells cotransfected with p1.2×HBVΔCore and Vps4A.wt, Vps4A.dn, Vps4B.wt, Vps4B.dn, or CHMP3.dn were analyzed by Western blotting with LHBs-specific antibody (MA18/7). Due to the insolubility of intracellular LHBs in denaturing reagent SDS, the detection of β-actin was used as a control. The signals on the blot were quantified by using Image Studio Lite. Values are normalized to the untreated panel. The data represent the mean value from three independent experiments. (C) Supernatants from Huh 7.5 cells were cotransfected with pSHBs and Vps4A.wt, Vps4A.dn, Vps4B.wt, Vps4B.dn, or CHMP3.dn were analyzed by Western blotting with the SHBs-specific antibody HB01 (left panel) and by HBsAg-specific ELISA (right panel). The signals on the blot were quantified by using Image Studio Lite. Values are normalized to the untreated panel. The data represent the mean value from three independent experiments. (D) Huh 7.5 cells first transfected with p1.2×HBV/A and then transfected with Vps4A.wt, Vps4A.dn, Vps4B.wt, Vps4B.dn, or CHMP3.dn 24 h after first transfection. At 48 h after the second transfection, the supernatants were analyzed by Western blotting with the SHBs-specific antibody HB01. The signals on the blot were quantified by using Image Studio Lite. Values are normalized to the untreated panel. The data represent the mean value from three independent experiments. (E) Supernatants from HepG2 cells cotransfected with p1.2×HBVΔCore and Vps4A.wt or Vps4A.dn were analyzed by Western blotting with LHBs-specific antibody MA18/7 (left panel) and HBsAg-specific ELISA (right panel). The signals on the blot were quantified by using Image Studio Lite. Values are normalized to the untreated panel. The data represent the mean value from three independent experiments. (F) Supernatants from HepG2 cells cotransfected with pSHBs and either Vps4A.wt or Vps4A.dn were analyzed by Western blotting with SHBs-specific antibody HB01 (left panel) and HBsAg-specific ELISA (right panel). The signals on the blot were quantified by using Image Studio Lite. Values are normalized to the untreated panel. The data represent the mean value from three independent experiments. (G) HepG2 cells first transfected with p1.2×HBV/A and then transfected with Vps4A.wt or Vps4A.dn 24 h after the first transfection. At 48 h after the second transfection, the supernatants were analyzed by Western blotting with the SHBs-specific antibody HB01. The signals on the blot were quantified by using Image Studio Lite. Values are normalized to the untreated panel. The data represent the mean value from three independent experiments.
To confirm that the observed effects do not depend on the Huh 7.5 cellular background, the key experiments were performed in addition in HepG2 cells (Fig. 6E to G). Coexpression of Vps4A.dn in cells expressing the core-deficient mutant impaired the release of filaments (Fig. 6E), while the release of spheres was not impaired; in contrast, a slight increase was observed (Fig. 6F), as demonstrated by Western blotting of the supernatants. Moreover, HepG2 cells expressing the wt genome (1.2×HBV/A) were cotransfected with Vps4A.dn or the corresponding control (Vps4A.wt). The supernatants were analyzed by Western blotting with the SHBs-specific antibody HB01. As observed for Huh 7.5 cells, the release of LHBs was significantly inhibited in HepG2 cells, whereas the secretion of SHBs was not affected in the expression of the dn mutant (Fig. 6G). These data indicate that the functionality of MVB is required for the efficient release of HBV filaments but not spheres.
DISCUSSION
Spherical subviral particles and infectious viral particles are released from the cells in two different ways. Spheres bud into the lumen of the ER/ERGIC and are secreted through the constitutive secretory pathway, thereby traversing the Golgi complex (8, 9). In contrast to this, virions are released via the ESCRT/MVB machinery (9, 10). Recently, α-taxilin was identified as an essential factor for the release of viral particles. On the one hand, α-taxilin harbors a YAEL-motive that acts as a late domain and mediates the interaction with the ESCRT component tsg101 and, on the other hand, α-taxilin binds to the PreS1-domain of LHBs. This contributes to recruit viral particles to the ESCRT machinery (19). A small interfering RNA-based study failed to observe an effect of silencing the tsg101 expression on the release of HBV particles in Huh 7.5 cells (42). However, it remains unclear whether there were still enough tsg101 molecules enabling the release of HBV.
Filaments are characterized by a significant higher content of LHBs than spheres, the question arose whether filaments are released ESCRT-dependently via MVBs like viral particles. To exclude any false signals due to interference with the release of viral particles, a mutated HBV genome was generated. This HBV core-deficient mutant fails to express core and therefore does not allow the formation of viral particles, whereas the production of subviral particles is not affected. Moreover, this approach does not affect the ratio of the amount of LHBs and SHBs. Based on examination of this mutant by a combination of biochemical, cell biological, and ultrastructural approaches, we found that in contrast to spheres, filaments are released like infectious viral particles via MVBs.
It is well established that SHBs in the absence of MHBs and LHBs can bud into a post-ER/pre-Golgi compartment to form 20-nm spheres that can be released through the constitutive secretory pathway (9, 41). In contrast to SHBs, the selective overexpression of LHBs leads to the formation of aggregates and is associated with the intracellular retention of HBsAg (43–45) that can lead to an intracellular accumulation (ground glass hepatocytes). Coexpression of SHBs can favor the secretion of LHBs (6). The formation and secretion efficiency of filaments depends on the relative amounts of SHBs and LHBs. Low levels of LHBs result in the production and secretion of spheres containing LHBs, whereas higher levels of LHBs lead to the formation of filamentous aggregates that are not secreted (46). These aggregates are recognized as “misfolded proteins” and therefore retained from secretion (47, 48). However, in cells transfected with an HBV genome, in HBV-infected cells and patients, filaments are formed and released in the supernatant and serum (49–51). This indicates that the proper ratio between LHBs and SHBs is crucial for the formation and release of filaments. A simple way to ensure this was to generate a core-deficient genome that allows the formation of the viral surface proteins in a comparable ratio, as found for the wild-type genome, but has no capacity to form viral particles, ensuring that an overlap between filament- and virion-specific signals can be excluded. Indeed, the lack of core protein in this mutant does not affect synthesis, intracellular distribution, and secretion of SHBs and LHBs compared to the corresponding wild type.
In cells producing wtHBV or the core-deficient HBV mutant, LHBs displays a diffuse extranuclear distribution, as observed for assembled HBV capsids, which differs from the SHBs-specific staining that was found to accumulate around the nucleus in the absence of MHBs and LHBs. A more detailed analysis using the MVB markers Rab7 (34) and Vps4A and Vps4B (40, 52, 53) revealed that LHBs enters MVBs. In contrast to this, LHBs weakly colocalizes with 1,4-galactosyltransferase (GalT), a marker of the trans-Golgi network (37, 38). The presence of filaments in MVBs is further supported by detection of LHBs in isolated MVBs and by immune electron microscopy analysis of ultrathin sections from p1.2×HBVΔCore-transfected Huh 7.5 cells.
Disrupting the morphogenesis of MBVs by a small molecule inhibitor U18666A strongly inhibits the release of LHBs/filaments from cells expressing the core-deficient HBV mutant, while the release of SHBs/spheres is not significantly affected. To impair ESCRT-MVBs in a more specific way, dn mutants of Vps4A/B or CHMP3 were used. In our study, disruption of ESCRT-MVBs by dn mutants entrapped LHBs in the classic E compartment and changed the distribution of LHBs. The intracellular LHBs hijacked by this aberrant endosomes could not be detected by Western blotting due to its insolubility in denaturing reagent SDS (data not shown), a finding consistent with previous reports (11, 14, 54). Disturbance of ESCRT/MVB pathway by dn mutants blocks the release of LHBs/filaments but not of SHBs/spheres. This demonstrates the specificity of the observed effect and argues against a general effect on the release of proteins. A cytotoxic effect of the applied concentrations of the inhibitor was excluded by cell viability assays. Moreover, if the morphogenesis of MVBs is impaired, the strong reduction of the amount of LHBs in the supernatant demonstrates that the MVB pathway is central for the release of filaments.
A recent study shows that HBV subviral particles based on SHBs self-assemble in the lumen of the ER into branched filaments (9). These SHBs-based filaments are then folded into crystal-like structures, which are transported to the ERGIC by ER-derived vesicles. Since their size and shape might limit further transport through the secretory pathway, they are unpacked at the ERGIC. Their further progression through the secretory pathway requires their conversion into spherical particles. It can be speculated that in the case of filaments characterized by a higher content of LHBs, chaperones less efficiently mediate the disassembly, and that therefore the filaments are excluded from the classic secretory pathway due to their size. The dual topology of the PreS1PreS2 domain of the LHBs integrated (55, 56) into these filaments enables the interaction with cytoplasmic factors, which means that, i.e., α-taxilin that mediates the binding to the ESCRT machinery can detour filaments into the MVB system.
Based on the data from our study, we demonstrate that the HBV subviral particles spheres and filaments leave the infected cell following different routes. Spheres are secreted via the secretory pathway, filaments are released like viral particles via the ESCRT/MVB pathway. A detailed understanding of these mechanisms might be helpful for the design of novel antiviral strategies. Although a general block in the release of filaments can lead to ER stress and a storage disease analogous situation (43, 44), a selective interference with the release can be used to moderately increase the intracellular amounts of HBV-specific antigens, leading to larger amounts of HBV-specific peptides presented on the surfaces of HBV-infected cells, facilitating their recognition and elimination by the immune system.
ACKNOWLEDGMENTS
We thank Regina Eberle for excellent technical assistance and Dagmar Fecht-Schwarz for critical reading of the manuscript.
B.J. obtained a grant from the China Scholarship Council. E.H. was funded by the DZIF–German Center of Infection Research.
REFERENCES
- 1.Schadler S, Hildt E. 2009. HBV life cycle: entry and morphogenesis. Viruses 1:185–209. doi: 10.3390/v1020185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerlich WH, Heermann KH, Lu X. 1992. Functions of hepatitis B surface proteins. Arch Virol Suppl 4:129–132. doi: 10.1007/978-3-7091-5633-9_28. [DOI] [PubMed] [Google Scholar]
- 3.Heermann KH, Goldmann U, Schwartz W, Seyffarth T, Baumgarten H, Gerlich WH. 1984. Large surface proteins of hepatitis B virus containing the pre-s sequence. J Virol 52:396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruss V. 2007. Hepatitis B virus morphogenesis. World J Gastroenterol 13:65–73. doi: 10.3748/wjg.v13.i1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garcia T, Li J, Sureau C, Ito K, Qin Y, Wands J, Tong S. 2009. Drastic reduction in the production of subviral particles does not impair hepatitis B virus virion secretion. J Virol 83:11152–11165. doi: 10.1128/JVI.00905-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peiffer KH, Akhras S, Himmelsbach K, Hassemer M, Finkernagel M, Carra G, Nuebling M, Chudy M, Niekamp H, Glebe D, Sarrazin C, Zeuzem S, Hildt E. 2014. Intracellular accumulation of subviral HBsAg particles and diminished Nrf2 activation in HBV genotype G expressing cells lead to an increased ROI level. J Hepatol 62:791–798. doi: 10.1016/j.jhep.2014.11.028. [DOI] [PubMed] [Google Scholar]
- 7.Bruns M, Miska S, Chassot S, Will H. 1998. Enhancement of hepatitis B virus infection by noninfectious subviral particles. J Virol 72:1462–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patient R, Hourioux C, Roingeard P. 2009. Morphogenesis of hepatitis B virus and its subviral envelope particles. Cell Microbiol 11:1561–1570. doi: 10.1111/j.1462-5822.2009.01363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patient R, Hourioux C, Sizaret PY, Trassard S, Sureau C, Roingeard P. 2007. Hepatitis B virus subviral envelope particle morphogenesis and intracellular trafficking. J Virol 81:3842–3851. doi: 10.1128/JVI.02741-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watanabe T, Sorensen EM, Naito A, Schott M, Kim S, Ahlquist P. 2007. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc Natl Acad Sci U S A 104:10205–10210. doi: 10.1073/pnas.0704000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lambert C, Doring T, Prange R. 2007. Hepatitis B virus maturation is sensitive to functional inhibition of ESCRT-III, Vps4, and gamma 2-adaptin. J Virol 81:9050–9060. doi: 10.1128/JVI.00479-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Votteler J, Sundquist WI. 2013. Virus udding and the ESCRT pathway. Cell Host Microbe 14:232–241. doi: 10.1016/j.chom.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin-Serrano J, Yarovoy A, Perez-Caballero D, Bieniasz PD. 2003. Divergent retroviral late-budding domains recruit vacuolar protein sorting factors by using alternative adaptor proteins. Proc Natl Acad Sci U S A 100:12414–12419. doi: 10.1073/pnas.2133846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Strack B, Calistri A, Craig S, Popova E, Gottlinger HG. 2003. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114:689–699. doi: 10.1016/S0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- 15.Carlson LA, Hurley JH. 2012. In vitro reconstitution of the ordered assembly of the endosomal sorting complex required for transport at membrane-bound HIV-1 Gag clusters. Proc Natl Acad Sci U S A 109:16928–16933. doi: 10.1073/pnas.1211759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bishop N, Woodman P. 2000. ATPase-defective mammalian VPS4 localizes to aberrant endosomes and impairs cholesterol trafficking. Mol Biol Cell 11:227–239. doi: 10.1091/mbc.11.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujita H, Yamanaka M, Imamura K, Tanaka Y, Nara A, Yoshimori T, Yokota S, Himeno M. 2003. A dominant negative form of the AAA ATPase SKD1/VPS4 impairs membrane trafficking out of endosomal/lysosomal compartments: class E vps phenotype in mammalian cells. J Cell Sci 116:401–414. doi: 10.1242/jcs.00213. [DOI] [PubMed] [Google Scholar]
- 18.Rost M, Mann S, Lambert C, Doring T, Thome N, Prange R. 2006. Gamma-adaptin, a novel ubiquitin-interacting adaptor, and Nedd4 ubiquitin ligase control hepatitis B virus maturation. J Biol Chem 281:29297–29308. doi: 10.1074/jbc.M603517200. [DOI] [PubMed] [Google Scholar]
- 19.Hoffmann J, Boehm C, Himmelsbach K, Donnerhak C, Roettger H, Weiss TS, Ploen D, Hildt E. 2013. Identification of alpha-taxilin as an essential factor for the life cycle of hepatitis B virus. J Hepatol 59:934–941. doi: 10.1016/j.jhep.2013.06.020. [DOI] [PubMed] [Google Scholar]
- 20.Roingeard P. 2003. Vacuolization in hepatitis B virus-infected hepatocytes. Hepatology 37:1223–1224. doi: 10.1053/jhep.2003.50168. [DOI] [PubMed] [Google Scholar]
- 21.Roingeard P, Sureau C. 1998. Ultrastructural analysis of hepatitis B virus in HepG2-transfected cells with special emphasis on subviral filament morphogenesis. Hepatology 28:1128–1133. doi: 10.1002/hep.510280431. [DOI] [PubMed] [Google Scholar]
- 22.Ehrhardt C, Schmolke M, Matzke A, Knoblauch A, Will C, Wixler V, Ludwig S. 2006. Polyethylenimine, a cost-effective transfection reagent. Signal Transduction 6:179–184. doi: 10.1002/sita.200500073. [DOI] [Google Scholar]
- 23.Kucinskaite-Kodze I, Pleckaityte M, Bremer CM, Seiz PL, Zilnyte M, Bulavaite A, Mickiene G, Zvirblis G, Sasnauskas K, Glebe D, Zvirbliene A. 2015. New broadly reactive neutralizing antibodies against hepatitis B virus surface antigen. Virus Res 211:209–221. doi: 10.1016/j.virusres.2015.10.024. [DOI] [PubMed] [Google Scholar]
- 24.Dahl NK, Reed KL, Daunais MA, Faust JR, Liscum L. 1992. Isolation and characterization of Chinese hamster ovary cells defective in the intracellular metabolism of low density lipoprotein-derived cholesterol. J Biol Chem 267:4889–4896. [PubMed] [Google Scholar]
- 25.Higgins ME, Davies JP, Chen FW, Ioannou YA. 1999. Niemann-Pick C1 is a late endosome-resident protein that transiently associates with lysosomes and the trans-Golgi network. Mol Genet Metab 68:1–13. doi: 10.1006/mgme.1999.2882. [DOI] [PubMed] [Google Scholar]
- 26.Kobayashi T, Beuchat MH, Chevallier J, Makino A, Mayran N, Escola JM, Lebrand C, Cosson P, Kobayashi T, Gruenberg J. 2002. Separation and characterization of late endosomal membrane domains. J Biol Chem 277:32157–32164. doi: 10.1074/jbc.M202838200. [DOI] [PubMed] [Google Scholar]
- 27.Chudy M, Hanschmann KM, Kress J, Nick S, Campos R, Wend U, Gerlich W, Nubling CM. 2012. First WHO International Reference Panel containing hepatitis B virus genotypes A-G for assays of the viral DNA. J Clin Virol 55:303–309. doi: 10.1016/j.jcv.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 28.Huber LA, Pfaller K, Vietor I. 2003. Organelle proteomics: implications for subcellular fractionation in proteomics. Circ Res 92:962–968. doi: 10.1161/01.RES.0000071748.48338.25. [DOI] [PubMed] [Google Scholar]
- 29.Burckstummer T, Kriegs M, Lupberger J, Pauli EK, Schmittel S, Hildt E. 2006. Raf-1 kinase associates with Hepatitis C virus NS5A and regulates viral replication. FEBS Lett 580:575–580. doi: 10.1016/j.febslet.2005.12.071. [DOI] [PubMed] [Google Scholar]
- 30.Brandenburg B, Stockl L, Gutzeit C, Roos M, Lupberger J, Schwartlander R, Gelderblom H, Sauer IM, Hofschneider PH, Hildt E. 2005. A novel system for efficient gene transfer into primary human hepatocytes via cell-permeable hepatitis B virus-like particle. Hepatology 42:1300–1309. doi: 10.1002/hep.20950. [DOI] [PubMed] [Google Scholar]
- 31.Chai N, Chang HE, Nicolas E, Han Z, Jarnik M, Taylor J. 2008. Properties of subviral particles of hepatitis B virus. J Virol 82:7812–7817. doi: 10.1128/JVI.00561-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayat FM. 1970. Principles and techniques of electron microscopy: biological applications. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 33.Lupberger J, Schaedler S, Peiran A, Hildt E. 2013. Identification and characterization of a novel bipartite nuclear localization signal in the hepatitis B virus polymerase. World J Gastroenterol 19:8000–8010. doi: 10.3748/wjg.v19.i44.8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chavrier P, Parton RG, Hauri HP, Simons K, Zerial M. 1990. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell 62:317–329. doi: 10.1016/0092-8674(90)90369-P. [DOI] [PubMed] [Google Scholar]
- 35.Vanlandingham PA, Ceresa BP. 2009. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J Biol Chem 284:12110–12124. doi: 10.1074/jbc.M809277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng Y, Press B, Wandinger-Ness A. 1995. Rab 7: an important regulator of late endocytic membrane traffic. J Cell Biol 131:1435–1452. doi: 10.1083/jcb.131.6.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roth J, Berger EG. 1982. Immunocytochemical localization of galactosyltransferase in HeLa cells: codistribution with thiamine pyrophosphatase in trans-Golgi cisternae. J Cell Biol 93:223–229. doi: 10.1083/jcb.93.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lucocq JM, Berger EG, Warren G. 1989. Mitotic Golgi fragments in HeLa cells and their role in the reassembly pathway. J Cell Biol 109:463–474. doi: 10.1083/jcb.109.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fialka I, Pasquali C, Lottspeich F, Ahorn H, Huber LA. 1997. Subcellular fractionation of polarized epithelial cells and identification of organelle-specific proteins by two-dimensional gel electrophoresis. Electrophoresis 18:2582–2590. doi: 10.1002/elps.1150181414. [DOI] [PubMed] [Google Scholar]
- 40.Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, Myszka DG, Sundquist WI. 2001. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107:55–65. doi: 10.1016/S0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- 41.Huovila AP, Eder AM, Fuller SD. 1992. Hepatitis B surface antigen assembles in a post-ER, pre-Golgi compartment. J Cell Biol 118:1305–1320. doi: 10.1083/jcb.118.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stieler JT, Prange R. 2014. Involvement of ESCRT-II in hepatitis B virus morphogenesis. PLoS One 9:e91279. doi: 10.1371/journal.pone.0091279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chisari FV, Klopchin K, Moriyama T, Pasquinelli C, Dunsford HA, Sell S, Pinkert CA, Brinster RL, Palmiter RD. 1989. Molecular pathogenesis of hepatocellular carcinoma in hepatitis B virus transgenic mice. Cell 59:1145–1156. doi: 10.1016/0092-8674(89)90770-8. [DOI] [PubMed] [Google Scholar]
- 44.Chisari FV, Filippi P, Buras J, McLachlan A, Popper H, Pinkert CA, Palmiter RD, Brinster RL. 1987. Structural and pathological effects of synthesis of hepatitis B virus large envelope polypeptide in transgenic mice. Proc Natl Acad Sci U S A 84:6909–6913. doi: 10.1073/pnas.84.19.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chisari FV, Filippi P, McLachlan A, Milich DR, Riggs M, Lee S, Palmiter RD, Pinkert CA, Brinster RL. 1986. Expression of hepatitis B virus large envelope polypeptide inhibits hepatitis B surface antigen secretion in transgenic mice. J Virol 60:880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Standring DN, Ou JH, Rutter WJ. 1986. Assembly of viral particles in Xenopus oocytes: pre-surface antigens regulate secretion of the hepatitis B viral surface envelope particle. Proc Natl Acad Sci U S A 83:9338–9342. doi: 10.1073/pnas.83.24.9338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hurtley SM, Helenius A. 1989. Protein oligomerization in the endoplasmic reticulum. Annu Rev Cell Biol 5:277–307. doi: 10.1146/annurev.cb.05.110189.001425. [DOI] [PubMed] [Google Scholar]
- 48.Ellgaard L, Molinari M, Helenius A. 1999. Setting the standards: quality control in the secretory pathway. Science 286:1882–1888. doi: 10.1126/science.286.5446.1882. [DOI] [PubMed] [Google Scholar]
- 49.Bayer ME, Blumberg BS, Werner B. 1968. Particles associated with Australia antigen in the sera of patients with leukaemia, Down's syndrome and hepatitis. Nature 218:1057–1059. doi: 10.1038/2181057a0. [DOI] [PubMed] [Google Scholar]
- 50.Sureau C, Romet-Lemonne JL, Mullins JI, Essex M. 1986. Production of hepatitis B virus by a differentiated human hepatoma cell line after transfection with cloned circular HBV DNA. Cell 47:37–47. doi: 10.1016/0092-8674(86)90364-8. [DOI] [PubMed] [Google Scholar]
- 51.Sells MA, Chen ML, Acs G. 1987. Production of hepatitis B virus particles in HepG2 cells transfected with cloned hepatitis B virus DNA. Proc Natl Acad Sci U S A 84:1005–1009. doi: 10.1073/pnas.84.4.1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scheuring S, Rohricht RA, Schoning-Burkhardt B, Beyer A, Muller S, Abts HF, Kohrer K. 2001. Mammalian cells express two VPS4 proteins both of which are involved in intracellular protein trafficking. J Mol Biol 312:469–480. doi: 10.1006/jmbi.2001.4917. [DOI] [PubMed] [Google Scholar]
- 53.von Schwedler UK, Stuchell M, Muller B, Ward DM, Chung HY, Morita E, Wang HE, Davis T, He GP, Cimbora DM, Scott A, Krausslich HG, Kaplan J, Morham SG, Sundquist WI. 2003. The protein network of HIV budding. Cell 114:701–713. doi: 10.1016/S0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
- 54.Lin Y, Kimpler LA, Naismith TV, Lauer JM, Hanson PI. 2005. Interaction of the mammalian endosomal sorting complex required for transport (ESCRT) III protein hSnf7-1 with itself, membranes, and the AAA+ ATPase SKD1. J Biol Chem 280:12799–12809. doi: 10.1074/jbc.M413968200. [DOI] [PubMed] [Google Scholar]
- 55.Bruss V, Lu X, Thomssen R, Gerlich WH. 1994. Posttranslational alterations in transmembrane topology of the hepatitis B virus large envelope protein. EMBO J 13:2273–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prange R, Streeck RE. 1995. Novel transmembrane topology of the hepatitis B virus envelope proteins. EMBO J 14:247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]