

Abstract
This letter describes the further lead optimization of the VU0486321 series of mGlu1 positive allosteric modulators (PAMs), driven by recent genetic data linking loss of function GRM1 to schizophrenia. Steep and caveat-laden SAR plagues the series, but ultimately potent mGlu1 PAMs (EC50s ~ 5 nM) have resulted with good DMPK properties (low intrinsic clearance, clean CYP profile, modest Fu) and CNS penetration (Kps 0.25 to 0.97), along with up to >450-fold selectivity versus mGlu4 and mGlu5.
Keywords: mGlu1, Metabotropic glutamate receptor, Positive allosteric modulator (PAM), Schizophrenia, Structure-Activity Relationship (SAR)
Graphical Abstract

Driven by the recent reports of deleterious non-synonymous single nucleotide polymorphisms (nsSNPS) in the GRM1 gene, which encodes the metabotropic glutamate receptor subtype 1 (mGlu1), that correlated with a higher incidence of neuropsychiatric disease,1,2,3 interest in mGlu1 PAMs has increased.3 In vitro, we have shown that mGlu1 PAMs can potentiate, and in some cases restore activity to wild-type levels in these mutants.3 However, historical tools lacked the DMPK profiles to serve as robust in vivo tools.3-6 En route to the ideal in vivo tool, our lab has published many advancements in the mGlu1 PAM ligand field,3,5,6 as well as demonstrating that the adverse effect of epilitform discharges and seizure liability of Group I mGluR agonists, such as DHPG, is not mGlu1 mediated,5 and therefore widening the therapeutic window for mGlu1 PAMs (Figure 1).
Figure 1.

Structures of representative mGlu1 PAMs 1-5.
As previously discussed, our entry into the VU0486321 (4) series of mGlu1 PAMs was via a ‘double molecular switch’ of an mGlu4 PAM ligand.3,7,8 While surveying a diverse array of 5-membered heterocyclic amides in the optimization effort, only a furyl amide was active, but substitution with a 3-methyl group, as in 4 and 5, greatly enhanced mGlu1 PAM potency.5,6 However, we never went back and surveyed the impact of incorporation of a methyl moiety in the context of other 5-membered heterocycles, with more desirerable physiochemical and DMPK properties than a furyl ring (Figure 2). In this Letter, we will detail the steep and caveat-laden SAR en route to an in vivo tool compound within the VU0486321 (4) series of mGlu1 PAMs.
Figure 2.

Chemical optimization plan to access multi-dimensional SAR around analogs 6.
In order to access analogs 6 and survey the SAR for the three regions highlighted in Figure 2, a general three step synthetic route was developed. As shown in Scheme 1, commercial, functionalized p-amino nitroarenes/heteroarenes 7 were condensed with various phthalic anhydrides to afford analogs 8. The nitro group was reduced to the aniline 9 via hydrogenation conditons, and final analogs 6 were afforded by standard amide coupling conditions with a diverse array of 3-methyl substituted 5-membered heterocyclic acids.
Scheme 1.

Reagents and conditions: (a) phthalic anhydrides, AcOH, reflux, 53-94%; (b) H2, Pd/C, EtOH, rt, 94-99-%; (c) methyl substituted 5-membered heterocyclic acids, HATU, DCM, r.t., 39-98%.
For the initial library to survey alternative, 5-membered heterocyclic amides with a methyl group adjacent to the amide, we employed an unsubstituted phthalimide moiety and held the 3-chlorophenyl moiety constant. As shown in Table 1, this library afforded active mGlu1 PAMs, and further highlights the impact of a methyl substituent (as des-methyl congeners were all inactive, EC50s >10 μM).6 However, not all analogs 10 were active, and even regioisomeric congeners displayed divergent SAR. In case of regioisomeric thiophenes, both 10d and 10e were equipotent, but weak mGlu1 PAMs (EC50s 1.5 to 1.8 μM); however, both oxadiazoles (10f and 10g) and thiazoles (10h and 10i) displayed divergent SAR, with the 5-methyl regioisomers 10g and 10i displaying potent PAM activity (EC50s of 1.47 μM and 56 nM, respectively), while the 4-methyl regioisomers 10f and 10h were inactive (EC50s >10 μM). An imidazole analog 10j was also active (EC50 = 374 nM), but strongly basic amines, such as the two enantiomeric, N-methyl prolines 10l and 10m, were inactive. These were very exciting findings overall, especially in the case of 10i, a 56 nM mGlu1 PAM, where a single methyl group increased potency almost 200-fold relative to the unsubstituted thiazole.6 These data narrowed down the field to four methyl-substituted heterocycles (10g, 10i, 10j and 10k) for further optimization in the context of functionalized phthalimides, and determine if SAR developed within the 3-furylamide series (4 and 5) would translate.
Table 1.
Structures and activities for analogs 10.

| |||
|---|---|---|---|
| Cpd | Het | hmGlu1 EC50 (μM)a [% Glu Max ±SEM] | mGlu1 pEC50 (±SEM) |
| 10a |
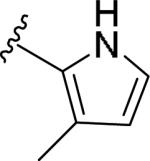
|
3.42 [95±8] | 5.46±0.13 |
| 10b |
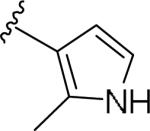
|
>10 [-] | >5 |
| 10c |
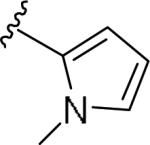
|
5.26 [95±4] | 5.28±0.10 |
| 10d |
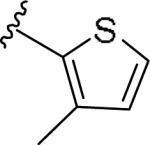
|
1.89 [105±11] | 5.72±0.27 |
| 10e |
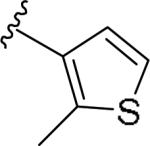
|
1.51 [105±8] | 5.82±0.17 |
| 10f |
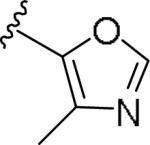
|
>10 [-] | >5 |
| 10g |
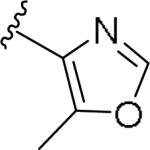
|
1.47 [100±17] | 5.83±0.02 |
| 10h |
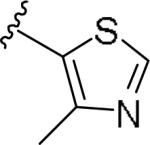
|
>10 [-] | >5 |
| 10i |
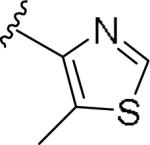
|
0.056 [96±7] | 7.23±0.13 |
| 10j |
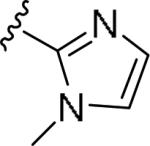
|
0.374 [59±1] | 6.45±0.13 |
| 10k |
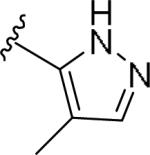
|
1.24 [107±11] | 5.91±0.21 |
| 10l |
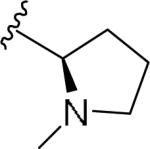
|
>10 [-] | >5 |
| 10m |
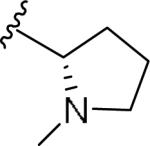
|
>10 [-] | >5 |
Calcium mobilization mGlu1 assays, values are average of three (n=3) independent experiments performed in triplicate.
Next, we prepared a 4 × 7 matrix library to assess SAR of the four methyl-substituted heterocycles (10g, 10i, 10j and 10k) in the context of seven differentially substituted (3-Me, 4-Me, 3-Cl, 4-Cl, 3-F, 4-F and 4-aza) phthalimide moieties (Table 2) to provide analogs 11. As mGlu4 has been a pervasive anti-target, we also counter-screened the 28-membered library against mGlu4, in singlicate, to understand any undesired off-target activity.
Table 2.
Structures and activities for analogs 11.
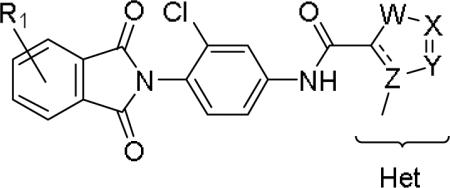
| ||||||
|---|---|---|---|---|---|---|
| Cpd | R1 | Het | hmGlu1 EC50 (μM)a [% Glu Max ±SEM] | mGlu1 pEC50 (±SEM) | hmGlu4 EC50 (μM) [% Glu Maxb] | Fold versus mGlu4 |
| 11a | 3-Me |
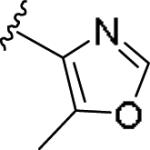
|
0.041 [98±2] | 7.38±0.11 | 0.198 [75] | 4.8 |
| 11b | 4-Me |
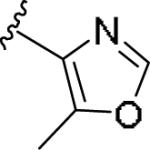
|
0.469 [91±5] | 6.32±0.13 | 0.519 [39] | 0.9 |
| 11c | 3-Cl |
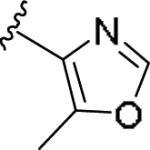
|
0.054 [104±4] | 7.26±0.13 | 0.405 [67] | 7.4 |
| 11d | 4-Cl |
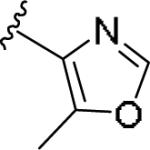
|
1.29 [112±15] | 5.88±0.26 | 0.983 [24] | 0.7 |
| 11e | 3-F |
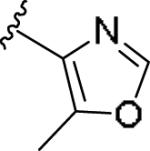
|
0.141 [93±3] | 6.85±0.09 | 0.642 [36] | 4.6 |
| 11f | 4-F |
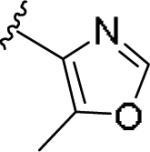
|
0.241 [98±7] | 5.77±0.18 | >10 [-] | 1.7 |
| 11g | 4-Aza |
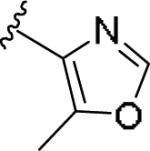
|
1.70 [91±8] | 5.77±0.18 | 2.53 [45] | >5.9 |
| 11h | 3-Me |
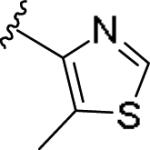
|
0.022 [86±2] | 7.66±0.12 | 1.12 [161] | 51.1 |
| 11i | 4-Me |
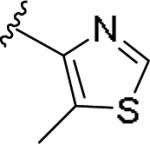
|
0.232 [104±5] | 6.64±0.14 | 1.41 [75] | 6.1 |
| 11j | 3-Cl |
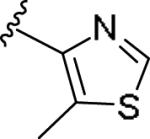
|
0.051 [91±3] | 7.30±0.15 | 1.98 [169] | 39.1 |
| 11k | 4-Cl |
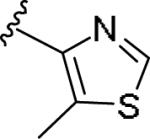
|
1.37 [121±12] | 5.86±0.28 | >10 [59] | >7.3 |
| 111 | 3-F |
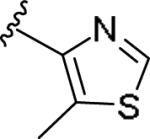
|
0.063 [98±4] | 7.20±0.14 | 0.306 [52] | 4.9 |
| 11m | 4-F |
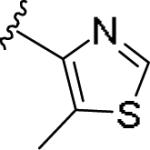
|
0.187 [111±6] | 6.73±0.16 | 0.474 [52] | 2.5 |
| 11n | 4-Aza |
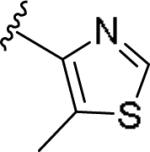
|
0.191 [98±4] | 6.72±0.11 | >10 [-] | >52 |
| 11o | 3-Me |
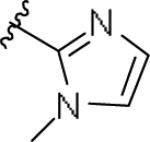
|
0.254 [108±8] | 6.60±0.20 | 0.602 [124] | 2.4 |
| 11p | 4-Me |
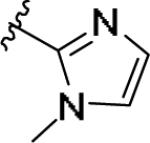
|
±10 [30±1] | >5 | 0.506 [32] | - |
| 11q | 3-Cl |
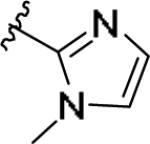
|
0.265 [103±5] | 6.58±0.13 | 1.573 [145] | 5.9 |
| 11r | 4-Cl |
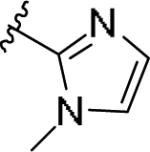
|
±10 [30±1] | >5 | >10 [30] | - |
| 11s | 3-F |
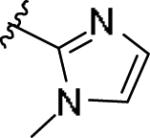
|
0.673 [87±4] | 6.17±0.12 | 1.07 [63] | 1.5 |
| 11t | 4-F |
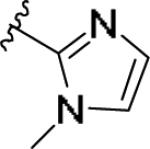
|
±10 [43±9] | >5 | 1.09 [42] | - |
| 11u | 4-Aza |
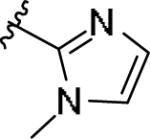
|
±10 [44±8] | >5 | >10 [-] | - |
| 11v | 3-Me |
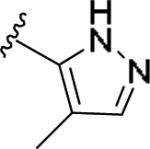
|
0.557 [106±7] | 6.25±0.16 | 2.75 [133] | 4.9 |
| 11w | 4-Me |
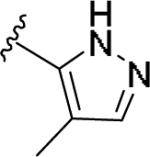
|
4.94 [110±15] | 5.36±0.22 | >10 [-] | >2 |
| 11x | 3-Cl |
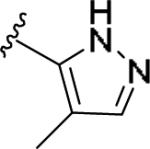
|
0.855 [113±5] | 6.07±0.09 | 3.38 [117] | 3.9 |
| 11y | 4-Cl |
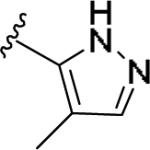
|
±10 [94±2] | >5 | >10 [-] | - |
| 11z | 3-F |
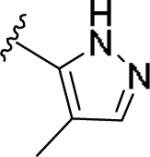
|
0.632 [106±5] | 6.20±0.17 | 6.20 [59] | 9.8 |
| 11aa | 4-F |
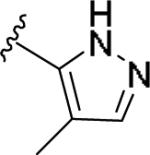
|
0.599 [83±6] | 7.65±0.13 | >10 [-] | >16 |
| 11bb | 4-Aza |
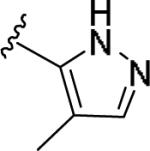
|
>10 [37±9] | >5 | >10 [27] | - |
Calcium mobilization mGlu1 assays, values are average of three (n=3) independent experiments performed in triplicate.
Glu Max is expressed as % of PHCCC response.
While the SAR was steep amongst the imidazole (11o-u) and pyrazole (11v-bb) congeners, the oxazole (11a-g) and thiazole (11h-n) uniformly provided potent mGlu1 PAMs (EC50s down to 22 nM) with a dynamic range of selectivity versus mGlu4 (from 0.7- to > 52-fold). This lack of mGlu4 selectivity was not unexpected based on the central Cl-phenyl core from earlier SAR efforts. However, we were pleased to see that thiazoles and oxazoles could effectively replace the furyl moiety, even those analogs 11 could not advance as in vivo tools. Previously, we demonstrated that replacement of the Cl-phenyl core as in 4 and analogs 11, with a fluorine atom in the 3-position (relative to the phthalimide moiety, as in 5) maintained mGlu1 PAM potency, while eliminating mGlu4 activity (>793-fold selective).6 Therefore, we synthesized analogs of 11a-n to survey the impact of the regioisomer fluorine core (analogs 12, Table 3) in a 10 μM single-point assay prior to running full CRCs. Surprisingly, none of these analogs were strong active mGlu1 PAMs (<50% potentiation of EC20 glutamate at 10 μM), highlighting once again the steep SAR challenges with allosteric ligands. Thus, it was clear that the more basic thiazole analogs could not be advanced due to the lack of selectivity versus mGlu4, and that the SAR developed to abolish activity at mGlu4 did not translate to the thiazoles.
Table 3.
Structures and activities for analogs 12.
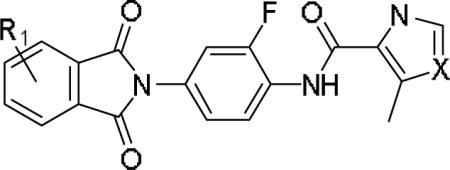
| |||||||
|---|---|---|---|---|---|---|---|
| Cpd | R1 | X | % Glu Max (10 μM)a | Cpd | R1 | X | % Glu Max (10 μM)a |
| 12a | H | O | 18 | 12i | H | S | 45 |
| 12b | 3-Me | O | 14 | 12j | 3-Me | S | 36 |
| 12c | 4-Me | O | 20 | 12k | 4-Me | S | 17 |
| 12d | 3-Cl | O | 25 | 12l | 3-Cl | S | 40 |
| 12e | 4-Cl | O | 27 | 12m | 4-Cl | S | 22 |
| 12f | 3-F | O | 16 | 12n | 3-F | S | 45 |
| 12g | 4-F | O | 36 | 12o | 4-F | S | 43 |
| 12h | 4-Aza | O | 9 | 12p | 4-Aza | S | 42 |
Calcium mobilization mGlu1 assays, single point at 10 μM.
The highly potent and selective mGlu1 PAM 5, was only prepared and evaluated in the context of an unsubstituted phthalimide moiety; therefore, it seemed prudent to further explore functionalized phthalimide analogs of 5, and assess physiochemical properties and selectivity in hopes of developing a robust in vivo tool compound. Following the route outlined in Scheme 1, we synthesized five functionalized analogs 13a-e (Table 4). Unlike the oxazole and thiazole congeners 12, the furyl analogs 13 proved to be very potent mGlu1 PAMs (EC50s 5.3 to 25.7 nM), and both electron donating and electron withdrawing substituents were tolerated. As these new analogs 13 were equipotent or more potent than 5, we assessed their disposition in a battery of in vitro and in vivo DMPK assays (Table 5).9 All of the analogs displayed excellent CYP profiles (most IC50s >30 μM against 3A4, 2C9, 2D6 and 1A2), low to moderate hepatic clearance in both rat (28.9 mL/min/kg to 52 mL/min/kg) and human (4.4 mL/min/kg to 11.9 mL/min/kg) microsomal incubations and exceptional free fraction in rat brain homogenate binding studies (Fu 0.034 to 0.29), the latter suggesting high free drug levels in the CNS. Analogs 13 displayed high protein binding in both rat and human plasma (rapid equilibrium dialysis binding assay), and low recovery suggested modest instability in rat plasma in vitro (as noted previously due hydrolysis of the phthalimide).5 However, the compounds were stable in human plasma, as well as rat brain homogenate, and importantly, in vivo. Analogs 13 were also CNS penetrant, with Kps of 0.25 to 0.95 in rat PBL cassette studies. Therefore, all of these new analogs 13 were attractive and were potential in vivo mGlu1 PAM tools compounds. In vivo rat PK after IV administration showed a wide range of clearance values (4.61 mL/min/kg to 65.5 mL/min/kg) and a disconnection from the in vitro predicted values (e.g., lack of IVIVC). Especially in the case of compound 13a and 13b, were clearance was, respectively, 8 and 4 times lower than the in vitro predicted value. It was also interesting to find a more rational trend in the in vivo clearance, where the electronic character and the position of the substituents on the phthalimide moiety impacts the disposition of the compounds. From this study, compound 13b emerged as the mGlu1 PAM with the best pharmacokinetic profile to date (CLp = 6.94 mL/min/kg, t1/2 = 4.75 h, Vss = 1.29 K/kg) and with high CNS penetration (Kp = 0.95). Finally, we assessed selectivity versus mGlu4 and mGlu5, key anti-targets for this chemotype and found 13a-13e were all uniformly inactive (>450 to >2,000-fold selective) against both mGlu4 and mGlu5 (EC50s >>10 μM). Thus, potent, selective and CNS penetrant mGlu1 PAMs were developed.
Table 4.
Structures and activities for analogs 13.
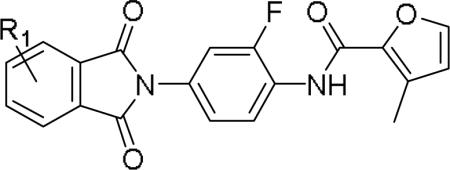
| |||
|---|---|---|---|
| Cpd | R1 | hmGlu1 EC50 (nM)a [% Glu Max ±SEM] | mGlu1 pEC50 (±SEM) |
| 13a | 3-Me | 11.4 [81±2] | 7.94±0.05 |
| 13b | 4-Me | 25.7 [70±2] | 7.59±0.04 |
| 13c | 3-Cl | 5.3 [60±2] | 8.27±0.02 |
| 13d | 3-F | 19.3 [81±2] | 7.71±0.06 |
| 13e | 4-F | 22.0 [67±2] | 7.65±0.07 |
Calcium mobilization mGlu1 assays, values are average of three (n=3) independent experiments performed in triplicate.
Table 5.
DMPK Characterization of mGlu1 PAMs 13a-e.
| Parameter | 13a | 13b | 13c | 13d | 13e |
|---|---|---|---|---|---|
| Hum CLhep (ml/min/kg) | 4.40 | 6.64 | 11.9 | 6.72 | 4.48 |
| Rat CLhep (ml/min/kg) | 35.0 | 28.9 | 46.7 | 52.0 | 35.8 |
| Hum Fu plasma | 0.002 | 0.009 | <0.001 | 0.03 | 0.027 |
| Rat Fu plasmaa | 0.009 | 0.038 | 0.001 | 0.011 | 0.011 |
| Rat Fu brain | 0.193 | 0.298 | 0.272 | 0.17 | 0.034 |
| CYP450 IC50 (μM) | |||||
| 1A2 2C9 | 10 >30 | >30 >30 | 6.3 >30 | >30 >30 | >30 >30 |
| 2D6 3A4 | >30 >30 | >30 >30 | >30 >30 | >30 >30 | >30 >30 |
| Rat iv PK (0.25 mg/kg) | |||||
| t1/2 (min) | 296 | 285 | 51.5 | 38.9 | 76.6 |
| MRT (min) | 330 | 186 | 45.4 | 30.7 | 80.4 |
| Clp (mL min−1 kg−1) | 4.61 | 6.94 | 65.5 | 62.6 | 16.7 |
| Vss (L/kg) | 1.52 | 1.29 | 2.97 | 1.92 | 1.34 |
| Rat iv PBL (0.25 mg/kg) | |||||
| Cn plasma (ng/mL) | 699 | 191 | 217 | 0.97 | 151 |
| Cn Brain (ng/g) | 177 | 182 | 120 | 147 | 97.2 |
| Kp (at 0.25 h) | 0.25 | 0.95 | 0.55 | 0.95 | 0.64 |
Indicates moderate compound instability in rat plasma in vitro.
In conclusion, the continued optimization of the VU0486321 series of mGlu1 PAMs has provided unique SAR, and highlighted the critical value of a single methyl group, a ‘magic methyl’ effect to engender PAM activity across a broad array of 5-member heterocycles. While we encountered instances of robust SAR, the classical steep SAR of allosteric modulators was noted, with key mGluR selectivity handles not translating to structurally similar chemotypes. However, revisiting the furyl amide congeners in the context of functionalized phthalimides, led to a sub-series of highly potent and CNS penetrant (Kps of 0.25 to 0.95) mGlu1 PAMs, with favorable DMPK profiles (low CLp, t1/2s up to 4.9 hours and desirable volumes of distribution) and excellent selectivity profiles versus mGlu4 and mGlu5 (EC50s >>10 μM, >450- to >2,000-fold selective). Of these, VU6004909 (13b) emerged as a near ideal rodent in vivo tool compound to probe selective mGlu1 activation.
Acknowledgments
We thank William K. Warren, Jr. and the William K. Warren Foundation who funded the William K. Warren, Jr. Chair in Medicine (to C.W.L.). P.M.G. would like to acknowledge the VISP program for its support. This work was funded by the William K. Warren, Jr. Chair in Medicine and the NIH (U54MH084659).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Frank RAW, McRae AF, Pocklington AJ, van de Lagemaat LN, Navarro P, Croning MDR, Komiyama NH, Bradley SJ, Challiss RAJ, Armstrong JD, Finn RD, Malloy MP, MacLean AW, Harris SE, Starr JM, Bhaskar SS, Howard EK, Hunt SE, Coffey AJ, Raganath V, Deloukas P, Rogers J, Muir WJ, Deary IJ, Blackwood DH, Visscher PM, Grant SGN. PLoS One. 2011;6:e19011. doi: 10.1371/journal.pone.0019011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ayoub MA, Angelicheva D, Vile D, Chandler D, Morar B, Cavanaugh JA, Visscher PM, Jablensky A, Pfleger KDG, Kalaydijeva L. PLoS One. 2012;7:e32849. doi: 10.1371/journal.pone.0032849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cho HP, Garcia-Barrantes PM, Brogan JT, Hopkins CR, Niswender CM, Rodriguez AL, Venable D, Morrison RD, Bubser M, Daniels JS, Jones CK, Conn PJ, Lindsley CW. ACS Chem. Bio. 2014;9:2334–2346. doi: 10.1021/cb500560h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vieira E, Huwyler J, Jolidon S, Knoflach F, Mutel V, Wichmann J. Bioorg. Med. Chem. Lett. 2009;19:1666–1669. doi: 10.1016/j.bmcl.2009.01.108. [DOI] [PubMed] [Google Scholar]
- 5.Garcia-Barrantes PM, Cho HP, Niswender CM, Byers FW, Locuson CW, Blobaum AL, Xiang Z, Rook JM, Conn PJ, Lindsley CW. J. Med. Chem. 2015;58:7959–7571. doi: 10.1021/acs.jmedchem.5b00727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcia-Barrantes PM, Cho HP, Niswender CM, Blobaum AL, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2015;25:5107–5110. doi: 10.1016/j.bmcl.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jones CK, Engers DW, Thompson AD, Field JR, Blobaum AL, Lindsley SR, Zhou Y, Gogliotti RD, Jadhav S, Zamorano R, Daniels JS, Morrison R, Weaver CD, Conn PJ, Lindsley CW, Niswender CM, Hopkins CR. J. Med. Chem. 2011;54:7639–7647. doi: 10.1021/jm200956q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wood MR, Hopkins CR, Brogan JT, Conn PJ, Lindsley CW. Biochemistry. 2011;50:2403–2410. doi: 10.1021/bi200129s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gentry PR, Kokubo M, Bridges TM, Byun N, Cho HP, Smith E, Hodder PS, Niswender CM, Daniels JS, Conn PJ, Lindsley CW, Wood MR. J. Med. Chem. 2014;57:7804–7810. doi: 10.1021/jm500995y. [DOI] [PMC free article] [PubMed] [Google Scholar]