

Abstract
Pheochromocytomas (PCC) and abdominal paragangliomas (PGL) display a highly diverse genetic background and recent gene expression profiling studies have shown that PCC and PGL (together PPGL) alter either kinase signaling pathways or the pseudo‐hypoxia response pathway dependent of the genetic composition. Recurrent mutations in the Harvey rat sarcoma viral oncogene homolog (HRAS) have recently been verified in sporadic PPGLs. In order to further establish the HRAS mutation frequency and to characterize the associated expression profiles of HRAS mutated tumors, 156 PPGLs for exon 2 and 3 hotspot mutations in the HRAS gene was screened, and compared with microarray‐based gene expression profiles for 93 of the cases. The activating HRAS mutations G13R, Q61R, and Q61K were found in 10/142 PCC (7.0%) and a Q61L mutation was revealed in 1/14 PGL (7.1%). All HRAS mutated cases included in the mRNA expression profiling grouped in Cluster 2, and 21 transcripts were identified as altered when comparing the mutated tumors with 91 HRAS wild‐type PPGL. Somatic HRAS mutations were not revealed in cases with known PPGL susceptibility gene mutations and all HRAS mutated cases were benign. The HRAS mutation prevalence of all PPGL published up to date is 5.2% (49/950), and 8.8% (48/548) among cases without a known PPGL susceptibility gene mutation. The findings support a role of HRAS mutations as a somatic driver event in benign PPGL without other known susceptibility gene mutations. HRAS mutated PPGL cluster together with NF1‐ and RET‐mutated tumors associated with activation of kinase‐signaling pathways. © 2016 The Authors Genes, Chromosomes & Cancer Published by Wiley Periodicals, Inc.
INTRODUCTION
Pheochromocytomas (PCCs) and abdominal paragangliomas (PGLs), together abbreviated PPGL, are neuroendocrine tumors of the adrenal medulla and extra‐adrenal paraganglia, respectively, displaying a highly heterogeneous genetic background (Dahia, 2014). Although the majority of cases are benign, significant subsets of PGLs are malignant and often associated with inactivating SDHB gene mutations. Recent studies have revealed that approximately 40% of PPGL patients carry a constitutional mutation in a susceptibility gene, and somatic mutations are found in an additional 30% of the tumors (Dahia, 2014). The currently known susceptibility genes include NF1, RET, VHL, SDHA, SDHB, SDHC, SDHD, SDHAF2, EGLN1, EPAS1, FH (Letouzé et al., 2013), KIF1Bb (Schlisio et al., 2008), MAX (Comino‐Méndez et al., 2011), and TMEM127 (Dahia, 2014). Single families with PPGL and a constitutional mutation in one of the genes BAP1 (Wadt et al., 2012) and MDH2 (Cascón et al., 2015) have also been reported. The known genetic background of PPGL further includes a set of genes that are recurrently mutated in PPGL tumors such as ATRX (Fishbein et al., 2015), KMT2D (Juhlin et al., 2015), MET (Castro‐Vega et al., 2015), BRAF (Luchetti et al., 2015), the TERT promoter (Liu et al., 2014), and HRAS (Yoshimoto et al., 1992; Crona et al., 2013). Expressional profiling studies of PPGL have shown that tumors fall into two main clusters depending on their genetic composition (Dahia et al., 2005; Burnichon et al., 2011). Cluster 1 with VHL, SDHx and EPAS1 mutated tumors is characterized by a pseudo‐hypoxic response and Cluster 2 includes tumors with mutations in MAX, NF1, RET, and TMEM127 that are associated with active kinase‐signaling pathways (Dahia et al., 2005).
Somatic mutations in the Harvey rat sarcoma viral oncogene homolog (HRAS) gene were first reported in a single pheochromocytoma (Yoshimoto et al., 1992), and HRAS was more recently verified as a recurrently mutated gene in PCC. However, the two other members of the RAS family, that is, NRAS and KRAS have not been reported to be mutated in PPGL. Crona et al. identified HRAS mutations via exome sequencing and reported 3 mutated PCCs and 1 PGL (Crona et al., 2013). Oudijk and co‐workers subsequently detected HRAS mutations in 5.2% of cases (14/271 PCCs) and proposed that the mutations are restricted to sporadic PCCs (10%, 14/140) (Oudijk et al., 2014) and Luchetti et al. published HRAS mutations in 6/65 PPGL (9.2%) (Luchetti et al., 2015). Recently, in a multiomics study by Castro‐Vega et al. the authors screened 193 PPGL for HRAS mutations and found 10 mutated cases, all in benign, sporadic PPGL (Castro‐Vega et al., 2015). Additionally, de Cubas et al. have mentioned 4 HRAS‐mutated PPGL among 156 cases screened, whereof one mutation was found in a metastatic PPGL (de Cubas et al., 2015). Mutations at the hotspots codons 13 and 61 activate the transforming properties of various tumor types, and hence these recurrent mutations are thought to propagate PPGL tumorigenesis for a subset of cases. Germ‐line HRAS mutations have been associated with the Costello syndrome, but to date no co‐occurrence of this syndrome and PPGL has been reported (Crona et al., 2013; Luchetti et al., 2015). In this study, we aimed to further establish the HRAS mutation prevalence as well as its possible impact on global mRNA expression profiles in HRAS mutated PPGL.
MATERIALS AND METHODS
Pheochromocytoma and Paraganglioma (PPGL) Tumor Samples
A total of 156 PPGL (142 PCCs and 14 PGLs) were collected from Karolinska University Hospital, Stockholm, Sweden (Series A; n = 75), University de Lorraine, Vandoeuvre‐les‐Nancy, France (Series B, n = 60), Linköping University Hospital, Sweden (Series C, n = 12), and Haukeland University Hospital in Bergen, Norway (Series D, n = 9), (Supporting Information Table 1). Samples were obtained with informed patient consent and with approval from the local ethics committee of the respective centers. Tumors were classified as benign or malignant following the WHO criteria (DeLellis et al., 2004). For Series A, a subset of the tumors (n = 54) had been characterized for mutations in 14 proposed PPGL susceptibility genes (EGLN1, EPAS1 KIF1Bβ, MAX, MEN1, NF1, RET, SDHA, SDHB, SDHC, SDHD, SDHAF2, TMEM127, and VHL) (Welander et al., 2014a) and the remaining tumors (n = 21) were screened for mutations in 8 of these genes (EPAS1, MAX, NF1, SDHB, SDHD, RET, TMEM127, and VHL) (Welander et al., 2014b) (Supporting Information Table 1). Furthermore, all tumors in Series C and D were previously analyzed for mutations in the 8 genes (EPAS1, MAX, NF1, SDHB, SDHD, RET, TMEM127, and VHL) (Welander et al., 2014b) (Supporting Information Table 1). For Series B, a subset of patients exhibited established PPGL syndromes with associated mutations (Supporting Information Table 1).
HRAS Mutation Analysis
Genomic DNA isolated from fresh frozen tumor samples was used for amplification of fragments of exon 2 and 3 covering codons 13 and 61 of the HRAS gene (NM_001130442) with primer sequences available upon request. Sanger sequencing was carried out at the KIGene core facility at Karolinska Institutet for 113 cases and at Linköping University for 42 cases using previously described methodology (Welander et al., 2014a). All samples showing chromatogram alterations were re‐analyzed with the reverse primer. One HRAS mutation (case 88) has been previously reported and was found via whole‐exome sequencing (Supporting Information Table 1) (Juhlin et al., 2015).
Gene Expression Profiling
Total RNA was extracted from 53 PPGLs from Series A (Supporting Information Table 1), using the mirVana Isolation Kit (Ambion, Austin, TX) and subsequently analyzed in an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA). As previously reported, RNA preparations from all cases were of sufficient quality as measured by RIN values (Andreasson et al., 2013a, 2013b). RNA samples (250 ng) were used for whole‐transcriptome analysis with GeneChip Human Gene 1.0 ST arrays (Affymetrix), covering approximately 29K annotated genes as previously described (Welander et al., 2014b). Tumor RNA from 40 cases in Series A–C (detailed in Supporting Information Table 1) had previously been analyzed with the GeneChip Human 1.0 ST array (Affymetrix) (Welander et al., 2014b). HRAS mutation status from the current study was implemented into the dataset and after normalization using the robust multiarray average (RMA) algorithm, hierarchical clustering of the microarray expression data for all 93 PPGLs was performed as previously described (Welander et al., 2014b) using a set of genes that has been shown to separate the clusters (Burnichon et al., 2011). These genes overlapped with 454 of the probe sets in our analysis which were used to perform the hierarchical clustering. Moreover, gene expression profiles based on the entire probe sets on the array were compared between the 7 HRAS mutated cases and the 91 HRAS wild‐type cases included. Given their involvement in PPGL, normalized signal intensities for the HRAS, vascular endothelial growth factor A (VEGFA) and phenylethanolamine N‐methyltransferase (PNMT) genes were exported for separate statistical analysis.
Within the cohort, tumors with known somatic mutations in EPAS1, KIF1Bb, MAX, NF1, RET, SDHA, SDHB, TMEM127, and VHL were included (Supporting Information Table 1). Additionally, five cases from patients with known PPGL syndromes (2 MEN2, 1 NF1, 1 PGL5, and 1 VHL) were included as internal controls and were also included in the hierarchical clustering. One identical sample was analyzed at both time points in (Welander et al., 2014b) and in the current study as an internal control between the GeneChip arrays. This sample did not show any difference in clustering behavior as evaluated with a principal component analysis quality control in the GeneSpring software (data not shown).
Statistical Analyses
Transcriptome‐wide statistical analyses and clustering were performed as previously described (Welander et al., 2014b) using the GeneSpring GX v. 12.6 (Agilent, Santa Clara, CA) software and the Benjamini–Hochberg method (Benjamini and Hochberg, 1995) was used to control for multiple testing. When comparing the gene expression profiles between the 7 HRAS mutated cases and the 91 HRAS wild‐type cases based on the entire probe sets on the array, a Benjamini–Hochberg corrected false discovery rate (FDR) of less than 0.1 was applied. Gene expression levels for HRAS, VEGFA, and PNMT were compared between sporadic HRAS‐mutated and HRAS wild‐type tumors using two‐tailed Student's t‐test. Two‐tailed Mann–Whitney U or Fisher's exact tests were used to analyze potential significant correlations between the clinical parameters and HRAS mutational status. P‐values of less than 0.05 were considered as statistically significant.
RESULTS
Detection of HRAS Mutations
A HRAS mutation was found in 11 out of 156 tumors screened (142 PCCs and 14 PGLs), equaling a total frequency of 7.1% (11/156) in our cohort (Table 1). One mutation was found in exon 2 (G13R) and ten mutations were found in exon 3 (six Q61R, three Q61K, and one Q61L) (Table 1, Fig. 1A).
Table 1.
HRAS Gene Mutations and Clinical Characteristics of the PPGL Included in the Study
| Series | Series | Series | Series | Series | |
|---|---|---|---|---|---|
| Parameter | A | B | C | D | A, B, C, and D |
| Gender | |||||
| Male:Female | 32:43 | 26:34 | 5:7 | 3:6 | 66:90 |
| Age at diagnosis | |||||
| Mean years | 55 | 53 | 63 | 58 | 55 |
| Median (range) years | 57 (14–83) | 52 (23–84) | 66 (39–76) | 58 (42–80) | 57 (14–84) |
| Tumor type | |||||
| Total | 75 | 60 | 12 | 9 | 156 |
| PCC | 64 | 57 | 12 | 9 | 142 |
| PGL | 11 | 3 | 0 | 0 | 14 |
| Tumor size | |||||
| Mean mm | 52 | 41 | 34 | 47 | 46 |
| Median (range) mm | 45 (20–160) | 40 (10–100) | 30 (17–60) | 50 (10–90) | 40 (10–160) |
| Malignancy | |||||
| Benign | 69 | 59 | 12 | 9 | 149 |
| Malignant | 6 | 1 | 0 | 0 | 7 |
| PPGL susceptibility gene | |||||
| Mutated | 37 | 9 | 3 | 6 | 55 |
| Unknown mutation (sporadic) | 38 | 51 | 9 | 3 | 101 |
| HRAS codon 13 and 61 | |||||
| Wild‐type | 70 | 57 | 10 | 8 | 145 |
| Mutated | 5 | 3 | 2 | 1 | 11 |
| G13R | 0 | 0 | 1 | 0 | 1 |
| Q61R | 3 | 1 | 1 | 1 | 6 |
| Q61K | 2 | 1 | 0 | 0 | 3 |
| Q61L | 0 | 1 (PGL) | 0 | 0 | 1 |
| HRAS mutation frequencies | |||||
| Total | 6.7% | 5.0% | 16.7% | 11.1% | 11/156 (7.1%) |
| PCC | 7.8% | 1.5% | 16.7% | 11.1% | 10/142 (7.0%) |
| PGL | 0% | 33.3% | 0% | 0% | 1/14 (7.1%) |
| According to gender | |||||
| Male | 6.3% | 7.7% | 0% | 0% | 4/66 (6.1%) |
| Female | 7.0% | 2.9% | 28.6% | 16.7% | 7/90 (7.8%) |
| According to malignancy | |||||
| benign | 7.2% | 5.1% | 16.7% | 11.1% | 11/149 (7.4%) |
| Malignant | 0% | 0% | 0% | 0% | 0/7 (0%) |
| According to susceptibility gene status | |||||
| Unknown mutation (sporadic) | 13.2% | 5.9% | 22.2% | 33.3% | 11/101 (10.9%) |
| Mutated | 0% | 0% | 0% | 0% | 0/55 (0%) |
Tumor size refer to the maximum diameter.
Series A = Karolinska University Hospital, Sweden.
Series B = University de Lorraine, Vandoeuvre‐les‐Nancy, France.
Series C = Linköping University Hospital, Linköping, Sweden.
Series D = Haukeland University Hospital, Bergen, Norway.
Figure 1.
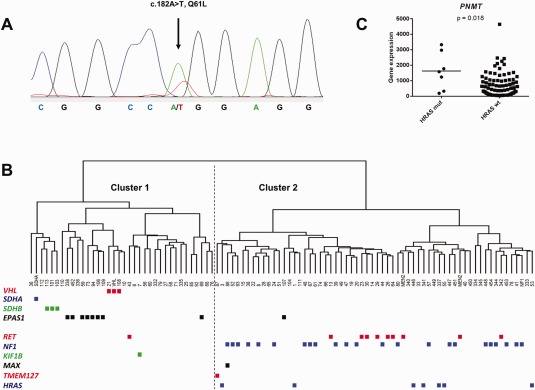
Detection of a HRAS Q61L mutation, hierarchical clustering of PPGLs and PNMT gene expression in relation to HRAS mutation status. (A) Chromatogram of case 227 (PGL) showing the Q61L mutation (c.182A>T, COSM498), which has previously not been reported in PPGL. A vertical arrow shows the heterozygous missense variant. (B) Hierarchical clustering of 93 tumors (indicated by their case numbers) and 5 control cases (indicated as MEN2, NF1, SDHA, and VHL) based on their expression levels for 454 genes according to Burnichon et al. 2011. The dendrogram shows separation of tumors into two distinct groups (Cluster 1 to the left and Cluster 2 to the right). The PPGL mutation status is indicated below. All 7 HRAS‐mutated cases clustered together with the tumors endowed with mutations in the NF1‐ and RET genes. (C) RNA levels of the PNMT gene compared between the PPGL with (HRAS mutated n = 7) and without (HRAS wild‐type n = 86) HRAS mutations. Horizontal bars represent mean values and the gene expression has been normalized to the mean value of cases endowed with constitutional NF1‐ and RET mutations. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
The HRAS mutation frequency in apparently sporadic PPGL (non‐familial and without known susceptibility gene mutation) was 10.9% (11/101; Table 1). The HRAS mutation status was compared with clinical and genetic characteristics of the present cohort and in combination with published studies (Table 1). No HRAS mutation was found in any PPGL endowed with a known PPGL susceptibility gene mutation (Table 1, Supporting Information Table 1). Hence, HRAS mutations were associated with the PPGL group without a known susceptibility gene mutation both in our study (Fisher's exact test, P = 0.017) (Table 1) and in all available studies combined (Fisher's exact test, P < 0.0001) (Table 2). Regarding clinical parameters, no mutations were found in PPGLs classified as malignant according to the current WHO criteria and the patients endowed with a HRAS mutation tended to have higher age at diagnosis (mean 63 ±10 years) compared with those without HRAS mutation (mean 54 ±16 years) however this association did not reach statistical significance (two‐tailed Mann–Whitney U‐test, P = 0.08). In our series of 11 mutated PPGLs there were four men and seven women, and no gender‐related difference in HRAS mutation frequency was observed (Fisher's exact test, P = 0.76). The mean tumor sizes of HRAS‐mutated and wild‐type cases were 58 ± 41 mm and 45 ± 22 mm, respectively. This difference was not statistically significant (Two‐tailed Mann–Whitney U‐test, P = 0.37).
Table 2.
Summary of HRAS Mutation Studies in PPGL
| HRAS gene status | PPGL susceptibility gene | |||||||
|---|---|---|---|---|---|---|---|---|
| HRAS mutated | Codon 13 G13R | Codon 61 Q61R | Codon 61 Q61K | Codon 61 Q61L | Wild‐type codon 13/61 | Known mutation | Unknown mutation (sporadic) | |
| This study a | ||||||||
| PCC (n = 141) | 9 | 1 | 5 | 3 | 0 | 132 | 48 | 93 |
| PGL (n = 14) | 1 | 0 | 0 | 0 | 1 | 13 | 7 | 7 |
| Total (n = 155) | 10 | 1 | 5 | 3 | 1 | 145 | 55 | 100 |
| Moley et al. 1991 | ||||||||
| PCC (n = 10) | 0 | 0 | 0 | 0 | 0 | 10 | 0 | 10 |
| Total (n = 10) | 0 | 0 | 0 | 0 | 0 | 10 | 0 | 10 |
| Yoshimoto et al. 1992 | ||||||||
| PCC (n = 19) | 1 | 1 | 0 | 0 | 0 | 18 | 0 | 19 |
| Total (n = 19) | 1 | 1 | 0 | 0 | 0 | 18 | 0 | 19 |
| Crona et al. 2013 , b | ||||||||
| PCC (n = 72) | 3 | 1 | 1 | 1 | 0 | 69 | 22 | 50 |
| PGL (n = 9) | 1 | 0 | 1 | 0 | 0 | 8 | 3 | 6 |
| Total (n = 81) | 4 | 1 | 2 | 1 | 0 | 77 | 25 | 56 |
| Oudijk et al. 2013 | ||||||||
| PCC (n = 216) | 14 | 1 | 12 | 1 | 0 | 202 | 76 | 140 |
| PGL (n = 55) | 0 | 0 | 0 | 0 | 0 | 55 | 31 | 24 |
| Total (n = 271) | 14 | 1 | 12 | 1 | 0 | 257 | 107 | 164 |
| Luchetti et al. 2015 , c | ||||||||
| PCC (n = 60) | 6 | 1 | 5 | 0 | 0 | 54 | 16 | 44 |
| PGL (n = 5) | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 5 |
| Total (n = 65) | 6 | 1 | 5 | 0 | 0 | 59 | 16 | 49 |
| Castro‐Vega et al. 2015 , d | ||||||||
| PCC (n = 168) | 10 | 1 | 4 | 2 | 0 | 158 | 100 | 68 |
| PGL (n = 25) | 0 | 0 | 0 | 0 | 0 | 25 | 16 | 9 |
| Total (n = 193) | 10 | 1 | 4 | 2 | 0 | 183 | 116 | 77 |
| de Cubas et al. 2015 | ||||||||
| PCC (n = 128) | 3 | 0 | 3 | 0 | 0 | 125 | 68 | 60 |
| PGL (n = 28) | 1 | 0 | 0 | 1 | 0 | 27 | 15 | 13 |
| Total (n = 156) | 4 | 0 | 3 | 1 | 0 | 152 | 83 | 73 |
| HRAS mutations in the eight studies | ||||||||
| PCC (n = 814) | 46 (5.7%) | 6 (0.7%) | 30 (3.7%) | 7 (0.9%) | 0 | 768 (94.3%) | 1/330 (0.3%) | 45/484 (9.3%) |
| PGL (n = 136) | 3 (2.2%) | 0 | 1 (0.7%) | 1 (0.7%) | 1 (0.7%) | 133 (97.8%) | 0/72 | 3/64 (4.7%) |
| Total (n = 950) | 49 (5.2%) | 6 (0.6%) | 31 (3.3%) | 8 (0.8%) | 1 (0.1%) | 901 (94.8%) | 1/402 (0.2%) | 48/548 (8.8%) |
One PCC with a Q61R HRAS mutation has been previously published (Juhlin et al., 2015) and is excluded.
One head and neck paraganglioma is excluded.
Twenty head and neck paragangliomas are excluded.
Six metastases and 3 thoracic paragangliomas are excluded. Three HRAS mutations from this study are not reported in the table: G12R (n = 1), S145L (n = 1), and A146T (n = 1).
mRNA Expression Profiles of HRAS Mutated PPGLs
Two approaches were taken to reveal gene expression profiles associated with the HRAS mutational status. First, hierarchical clustering was performed using the previously defined set of 454 probe sets (Burnichon et al., 2011). This showed that the seven HRAS mutated PPGL clustered together with the tumors endowed with mutations in the NF1 and RET genes, associated with PIK3/AKT/mTOR and RAS/RAF activation in Cluster 2 (Fig. 1B). When HRAS mutated and HRAS wild‐type cases were separately compared for the individual genes HRAS, VEGFA, and PNMT, the latter was found to have significantly higher expression in HRAS‐mutated cases (Student's t‐test, P = 0.018) (Fig. 1C) whereas HRAS and VEGFA did not show statistically significant differences between groups (Student's t‐test, P = 0.061 and P = 0.29, respectively).
Subsequently, the 7 HRAS mutated tumors were compared with 91 HRAS wild‐type tumors using the complete set of probe sets on the array. Based on this, 21 differentially expressed transcripts were identified as detailed in Table 3. Thirteen of the identified transcripts correspond to a known gene, including the receptor‐type tyrosine‐protein phosphatase zeta (PTPRZ1) and the transmembrane protein 195 (TMEM195) as the two most up‐regulated genes.
Table 3.
Genes with Altered Expression in HRAS Mutated Tumors (n = 7) Compared with HRAS Wild‐Type Cases (n = 91) using a Benjamini–Hochberg Corrected FDR of 10 %
| Transcript cluster id | Corrected P‐value | Fold change | Fold change log | Gene symbol |
|---|---|---|---|---|
| Up‐regulated in HRAS mutated vs. wild‐type | ||||
| 8135774 | 0.0632 | 2.7526 | 1.4608 | PTPRZ1 |
| 8138337 | 0.0000 | 2.3660 | 1.2424 | TMEM195 |
| 7928907 | 0.0253 | 2.0682 | 1.0484 | |
| 8000963 | 0.0025 | 1.9834 | 0.9880 | STX1B |
| 7921852 | 0.0532 | 1.7622 | 0.8174 | MPZ |
| 8107518 | 0.0532 | 1.6164 | 0.6928 | |
| 8103374 | 0.0889 | 1.5410 | 0.6239 | |
| 8152863 | 0.0253 | 1.5150 | 0.5993 | |
| 8156110 | 0.0253 | 1.5148 | 0.5991 | |
| 8000757 | 0.0005 | 1.4531 | 0.5391 | DOC2A |
| 8157027 | 0.0253 | 1.4506 | 0.5367 | NIPSNAP3B |
| 7998053 | 0.0854 | 1.3622 | 0.4459 | |
| 7948037 | 0.0253 | 1.3206 | 0.4012 | |
| 7965838 | 0.0300 | 1.1981 | 0.2608 | |
| 8040672 | 0.0832 | 1.1767 | 0.2347 | DRC1 |
| Down‐regulated in HRAS mutated vs. wild‐type | ||||
| 8036483 | 0.0909 | −1.3839 | −0.4687 | YIF1B |
| 8098705 | 0.0604 | −1.3403 | −0.4225 | MTRF1L |
| 8061542 | 0.0832 | −1.3147 | −0.3948 | HM13 |
| 7989619 | 0.0419 | −1.3038 | −0.3827 | PPIB |
| 7983290 | 0.0253 | −1.3018 | −0.3805 | SERF2 |
| 7924230 | 0.0832 | −1.2333 | −0.3025 | ABHD17A |
DISCUSSION
In this study we aimed to further establish the HRAS mutation frequency in PPGL and examine the impact on global expressional profiles in HRAS mutated tumors. We consequently screened 156 PPGLs for mutations in the HRAS gene and compared the results with microarray‐based gene expression profiles for 93 (60%) of the cases.
Eleven out of 156 cases were found endowed with HRAS mutations equaling a total frequency of 7.1%. This prevalence is in line with previously published results (Table 2) (Yoshimoto et al., 1992; Crona et al., 2013; Oudijk et al., 2014; Castro‐Vega et al., 2015; Luchetti et al., 2015). One single mutation was found in exon 2 (G13R) and ten mutations were found in exon 3 (six Q61R, three Q61K and one Q61L). The Q61L mutation at c.182 A>T (COSM498), which has previously not been reported in PPGL, was the only mutation found in a PGL in our cohort. This alteration has previously been reported in cutaneous squamous cell carcinoma (Su et al., 2012) and in penile cancer (Andersson et al., 2008).
No HRAS mutations were found in PPGLs classified as malignant according to the current WHO criteria, which is in line with previous findings (Yoshimoto et al., 1992; Crona et al., 2013; Oudijk et al., 2014; Castro‐Vega et al., 2015), however one single metastatic case with a HRAS mutation has been previously reported (de Cubas et al., 2015). The observed male:female proportion is in line with the results shown in two studies (Oudijk et al., 2014; Castro‐Vega et al., 2015), but conflicting the results shown in an earlier study (Crona et al., 2013) where 4/4 patients with HRAS mutations were men. The mean tumor size of HRAS‐mutated cases tended to be slightly increased as compared with HRAS wild‐type cases (58 mm vs. 45 mm), however the difference was not statistically significant which is in agreement with the results of three preceding studies where the parameter was included (Crona et al., 2013; Oudijk et al., 2014).
None of the HRAS mutated tumors in our cohort were malignant according to the WHO criteria, suggesting a role of HRAS mutations as a somatic driver event in benign PPGL. As with the other genes associated with kinase signaling pathways in PPGL, HRAS mutations appear to be associated with a benign phenotype overall, although characterization and long term follow‐up in additional cohorts will be required to determine if they may be used as predictive markers.
Activating mutations in the HRAS gene are known to affect MAPK signaling (Balmain and Pragnell, 1983), and as might be expected, all seven HRAS mutated cases included in the microarray‐based profiling were grouped in Cluster 2 together with tumors harboring mutations in the NF1, MAX, RET and TMEM127 genes associated with PIK3/AKT/mTOR and RAS/RAF activation. Taken together with preceding findings (Castro‐Vega et al., 2015), these results support the notion that HRAS‐mutated cases segregate separately from Cluster 1 tumors. Tumors with HRAS mutations exhibited higher expression of the PNMT gene encoding the PNMT enzyme that catalyzes the conversion (methylation) between norepinephrine and epinephrine. This finding is in line with a previous study showing that Cluster 2 tumors have increased PNMT expression and hence higher epinephrine levels in the patient (Eisenhofer et al., 2004). Interestingly, several tumors without mutations in any of the so far known susceptibility genes appear to form a group within Cluster 1 (Fig. 1B). An underlying somatic VHL mutation was excluded in these cases based on previous mutation screenings in all cases included in the microarray (Supporting Information Table S1). One may speculate that these tumors might share unknown underlying genetic mechanisms that potentially involve regulation of the hypoxia response, which may be an interesting subject for future studies.
Based on the analyses using the entire probe set on the array, PTPRZ1 was found to be the most up‐regulated gene in HRAS mutated PPGL compared with HRAS wild‐type tumors (Table 3). This gene has been shown to regulate glioblastoma cell motility (Müller et al., 2003) and activation of PTPRZ1 via hypoxia inducible factor‐2 alpha (HIF‐2α) has also been suggested (Wang et al., 2010).
To summarize, we were able to establish a low HRAS mutation frequency (7.1%) in PPGL. Taken together with all other studies published up to date, the overall HRAS mutation prevalence in PPGL is 5.2% (49/950) and 8.8% (48/548) among apparently sporadic cases without a known PPGL susceptibility gene mutation. HRAS mutated cases were grouped into Cluster 2 and somatic HRAS mutations did not occur in patients with a known PPGL susceptibility gene mutation or in patients with malignant PPGL. Somatic HRAS mutations thus represent a possible driver event for a subset of benign PPGLs.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The authors wish to thank Ms. Lisa Ånfalk, Karolinska University Hospital, for excellent tissue handling and Professor Michael Brauckhoff at the Haukeland University Hospital, Norway, for providing material and clinical information regarding series D samples used in the study. The authors also want to thank Professor Anne‐Paule Gimenez‐Roqueplo and Dr. Luis‐Jaime Castro‐Vega for kindly providing the mutational data for the cases in the Paris study.
Supported by: Swedish Cancer Foundation, StratCan, the Swedish Research Council, the Cancer Research Foundations of Radiumhemmet, Karolinska Institutet, and the Stockholm County Council.
REFERENCES
- Andersson P, Kolaric A, Windahl T, Kirrander P, Söderkvist P, Karlsson MG. 2008. PIK3CA, HRAS and KRAS gene mutations in human penile cancer. J Urol 179:2030–2034. [DOI] [PubMed] [Google Scholar]
- Andreasson A, Kiss NB, Caramuta S, Sulaiman L, Svahn F, Bäckdahl M, Höög A, Juhlin CC, Larsson C. 2013a. The VHL gene is epigenetically inactivated in pheochromocytomas and abdominal paragangliomas. Epigenetics 8:1347–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasson A, Kiss NB, Juhlin CC, Höög A. 2013b. Long‐Term storage of endocrine tissues at −80°C does not adversely affect RNA quality or overall histomorphology. Biopreserv Biobank 11:366–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmain A, Pragnell IB. 1983. Mouse skin carcinomas induced in vivo by chemical carcinogens have a transforming Harvey‐ras oncogene. Nature 303:72–74. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc B Met 57:289–300. [Google Scholar]
- Burnichon N, Vescovo L, Amar L, Libé R, de Reynies A, Venisse A, Jouanno E, Laurendeau I, Parfait B, Bertherat J, Plouin PF, Jeunemaitre X, Favier J, Gimenez‐Roqueplo AP. 2011. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum Mol Genet 20:3974–3985. [DOI] [PubMed] [Google Scholar]
- Cascón A, Comino‐Méndez I, Currás‐Freixes M, de Cubas AA, Contreras L, Richter S, Peitzsch M, Mancikova V, Inglada‐Pérez L, Pérez‐Barrios A, Calatayud M, Azriel S, Villar‐Vicente R, Aller J, Setién F, Moran S, Garcia JF, Río‐Machín A, Letón R, Gómez‐Graña Á, Apellániz‐Ruiz M, Roncador G, Esteller M, Rodríguez‐Antona C, Satrústegui J, Eisenhofer G, Urioste M, Robledo M. 2015. Whole‐exome sequencing identifies MDH2 as a new familial paraganglioma gene. J Natl Cancer Inst 11:107. [DOI] [PubMed] [Google Scholar]
- Castro‐Vega LJ, Letouzé E, Burnichon N, Buffet A, Disderot PH, Khalifa E, Loriot C, Elarouci N, Morin A, Menara M, Lepoutre‐Lussey C, Badoual C, Sibony M, Dousset B, Libé R, Zinzindohoue F, Plouin PF, Bertherat J, Amar L, de Reyniès A, Favier J, Gimenez‐Roqueplo AP. 2015. Multi‐omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat Commun 6:6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comino‐Méndez I, Gracia‐Aznárez FJ, Schiavi F, Landa I, Leandro‐García LJ, Letón R, Honrado E, Ramos‐Medina R, Caronia D, Pita G, Gómez‐Graña A, de Cubas AA, Inglada‐Pérez L, Maliszewska A, Taschin E, Bobisse S, Pica G, Loli P, Hernández‐Lavado R, Díaz JA, Gómez‐Morales M, González‐Neira A, Roncador G, Rodríguez‐Antona C, Benítez J, Mannelli M, Opocher G, Robledo M, Cascón A. 2011. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet 43:663–667. [DOI] [PubMed] [Google Scholar]
- Crona J, Delgado Verdugo A, Maharjan R, Stålberg P, Granberg D, Hellman P, Björklund P. 2013. Somatic mutations in H‐RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. J Clin Endocrinol Metab 98:E1266–E1271. [DOI] [PubMed] [Google Scholar]
- de Cubas AA, Korpershoek E, Inglada‐Pérez L, Letouzé E, Currás‐Freixes M, Fernández AF, Comino‐Méndez I, Schiavi F, Mancikova V, Eisenhofer G, Mannelli M, Opocher G, Timmers H, Beuschlein F, de Krijger R, Cascon A, Rodríguez‐Antona C, Fraga MF, Favier J, Gimenez‐Roqueplo AP, Robledo M. 2015. DNA methylation profiling in pheochromocytoma and paraganglioma reveals diagnostic and prognostic markers. Clin Cancer Res 21:3020–3030. [DOI] [PubMed] [Google Scholar]
- Dahia PLM. 2014. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat Rev Cancer 14:108–119. [DOI] [PubMed] [Google Scholar]
- Dahia PL, Ross KN, Wright ME, Hayashida CY, Santagata S, Barontini M, Kung AL, Sanso G, Powers JF, Tischler AS, Hodin R, Heitritter S, Moore F, Dluhy R, Sosa JA, Ocal IT, Benn DE, Marsh DJ, Robinson BG, Schneider K, Garber J, Arum SM, Korbonits M, Grossman A, Pigny P, Toledo SP, Nosé V, Li C, Stiles CD. 2005. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet 1:72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLellis RA, Lloyd RV, Heitz PU, Eng C. 2004. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of Endocrine Organs. Lyon: IARC Press; pp. 147–166. [Google Scholar]
- Eisenhofer G, Huynh T‐T, Pacak K, Brouwers FM, Walther MM, Linehan WM, Munson PJ, Mannelli M, Goldstein DS, Elkahloun AG. 2004. Distinct gene expression profiles in norepinephrine‐ and epinephrine‐producing hereditary and sporadic pheochromocytomas: Activation of hypoxia‐driven angiogenic pathways in von Hippel‐Lindau syndrome. Endocr Relat Cancer 11:897–911. [DOI] [PubMed] [Google Scholar]
- Fishbein L, Khare S, Wubbenhorst B, DeSloover D, D'Andrea K, Merrill S, Cho NW, Greenberg RA, Else T, Montone K, LiVolsi V, Fraker D, Daber R, Cohen DL, Nathanson KL. 2015. Whole‐exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun 6:6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhlin CC, Stenman A, Haglund F, Clark VE, Brown TC, Baranoski J, Bilguvar K, Goh G, Welander J, Svahn F, Rubinstein JC, Caramuta S, Yasuno K, Günel M, Bäckdahl M, Gimm O, Söderkvist P, Prasad ML, Korah R, Lifton RP, Carling T. 2015. Whole‐exome sequencing defines the mutational landscape of pheochromocytoma and identifies KMT2D as a recurrently mutated gene. Genes Chromosomes Cancer 54:542–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letouzé E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, Buffet A, Marcaillou C, Bertherat J, Amar L, Rustin P, De Reyniès A, Gimenez‐Roqueplo AP, Favier J. 2013. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23:739–752. [DOI] [PubMed] [Google Scholar]
- Liu T, Brown TC, Juhlin CC, Andreasson A, Wang N, Bäckdahl M, Healy JM, Prasad ML, Korah R, Carling T, Xu D, Larsson C. 2014. The activating TERT promoter mutation C228T is recurrent in subsets of adrenal tumors. Endocr Relat Cancer 21:427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchetti A, Walsh D, Rodger F, Clark G, Martin T, Irving R, Sanna M, Yao M, Robledo M, Neumann HP, Woodward ER, Latif F, Abbs S, Martin H, Maher ER. 2015. Profiling of somatic mutations in phaeochromocytoma and paraganglioma by targeted next generation sequencing analysis. Int J Endocrinol 2015:138573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moley JF, Brother MB, Wells SA, Spengler BA, Biedler JL, Brodeur GM. 1991. Low frequency of ras gene mutations in neuroblastomas, pheochromocytomas, and medullary thyroid cancers. Cancer Res 51:1596–1599. [PubMed] [Google Scholar]
- Müller S, Kunkel P, Lamszus K, Ulbricht U, Lorente GA, Nelson AM, von Schack D, Chin DJ, Lohr SC, Westphal M, Melcher T. 2003. A role for receptor tyrosine phosphatase zeta in glioma cell migration. Oncogene 22:6661–6668. [DOI] [PubMed] [Google Scholar]
- Oudijk L, de Krijger RR, Rapa I, Beuschlein F, de Cubas AA, Dei Tos AP, Dinjens WN, Korpershoek E, Mancikova V, Mannelli M, Papotti M, Vatrano S, Robledo M, Volante M. 2014. H‐RAS mutations are restricted to sporadic pheochromocytomas lacking specific clinical or pathological features: Data from a multi‐institutional series. J Clin Endocrinol Metab 99:E1376–E1380. [DOI] [PubMed] [Google Scholar]
- Schlisio S, Kenchappa RS, Vredeveld LC, George RE, Stewart R, Greulich H, Shahriari K, Nguyen NV, Pigny P, Dahia PL, Pomeroy SL, Maris JM, Look AT, Meyerson M, Peeper DS, Carter BD, Kaelin WG. Jr. 2008. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev 22:884–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su F, Viros A, Milagre C, Trunzer K, Bollag G, Spleiss O, Reis‐Filho JS, Kong X, Koya RC, Flaherty KT, Chapman PB, Kim MJ, Hayward R, Martin M, Yang H, Wang Q, Hilton H, Hang JS, Noe J, Lambros M, Geyer F, Dhomen N, Niculescu‐Duvaz I, Zambon A, Niculescu‐Duvaz D, Preece N, Robert L, Otte NJ, Mok S, Kee D, Ma Y, Zhang C, Habets G, Burton EA, Wong B, Nguyen H, Kockx M, Andries L, Lestini B, Nolop KB, Lee RJ, Joe AK, Troy JL, Gonzalez R, Hutson TE, Puzanov I, Chmielowski B, Springer CJ, McArthur GA, Sosman JA, Lo RS, Ribas A, Marais R. 2012. RAS mutations in cutaneous squamous‐cell carcinomas in patients treated with BRAF inhibitors. N Engl J Med 366:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadt K, Choi J, Chung JY, Kiilgaard J, Heegaard S, Drzewiecki KT, Trent JM, Hewitt SM, Hayward NK, Gerdes AM, Brown KM. 2012. A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanoma, and paraganglioma. Pigment Cell Melanoma Res 25:815–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang V, Davis DA, Veeranna RP, Haque M, Yarchoan R. 2010. Characterization of the activation of protein tyrosine phosphatase, receptor‐type, Z polypeptide 1 (PTPRZ1) by hypoxia inducible factor‐2 alpha. PLoS One 5:9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welander J, Andreasson A, Juhlin CC, Wiseman RW, Bäckdahl M, Höög A, Larsson C, Gimm O, Söderkvist P. 2014a. Rare germline mutations identified by targeted next‐generation sequencing of susceptibility genes in pheochromocytoma and paraganglioma. J Clin Endocrinol Metab 99:1352–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welander J, Andreasson A, Brauckhoff M, Bäckdahl M, Larsson C, Gimm O, Söderkvist P. 2014b. Frequent EPAS1/HIF2α exons 9 and 12 mutations in non‐familial pheochromocytoma. Endocr Relat Cancer 21:495–504. [DOI] [PubMed] [Google Scholar]
- Yoshimoto K, Iwahana H, Fukuda A, Sano T, Katsuragi K, Kinoshita M, Saito S, Itakura M. 1992. ras mutations in endocrine tumors: Mutation detection by polymerase chain reaction‐single strand conformation polymorphism. Jpn J Cancer Res Gann 83:1057–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information