

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC), primary hypomagnesemia, renal hypomagnesemia (caused by variants in CLDN16).
FHHNC with severe ocular involvement, renal hypomagnesemia with severe ocular involvement (caused by variants in CLDN19).
1.2 OMIM# of the disease
248250 (FHHNC) and 248190 (FHHNC with severe ocular involvement).
1.3 Name of the analysed genes or DNA/chromosome segments
CLDN16 (previously known as PCLN1) (locus 3q27), CLDN19 (locus 1p34.2).
1.4 OMIM# of the gene(s)
603959 (CLDN16), 610036 (CLDN19).
1.5 Mutational spectrum
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC) and FHHNC with severe ocular involvement are autosomal recessive renal tubular disorders caused by variants in the CLDN16 (locus 3q27) and CLDN19 (locus 1p34.2) genes, respectively.1, 2 These genes encode the tight-junction proteins claudin-16 (paracellin-1) and claudin-19, respectively, which are important for the paracellular reabsorption of calcium and magnesium in the thick ascending limb of Henle's loop.3 Claudin-19 is also expressed in the retinal epithelium. FHHNC is characterized by excessive urinary losses of magnesium and calcium, nephrocalcinosis, kidney stones in about 1/3 of cases and progressive renal failure. Approximately one-third of patients progress to renal failure or end-stage renal disease (ESRD) during adolescence. In most cases, patients with CLDN19 variants additionally have severe ocular abnormalities (mainly myopia, nystagmus and macular colobomata).
Fifty-six CLDN16 disease-causing variants have been described so far.1, 2, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 (see also HGMD: http://www.hgmd.org/). These include missense variants (41), nonsense variants (5), splice site variants (5), small deletions (3) and small indels (2). The majority of CLDN16 variants are spread over all the five exons. Missense variants are located in or near the four transmembrane domains, in the two extracellular loops or in the C-terminal cytoplasmic region. Variants in affected individuals occur in either homozygous or compound-heterozygous state. The most frequent CLDN16 disease-causing variant, c.453G>T (p.L151F), occurring in almost 50% of the patients described so far, is due to a widespread founder effect (Germany and Eastern European countries).4 A founder effect has been also detected for the recurrent disease-causing c.416C>T (p.A139V) variant in North African families.10
Only 17 CLDN19 disease-causing variants are known,2, 3, 10, 15, 16, 17, 18 including missense variants (12), nonsense variants (3), one large deletion and one small deletion. Two missense variants affect the last nucleotide of exon 1 and are also predicted to disrupt a donor splice site.17 CLDN19 variants are spread over exons 1–4. Missense variants are located in the first extracellular loop or in the first three transmembrane domains. Most variants in affected individuals are homozygous, only a few are compound-heterozygous. A founder effect has been shown for the recurrent disease-causing variant c.59G>A (p.G20D) in Spanish and French families.2, 10, 17
The GeneBank accession numbers for CLDN16 and CLDN19 are NG_008149.1, NM_006580.3 and NG_008993.1, NM_148960.2, respectively. Reference CLDN16 (ENSG00000113946) and CLDN19 (ENSG00000164007) sequences can also be found in the Ensembl database (http://www.ensembl.org). A list of disease-causing variants for these two genes can be found in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php). A variation table with all the CLDN16 and CLDN19 variants can be found at http://www.ensembl.org.
1.6 Analytical methods
Direct bidirectional sequencing of coding exons and flanking intronic sequences of the CLDN16 and CLDN19 genes on genomic DNA. If no disease-causing variants or only one mutated allele was found, the next step would be the research of deletions of some or all exons of the gene by Quantitative Multiplex PCR of Short Fragments (QMPSF). Depending on the country of origin, it might be reasonable to start analysis specifically for frequent disease-causing variants, for instance, in patients from Germany and Eastern European countries for the CLDN16 c.453G>T (p.L151F) variant, in Spanish patients for the CLDN19 c.59G>A (p.G20D) variant or in North African families for the CLDN16 c.416C>T (p.A139V) variant. This might be done by variant-specific PCR methods (SNaPshot) or by sequencing the corresponding exon.
1.7 Analytical validation
Variants identified must be verified by a second independent test (direct sequencing or QMPSF) and further studied by analysing at least 100 chromosomes of the same ethnic origin, by bioinformatics prediction tools (SIFT, PolyPhen, Align-GVGD, MutationTaster), by comparison with data base entries, and, if possible, by segregation analysis. Intronic variants, missense and synonymous alterations that are suspected to affect pre-mRNA splicing should be characterized by RT-PCR analysis of an RNA sample.
1.8 Estimated frequency of the disease (incidence at birth (‘birth prevalence') or population prevalence. If known to be variable between ethnic groups, please report):
The population prevalence of FHHNC is unknown. FHHNC is a very rare disorder, and only about 131 affected families (178 patients) subjected to molecular-genetic testing have been described in the literature: 73 families (106 patients) with CLDN16 causative variant and 58 families (72 patients) with CLDN19 causative variant. However, FHHNC is likely to have been under-diagnosed due to a lack of awareness of this disorder. Because of the autosomal-recessive mode of inheritance, a significant proportion of patients affected by FHHNC originate from populations with a higher frequency of consanguineous marriages
1.9 Diagnostic setting:

Comment: Genetic testing is mainly used to confirm the clinical diagnosis, to assess genetic risk in relatives and in relation to genetic counselling. Prenatal diagnosis and pre-implantation genetic diagnosis are feasible for families with known disease-causing variants.
Hypomagnesemia, hypercalciuria and nephrocalcinosis are constant characteristics. Most patients with CLDN19 disease-causing variants also present with severe ocular defects (mainly myopia, nystagmus and macular colobomata).
2. Test characteristics
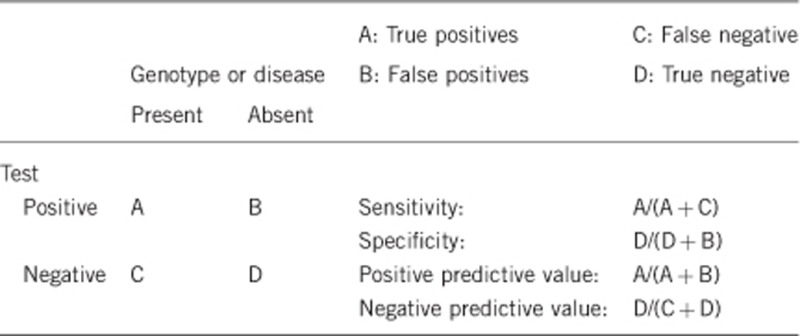
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
For both genes the analytical sensitivity of genomic sequencing, if necessary combined with QMPSF, should be nearly 100%. However, causative variants in non-coding regions are likely to be missed with these techniques, and these types of variants have not yet been reported in FHHNC.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
Analytical specificity is nearly 100% for both genes. False positives in genomic sequencing are rare.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors, such as age or family history. In such cases, a general statement should be given, even if quantification can only be made case by case.
Probably, it is close to 100%.
We may assume that variants exist that remain undetected by sequencing as well as by QMPSF (promoter variants and far intronic variants). In those cases where no CLDN16 or CLDN19 causative variants can be identified on one or both alleles, mRNA analysis would provide an alternative method of detecting a causative intronic variant.
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases, a general statement should be given, even if quantification can only be made case by case.
Clinical specificity is possibly 100%.
On the basis of our experience, it can be excluded that a healthy individual carries disease-causing CLDN16 or CLDN19 variants on both alleles.
2.5 Positive clinical predictive value
(lifetime risk to develop the disease if the test is positive)
As there is 100% penetrance, positive predictive value is 100%.
2.6 Negative clinical predictive value
(probability not to develop the disease if the test is negative).
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
Nearly 100%.
Index case in that family had not been tested:
Nearly 100%.
3. Clinical utility
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.9 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
The clinical and biochemical diagnosis of FHHNC is not invasive (primarily urine and blood tests). Nephrocalcinosis can be diagnosed by abdominal ultrasound. Ocular defects are detected by eye examination. So, the only burden is the drawing of a blood sample, which is also necessary for the genetic test.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
As far as we know, there are no studies available to determine the cost effectiveness of any diagnostic approach to FHHNC or FHHNC with ocular involvement. The differences in price between tests would depend on the laboratory and the country where testing is performed. In general, genetic testing is at the moment more expensive than clinical and biochemical diagnosis, but this may change in future. Furthermore, the two genes analysed, CLDN16 and CLDN19, are small, with only five exons. We believe that it is cheaper to exclude at-risk relatives using a DNA test than by using the full clinical investigation protocol. On the other hand, genetic testing is valuable for confirming a clinical diagnosis, informing genetic counselling, and facilitating prenatal diagnosis and pre-implantation genetic diagnosis.
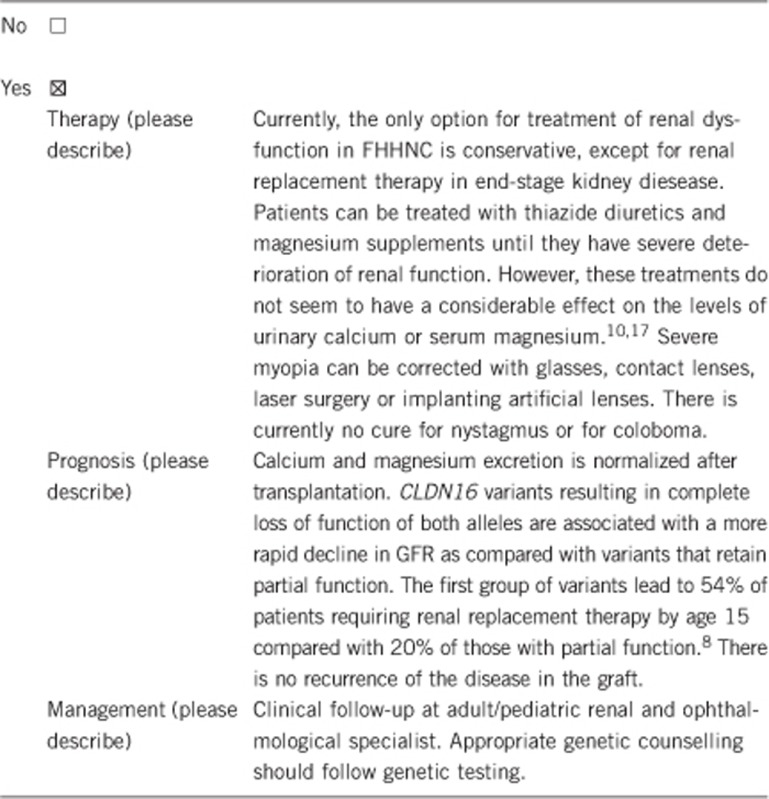
3.1.4 Will disease management be influenced by the result of a genetic test?
3.2 Predictive setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.9 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe):
Yes. Early diagnosis may allow early initiation of treatment and/or surveillance (potential medical interventions, such as pharmacological therapy of hypercalciuria and magnesium supplementation). The search for potential kidney donors for transplantation can be started. Monitoring of kidney function, as well as ophthalmological evaluation in case of CLDN19 causative variants, is needed continuously. Genetic counselling should be recommended. Prenatal or pre-implantation genetic testing can be offered.
Hypercalciuria and nephrolithiasis have been described in relatives harbouring heterozygous variants.4
If the test result is negative (please describe):
No. Clinical follow-up is not relevant if the test is negative for a known familial variant. In case of potential heterozygotes, the knowledge of not having an elevated carrier risk results in ‘relief' with regard to the family risk, and allows an informed decision on family planning and prenatal diagnosis.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
Urine and serum could be tested for the presence of hypercalciuria and hypomagnesemia, respectively, which are constant findings in FHHNC. The patient could also be examined for nephrocalcinosis and kidney function and secondary problems, due to deterioration of renal function. Regular ophthalmological examination in case a CLDN19 variant(s) has been identified in the family.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.9 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes, the genetic test confirms the autosomal recessive inheritance in the family and clarifies the recurrence risk in a future pregnancy. In case of non-affected parents with an affected child the recurrence risk is 25%. If causative variants have been identified in CLDN16 or CLDN19, it is possible to assess the carrier status of other healthy relatives and to offer genetic counselling to the family.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Yes, if a causative CLDN16 or CLDN19 variant has been identified in the index patient, only that gene or the specific variant needs to be tested in family members instead of complete screening of the entire genes. If a relative tests negative, then none of their offspring are at risk and they will not need to be tested.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes. Identifying the causal variants in the index case allows for carrier testing and pre-symptomatic testing in relatives.
3.4 Prenatal diagnosis
(To be answered if in 1.9 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes, provided that both disease-causing alleles have been identified and their segregation from carrier parents has been traced, it offers the possibility of early and reliable prenatal testing or even pre-implantation diagnostics.
4. If applicable, further consequences of testing
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
The genetic diagnosis establishes a precise diagnosis and allows precise genetic counselling of patients and relatives. Carriers' awareness of their genetic status is important for family planning. Genetic testing of relatives can also be important in relation to selection of potential family donors for kidney transplantation.
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics. RV-P was supported by European Community's Seventh Framework Programme grant 2012-305608 (EURenOmics). FC-M was supported by RenalTube, a project financed by Fondo de Investigación Sanitaria, Spain (grant PI11/00342) and co-financed by the European Regional Development Fund ‘A way to build Europe'.
The authors declare no conflict of interest.
References
- Simon DB, Lu Y, Choate KA et al: Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 1999; 285: 103–106. [DOI] [PubMed] [Google Scholar]
- Konrad M, Schaller A, Seelow D et al: Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet 2006; 79: 949–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Renigunta A, Konrad M et al: Claudin-16 and claudin-19 interact and form a cation-selective tight junction complex. J Clin Invest 2008; 118: 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber S, Schneider L, Peters M et al: Novel paracellin-1 mutations in 25 families with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol 2001; 12: 1872–1881. [DOI] [PubMed] [Google Scholar]
- Blanchard A, Jeunemaitre X, Coudol P et al: Paracellin-1 is critical for magnesium and calcium reabsorption in the human thick ascending limb of Henle. Kidney Int 2001; 59: 2206–2215. [DOI] [PubMed] [Google Scholar]
- Müller D, Kausalya PJ, Claverie-Martin F et al: A novel claudin 16 mutation associated with childhood hypercalciuria abolishes binding to ZO-1 and results in lysosomal mistargeting. Am J Hum Genet 2003; 73: 1293–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller D, Kausalya PJ, Bockenhauer D et al: Unusual clinical presentation and possible rescue of a novel claudin-16 mutation. J Clin Endocrinol Metab 2006; 91: 3076–3079. [DOI] [PubMed] [Google Scholar]
- Konrad M, Hou J, Weber S et al: CLDN16 genotype predicts renal decline in familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol 2008; 19: 171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasapkara CS, Tumer L, Okur I, Hasanoglu A: A novel mutation of the claudin 16 gene in familial hypomagnesemia with hypercalciuria and nephrocalcinosis mimicking rickets. Genet Couns 2011; 22: 187–192. [PubMed] [Google Scholar]
- Godron A, Harambat J, Boccio V et al: Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: phenotype-genotype correlation and outcome in 32 patients with CLDN16 or CLDN19 mutations. Clin J Am Soc Nephrol 2012; 7: 801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guran T, Akcay T, Bereket A et al: Clinical and molecular characterization of Turkish patients with familial hypomagnesaemia: novel mutations in TRPM6 and CLDN16 genes. Nephrol Dial Transplant 2012; 27: 667–673. [DOI] [PubMed] [Google Scholar]
- Seeley HH, Loomba-Albrecht LA, Nagel M, Butani L, Bremer AA: Familial hypomagnesemia with hypercalciuria and nephrocalcinosis in three siblings having the same genetic lesion but different clinical presentations. World J Pediatr 2012; 8: 177–180. [DOI] [PubMed] [Google Scholar]
- Deeb A, Abood SA, Simon J, Dastoor H, Pearce SH, Sayer JA: A novel CLDN16 mutation in a large family with familial hypomagnesaemia with hypercalciuria and nephrocalcinosis. BMC Res Notes 2013; 6: 527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadarajah L, Khosravi M, Dumitriu S et al: A novel claudin-16 mutation, severe bone disease, and nephrocalcinosis. Lancet 2014; 383: 98. [DOI] [PubMed] [Google Scholar]
- Naeem M, Hussain S, Akhtar N: Mutation in the tight-junction gene claudin 19 (CLDN19) and familial hypomagnesemia, hypercalciuria, nephrocalcinosis (FHHNC) and severe ocular disease. Am J Nephrol 2011; 34: 241–248. [DOI] [PubMed] [Google Scholar]
- Ekinci Z, Karabaş L, Konrad M: Hypomagnesemia-hypercalciuria-nephrocalcinosis and ocular findings: a new claudin-19 mutation. Turk J Pediatr 2012; 54: 168–170. [PubMed] [Google Scholar]
- Claverie-Martín F, García-Nieto V, Loris C et al: Claudin-19 mutations and clinical phenotype in Spanish patients with familial hypomagnesemia with hypercalciuria and nephrocalcinosis. PLoS One 2013; 8: e53151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Shibli A, Konrad M, Altay W, Al Masri O, Al-Gazali L, Al Attrach I: Familial hypomagnesemia with hypercalciuria and nephrocalcinosis (FHHNC): report of three cases with a novel mutation in CLDN19 gene. Saudi J Kidney Dis Transpl 2013; 24: 338–344. [DOI] [PubMed] [Google Scholar]