

Update to: European Journal of Human Genetics (2011) 19,; doi:10.1038/ejhg.2011.7; published online 2 February 2011
1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Familial adenomatous polyposis (FAP), adenomatous polyposis coli (APC), familial polyposis coli (FPC), attenuated adenomatous polyposis coli (AAPC); phenotypic variants: Gardner syndrome, Turcot syndrome.
1.2 OMIM# of the disease
175100.
1.3 Name of the analysed genes or DNA/chromosome segments
APC (5q22).
1.4 OMIM# of the gene(s)
611731.
1.5 Mutational spectrum
Reference sequences for variant description: LRG_130; NG_008481.4; NM_000038.5; NC_000005.10.
Broad spectrum of point mutations, >90% are truncating (nonsense, del/ins, splice sites).2
Mutational hot spots: c.3927_3931delAAAGA;p.(Glu1309Aspfs*4) (11%), c.3183_3187delACAAA; p.(Gln1062*) (7%), c.637C>T;p.(Arg213*) (3%), c.3202_3205delTCAA;p.(Ser1068Glyfs*57) (2%).
The vast majority of mutations is located in the 5' half of the gene, mutations 3' to codon 1700 are rare (1%).
Genomic rearrangements: large deletions <10–15% in classical FAP; large duplications are very rare.5, 6
In APC/MUTYH mutation-negative families, deep intronic mutations not covered by routine methods have been identified in up to 8% of unselected and up to 30% of familial cases.7
Post-zygotic mosaicism in 10–15% of de novo events.8
For the mutational spectrum, see locus-specific databases: http://databases.lovd.nl/shared/genes/APC; www.umd.be/APC/, and www.hgmd.cf.ac.uk/ac/. Novel mutations are still being reported.
For variants with no functional effect (‘polymorphisms'), see NCBI accession number NM_000038.5 (www.ncbi.nlm.nih.gov/nuccore/NM_000038.5).
1.6 Analytical methods
- Clinical selection:
- All patients with the clinical diagnosis of an attenuated or classical colorectal adenomatous polyposis (at least 10 synchronous adenomatous polyps).9
- In pedigrees consistent with an autosomal recessive mode of inheritance, screening for MUTYH mutations should be performed preferably before APC screening.
- In case of few colorectal adenomas diagnosed at a young age, tumour screening for microsatellite instability and immunohistochemical staining should be considered (see 3.1, differential diagnoses).
- A careful clinical examination including histology is a prerequisite for performing cost-effective mutation analysis.
- Germline mutation analysis:
- Direct sequencing of all 15 coding exons.10
- In some centres, screening of exons 3 to 15J (codon 1700) in all patients and exons 1, 2, 15J-W (codons 1700-ter) in case of extra-digestive manifestation only.
- In some centres, pre-screening of the gene by protein truncation test of exon 15 (genomic level) or of the whole gene (RNA level) and/or by DHPLC, SSCP, CSGE.
- Screening of the whole gene including promoter region for large genomic anomalies (deletions, duplications) by MLPA or QMPSF.
- Linkage analysis and functional tests for interpretation of unclassified APC variants.
- High-throughput techniques/massive parallele sequencing
- Sequencing of the coding regions or the whole gene by next generation sequencing (NGS) technologies.10
- This is usually done in the context of commercially available or customized gene panels including a number of genes related to the phenotype and its differential diagnoses. These panels vary by methods used and genes included (see, for example, NGS panel database; www.eurogentest.org/index.php?id=668).
- APC germline mutations (point mutations or large deletions/duplications) are also identified as incidental findings in the context of large-scale screening approaches (exome or genome sequencing, chromosome analysis, (array)-comparative genomic hybridization), which are performed in individuals with varying non-polyposis phenotypes or population-based healthy controls both in a diagnostic or research setting.
1.7 Analytical validation
As with other molecular genetic diagnostic tests, analytical results can be validated using standard procedures of internal and external quality assessment (EQA). These may include:
Internal validation through analysis of known mutations (positive controls).
Direct sequencing of both DNA strands (bidirectional sequencing).
Validation of NGS results by Sanger sequencing (if the read depth/coverage or other NGS quality parameters are below defined thresholds).
In case of assumed mosaicism confirmation of results with a second method, using different primers and additional tissues.
Confirmation of mutation in an independent biological sample of the index case or an affected relative.
In some cases (for example, single-exon deletions detected by MLPA), the results of semiquantitative methods should be confirmed by an independent technique (long-range PCR, RNA analysis, different MLPA kit).
External validation through exchange of DNA control samples with other diagnostic institutions and participation in EQA schemes (for example, www.emqn.org).
1.8 Estimated frequency of the disease (incidence at birth (‘birth prevalence') or population prevalence). If known to be variable between ethnic groups, please report)
1.9 Diagnostic setting

Comment:
Prenatal diagnosis and preimplantation genetic diagnosis (PGD) are rarely requested. An explanation might be that FAP is a relatively late-manifesting and treatable disease. Another reason might be that some FAP patients at child-bearing age are not informed about reproductive options. In general, prenatal diagnosis and PGD should be performed according to each countries' law, and only after appropriate, non-directive genetic counselling.13
2. TEST CHARACTERISTICS
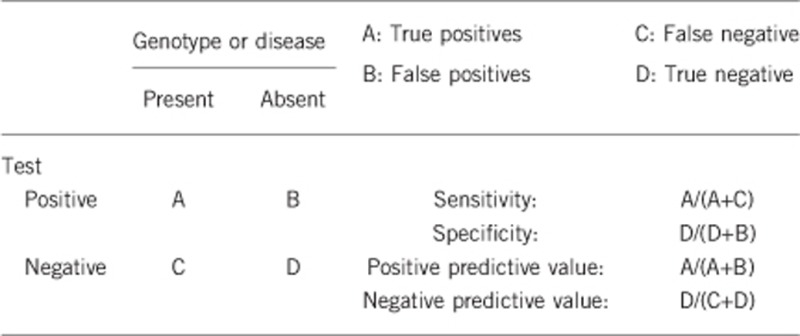
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
Almost 100% (by direct sequencing of all coding exons and deletion/duplication screening by methods such as MLPA.
Can be distinctly less in mosaic cases, depending on degree of mosaic and analysed tissue. In these cases, pre-screening methods appear to be more sensitive than direct sequencing.8
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
Almost 100%.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present) The clinical sensitivity dependent on variable factors such as age or family history. In such cases, a general statement should be given, even if a quantification can only be made case by case.
Dependent on colorectal phenotype (number of adenomas), age, and family history.2
Mutations deep within introns or in regulatory elements are missed with current standard methods,7 and the causal relevance of some missense or intronic mutations is unclear, so far (variants of uncertain clinical significance, VUS).
AFAP: about 20–30%.
2.4 Clinical specificity
(proportion of negative tests if the disease is not present) The clinical specificity can be dependent on variable factors such as age or family history. In such cases, a general statement should be given, even if a quantification can only be made case by case.
Almost 100% (except for variants of uncertain clinical significance).
2.5 Positive clinical predictive value
(life-time risk to develop the disease if the test is positive)
Penetrance in proven mutation carriers is almost complete. Due to the high clinical variability, clinically mildly affected persons may not be diagnosed or will be deceased for other reasons during presymptomatic (sub-clinical) stage of the disease.
2.6 Negative clinical predictive value
(probability not to develop the disease if the test is negative) Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested and a pathogenic germline mutation was identified:
Almost 100%.
Index case in that family had not been tested:
As the mutation-detection rates in APC and MUTYH strongly depend on the colorectal phenotype (mild versus more florid forms of polyposis) and the family history (dominant vs. recessive pedigree pattern), and – particularly in attenuated courses of adenomatous polyposis – more and still unknown genes may be involved (locus heterogeneity), no exact figures can be given.
To test people at risk without having identified the underlying germline mutations in a clearly affected index patient of the family is in general not a meaningful approach and should therefore be avoided, since persons at risk who are tested negative may still have a substantial risk and cannot be released from surveillance.
If no index patient is available in a family, mutation screening of risk persons might be performed with the intention to identify the familial mutation, which subsequently can be used for predictive testing of other family members at risk. However, to date, no guidelines or state-of-the art recommendations for this scenario exist.
3. CLINICAL UTILITY
3.1 (Differential) diagnosis: the tested person is clinically affected
(To be answered if in 1.9 ‘A' was marked)
Clinical diagnostic criteria.
According to polyp number and age at onset, the phenotype is usually classified as classical (typical) FAP or attenuted FAP (AFAP).14, 15 However, it should kept in mind that the formation of colorectal adenomas is a biological continuum without any clearly delineated features. In particular, AFAP is not well defined as a disease entity. Widely used clinical criteria are the following:
Classical FAP: more than 100 colorectal adenomas; early onset (polyp formation during second decade of life, gastrointestinal symptoms during third decade of life).9
AFAP: a milder course of the colorectal disease with a delay in onset of adenomatous polyposis and colorectal cancer of 10–25 years compared with classical FAP; <100 colorectal adenomas at 25 years of age or older and/or a late onset of disease (≥45 years of age) irrespective of polyp number.15
3.1.1 Can a diagnosis be made other than through a genetic test?

Comment:
The most relevant differential diagnoses of an attenuated/late-onset FAP is the autosomal recessive MAP caused by biallelic MUTYH germline mutations (see CUGC MAP16) and the autosomal dominant PPAP caused by specific heterozygous POLE or POLD1 germline missense mutations.17
In case of a low number of (synchronous) adenomas, Lynch syndrome (previously referred to as hereditary non-polyposis colorectal cancer) (see CUGC Lynch syndrome18) should be considered.
3.1.2 Describe the burden of alternative diagnostic methods to the patient
The diagnosis colorectal polyposis in a clinically affected person can only be established by colonoscopy and subsequent histological examination of removed polyps, which is a burdensome examination. Alternative burdenless diagnostic methods are ocular fundus examination and mandibular radiography, but these methods are helpful in only a few patients.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Very cost effective and time saving but not useful for predictive testing.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive Setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.9 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe)
Yes: Increase in compliance to participate in specific preventive surveillance programs (in particular colonoscopy and upper gastrointestinal endoscopy). In some cases, the position of the mutation might affect the procedure and time of surgical management (although the decision of colectomy should be based mainly on the clinical phenotype).
In some cases, family planning, choice of profession.
If the test result is negative (please describe).
Yes. Release from intensified screening program. Psychological relief.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
Same as for proven mutation carriers: Close-meshed early diagnosis and surveillance programs, colectomy when numerous polyps have been detected. Yet, these measures are taken in vain in half of the persons at risk (non-carriers).
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.9 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes (if the mutation is known in the family).
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Yes:
By securing the primary cause of the disease, extended diagnostic investigations in other symptomatic relatives can be avoided.
By exclusion of a carrier status in predictive diagnostics, superfluous preventive investigations can be avoided and psychological relief is obtained.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes.
3.4 Prenatal diagnosis
(To be answered if in 1.9 ‘D' was marked).
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Technically yes, after considering specific rules and ethical aspects.
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
Support for family life organization.
Efficiency of subsequent clinical management.
For many patients, proof of diagnosis is a value itself – irrespective of a medical benefit – because the disease and its cause can clearly be named.
When a genetic cause is verified, an assumption of ‘own fault' as cause of disease (exogenous poisons, ‘wrong conduct') often can be lapsed with relief.
The main benefits of genetic diagnostics in FAP are the differentiation from MAP (which, for example, do not have a risk for developing desmoid tumours), a precise recurrence risk calculation for close relatives, and relief of non-carriers during predictive testing, and a tailored surveillance program including prophylactic surgery options.20
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics, by the German Cancer Aid (Deutsche Krebshilfe e.V.; Grant Number 108421), and by the French National Cancer Institute (InCA Grant 2004-11).
The authors declare no conflict of interest.
References
- Lagarde A, Rouleau E, Ferrari A et al. Germline APC mutation spectrum derived from 863 genomic variations identified through a 15-years medical genetics service to French FAP patients. J Med Genet 2010; 47: 721–722. [DOI] [PubMed] [Google Scholar]
- Friedl W, Aretz S. Familial Adenomatous Polyposis: Experience from a Study of 1164 Unrelated German Polyposis Patients. Hered Cancer Clin Pract 2005; 3: 95–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgaard ML, Fenger K, Bulow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat 1994; 3: 121–125. [DOI] [PubMed] [Google Scholar]
- Aretz S, Uhlhaas S, Caspari R et al. Frequency and parental origin of de novo APC mutations in familial adenomatous Polyposis. Eur J Hum Genet 2004; 12: 52–58. [DOI] [PubMed] [Google Scholar]
- Aretz S, Stienen D, Uhlhaas S et al. Large submicroscopic genomic APC deletions are a common cause of typical familial adenomatous polyposis. J Med Genet 2005; 42: 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michils G, Tejpar S, Thoelen R et al. Large deletions of the APC gene in 15% of mutation-negative patients with classical polyposis (FAP): a Belgian study. Hum Mutat 2005; 25: 125–134. [DOI] [PubMed] [Google Scholar]
- Spier I, Horpaopan S, Vogt S et al. Deep intronic APC mutations explain a substantial proportion of patients with familial or early-onset adenomatous polyposis. Hum Mutat 2012; 33: 1045–1050. [DOI] [PubMed] [Google Scholar]
- Aretz S, Stienen D, Friedrichs N et al. Somatic APC mosaicism: a frequent cause of familial adenomatous polyposis (FAP). Hum Mutat 2007; 28: 985–992. [DOI] [PubMed] [Google Scholar]
- Jasperson KW, Burt RW et al. APC-Associated polyposis conditions, 18 December 1998 (updated 27 March 2004). In: Pagon RA, Adam MP, Ardinger HH (eds). GeneReviews [Internet]. Seattle, WA: University of Washington, Seattle, 1993-2014. Available at www.ncbi.nlm.nih.gov/books/NBK1345/. [PubMed] [Google Scholar]
- Hegde M, Ferber M, Mao R, Samowitz W, Ganguly A. Working Group of the American College of Medical Genetics and Genomics (ACMG) Laboratory Quality Assurance Committee.; ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYH-associated polyposis). Genet Med 2014; 16: 101–116. [DOI] [PubMed] [Google Scholar]
- Bülow S, Faurschou Nielsen T, Bülow C, Bisgaard ML, Karlsen L, Moesgaard F. The incidence rate of familial adenomatous polyposis. Results from the Danish Polyposis Register. Int J Colorectal Dis 1996; 11: 88–91. [DOI] [PubMed] [Google Scholar]
- Bülow S. Results of national registration of familial adenomatous polyposis. Gut 2003; 52: 742–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douma KF, Aaronson NK, Vasen HF, Verhoef S, Gundy CM, Bleiker EM. Attitudes toward genetic testing in childhood and reproductive decision-making for familial adenomatous polyposis. Eur J Hum Genet 2010; 18: 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol 2006; 101: 385–398. [DOI] [PubMed] [Google Scholar]
- Knudsen AL, Bülow S, Tomlinson I, Möslein G, Heinimann K, Christensen IJ. Attenuated familial adenomatous polyposis: results from an international collaborative study. Colorectal Dis 2010; 12: e243–e249. [DOI] [PubMed] [Google Scholar]
- Aretz S, Genuardi M, Hes FJ. Clinical utility gene card for: MUTYH-associated polyposis (MAP), Autosomal recessive colorectal adenomatous polyposis, Multiple colorectal adenomas, Multiple adenomatous polyps (MAP) - update 2012. Eur J Hum Genet 2013; 21: doi:10.1038/ejhg.2012.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palles C, Cazier JB, Howarth KM et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 2013; 45: 136–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahner N, Steinke V, Schlegelberger B, Eisinger F, Hutter P, Olschwang S. Clinical utility gene card for: Lynch syndrome (MLH1, MSH2, MSH6, PMS2, EPCAM) - update 2012. Eur J Hum Genet 2013; 21: doi:10.1038/ejhg.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwenhuis MH, Vasen HF. Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Crit Rev Oncol Hematol 2007; 61: 153–161. [DOI] [PubMed] [Google Scholar]
- Vasen HFA, Möslein G, Alonso A et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 2008; 57: 704–713. [DOI] [PubMed] [Google Scholar]