

1. Disease characteristics
1.1 Name of the disease (synonyms)
XMEN: X-linked immunodeficiency with magnesium defect, Epstein–Barr virus infection, and neoplasia.
1.2 OMIM# of the disease
300853.
1.3 Name of the analysed genes or DNA/chromosome segments
Major gene: MAGT1, Xq21.1.
1.4 OMIM# of the gene(s)
MAGT1 #300715.
1.5 Mutational spectrum
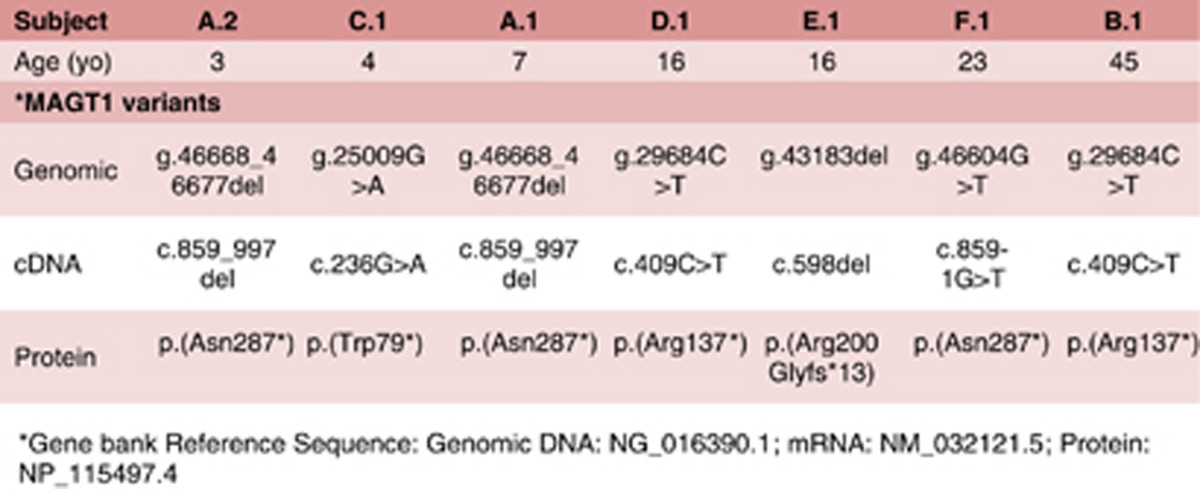
Among the seven patients we have characterized for this disease thus far, we have only found loss-of-function variants in MAGT1 leading to XMEN disease (Table 1): Patients A.1, A.2 and B.1,1 and patients C.1, D.1, E.1 and F.1.2 (Patients A.1 and A.2 are siblings).
Table 1. Variants in MAGT1 leading to XMEN disease.
1.6 Analytical methods
Different strategies can be used to identify XMEN patients:
Assessing for coding mutations in genomic DNA by polymerase chain reaction (PCR) amplification and DNA sequence determination of the 10 coding exons of MAGT1. 2,3
Assessing for loss of MAGT1 expression by real-time qRT-PCR, followed by targeted genomic sequencing of coding and non-coding gene regulatory regions of the MAGT1 locus.
Assessing large-scale deletions of the MAGT1 gene by comparative genomic hybridization.
1.7 Analytical validation
Genomic variants should be validated by investigating deficiencies in MAGT1 expression or function as indicated by one of more of the following means:
Decreased MAGT1 protein on immunoblotting. 1
Loss of T-cell receptor-triggered Mg2+ flux in T cells. 1
Poor responses in lymphocyte proliferation assays induced with T-cell-receptor-specific agonistic antibodies or lectins but not with phorbol-12-myristate-13-acetate and ionomycin.
Decreased intracellular free basal Mg2+ in patient cells loaded with MagFluo4. 2
Loss of surface expression of the NKG2D receptor on CD8 T lymphocytes and natural killer (NK) cells, as measured by flow cytometry. 2
1.8 Estimated frequency of the disease (incidence at birth (‘birth prevalence') or population prevalence)
If known to be variable between ethnic groups, please report:
Unknown.
Not applicable.
1.9 Diagnostic setting

Comment:
Diagnostic characteristics: As an X-linked disorder, XMEN disease will be mostly found in males, but it may also be found in females as male patients can often survive to reproductive age. Features strongly indicative of XMEN disease include persistent Epstein–Barr virus (EBV) viremia (>10 000 copies in viral load estimations), often with a history of EBV+ lymphoma, and/or a CD4:CD8 T-cell ratio of ~1 or less with a normal lymphocyte count or mild to moderate lymphopenia. Other nonspecific features that may be associated with XMEN disease include history of recurrent sinopulmonary and viral infections, hypogammaglobulinemia (IgM, IgG and/or IgA), variable antibody response to vaccinations (eg tetanus, diphtheria and pertussis) and neutropenia. XMEN disease should be suspected in any male patient with a history of lymphoma in multiple male family members and elevated titers of EBV in circulation (Table 2).
Table 2. Clinical phenotypes of XMEN patients5.

Differential diagnosis: The poor control of EBV in XMEN disease is also found in several other similar genetic disorders, including X-linked lymphoproliferative (XLP) syndrome secondary to signaling lymphocytic activation molecule (SLAM)-associated protein (SAP) deficiency, interleukin-2-inducible tyrosine kinase (ITK) deficiency, X-linked inhibitor of apoptosis (XIAP) deficiency secondary to mutations in BIRC4, CD27 deficiency, coronin 1A deficiency, familial hemophagocytic lymphohistiocytosis secondary to perforin 1 (PRF1) or syntaxin binding protein 2 (STXBP2) deficiency, or the as yet genetically unidentified cause(s) of chronic active EBV (CAEBV) disease.3 XMEN patients usually have a decreased CD4:CD8 ratio, and their absolute CD4+ T-cell count can sometimes fall below 400. Hence the differential diagnosis may also include idiopathic CD4 lymphopenia.
Predictive testing: This does not apply currently as all patients identified thus far with a MAGT1 variant have XMEN disease features.
Risk assessment in relatives: As this is an X-linked recessive genetic disorder, all female offspring of affected males will be carriers, while the carrier status of all other female relatives of the index case will need to be determined by genetic testing. Knowledge of carrier status will guide prenatal diagnostic testing or pre-implantation genetic diagnostic testing for family planning.
2. Test characteristics
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
Insufficient data to comment.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
Insufficient data to comment.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
Unknown, as the disease is clinically similar to many primary immunodeficiency disorders that suffer from chronic EBV infection as listed in the differential diagnosis in 1.9.
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
Unknown, only those with a consistent clinical history are tested so far.
2.5 Positive clinical predictive value
(life-time risk to develop the disease if the test is positive)
Unknown, but all XMEN patients found thus far have a history of chronic EBV viremia. Whether there are any patients with loss-of-function mutations in MAGT1 who do not exhibit any of the features of XMEN disease listed under 1.9 remains unknown.
2.6 Negative clinical predictive value
(probability not to develop the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
Not applicable, as XMEN disease is not uniquely defined clinically. There are several other genetic diseases listed under the differential diagnosis in 1.9 that have clinical features similar to XMEN disease. Thus, there is a possibility for any patient with no MAGT1 mutations to have one of these XMEN-like disorders. XMEN disease is currently defined by loss-of-function mutations in MAGT1.
Index case in that family had not been tested:
Not applicable, see above.
3. Clinical Utility
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.9 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?
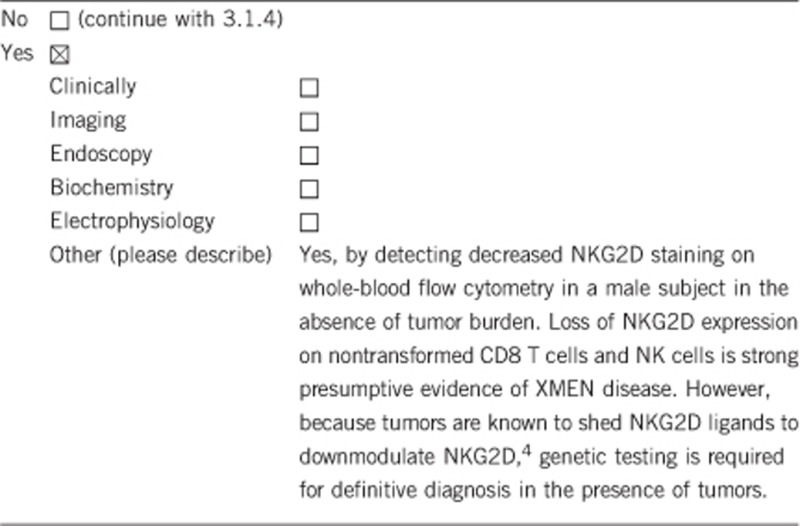
3.1.2 Describe the burden of alternative diagnostic methods to the patient
Assessment of NKG2D expression on flow cytometry requires at least 1 ml ACD whole blood from index males and normal control. This method of diagnosis cannot be used to determine the carrier status in females as they express normal NKG2D.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
This alternative method of diagnosis is more efficient and less labor intensive than the traditional genetic analysis. It can be completed in 1–2 hours as opposed to 1–2 weeks for definitive genetic testing. However, it does require the purchase of specific antibodies and access to a flow cytometer, both of which may outweigh the cost of traditional DNA sequencing.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.9 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe):
Not applicable.
If the test result is negative (please describe):
Not applicable.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
Not applicable.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.9 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Due to the X-linked nature of the disease, all female offspring of the index male with a positive test would be carriers. This may also avoid genetic testing on other clinically affected male family members. However, it does not preclude genetic testing in other potential female carriers in the family who want to know their carrier status.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes, female offspring of a male XMEN patient will be carriers and all male offspring will be unaffected.
3.4 Prenatal diagnosis
(To be answered if in 1.9 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes, female offspring of a male XMEN patient will be obligate carriers. Confirmed female carriers may consider prenatal genetic testing for male fetuses.
4. If applicable, further consequences of testing
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
Not applicable at this stage.
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469), the European Society of Human Genetics, the Division of Intramural Research, NIAID, NIH, through project ZO1AI001187-01, and by co-funding through the Office of Disease Prevention, NIH. We thank Drs Jeffrey Cohen, Steven Holland and the staff of the NIH Clinical Center for their roles in defining the clinical and laboratory profiles of this disease. We also thank the families of XMEN patients for their efforts to assist research on this disease.
The authors declare no conflict of interest.
References
- Li FY, Chaigne-Delalande B, Kanellopoulou C et al. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 2011; 475: 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaigne-Delalande B, Li FY, O'Connor GM et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science 2013; 341: 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvaneh N, Filipovich AH, Borkhardt A: Primary immunodeficiencies predisposed to Epstein-Barr virus-driven haematological diseases. Br J Haematol 2013; 162: 573–586. [DOI] [PubMed] [Google Scholar]
- Kaiser BK, Yim D, Chow IT et al. Disulphide-isomerase-enabled shedding of tumour-associated NKG2D ligands. Nature 2007; 447: 482–486. [DOI] [PubMed] [Google Scholar]
- Li FY, Chaigne-Delalande B, Su H, Uzel G, Matthews H, Lenardo MJ: XMEN disease: a new primary immunodeficiency affecting Mg2+ regulation of immunity against Epstein-Barr virus.(Blood Spotlight). Blood 2014; 123: 2148–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]