

Abstract
Druze individuals rarely marry outside their faith (often practicing consanguinity) and are thus believed to form a genetic isolate. To comprehensively characterize the genetic structure of the Druze population, we recruited and genotyped 40 parent-offspring trios from the Upper Galilee in Israel and the Golan Heights, attempting to capture different extended families (clans) across various geographical locations. Principal component (PC) and ADMIXTURE analyses demonstrated that Druze are close to, yet distinct from, other Middle-Eastern groups (Bedouins and Palestinians), supporting the Druze's Middle-Eastern origin and their recent genetic isolation. Reconstruction of the Druze demographic history using identical-by-descent (IBD) segments suggested an ≈15-fold reduction in population size taking place ≈22–47 generations ago, close to the documented time of the foundation of the Druze faith at the 11th century. Combining the Galilee and Golan Druze genotypes with previously published data on Druze from the Carmel (Israel) and Lebanon demonstrated that all four Druze communities are genetically distinct. The Lebanese group shared less IBD segments (within the group and with other groups) compared with the Israeli Druze and showed higher heterozygosity (suggesting less consanguinity), but was less diverse in PC space. These findings suggest complex recent and ancient demographic history of the Druze population.
INTRODUCTION
Druze individuals constitute a Middle-Eastern minority population. Traditionally, the Druze religion is believed to have formed as an Islamic reform movement, under the rule of the sixth caliph of the Fatimid Dynasty of Egypt, Hakim (AD 966–1020).1, 2 Worldwide, there are ≈1.5 million Druze, residing mainly in Syria (40–50%), Lebanon (30–40%), Israel (8%), Jordan (1–2%), and the United States (1–2%).3 For centuries, Druze have strictly prohibited marriage to non-Druze and limited conversion into the religion. These religious practices, combined with residence in isolated, mountainous regions, have made the Druze population a unique candidate for genetic research. The Druze communities in Israel are clustered in three major regions: the Carmel Mountain, the Upper Galilee, and the Golan Heights. These three Druze communities have, by historical accounts, distinct ancestral geographical origins, yet all these locales originated in the Middle East, primarily in Syria and Lebanon.4 Using family names and verbally transmitted information, these ancestral origins can be attributed to extended, multi-generational pedigrees – clans or Hamullas. The Druze community of the Golan Heights, numbering ≈22 000 individuals, resides in just four villages and is currently the most geographically isolated Druze group. For example, in the last four decades, only 1–2% of marriages were outside the community (Golan for Development Health Survey 1992–1993, unpublished data). Druze traditionally marry within the extended family (most marriages are pre-arranged), with ≈47% of unions estimated as consanguineous.5
Given the high prevalence of consanguineous marriages among Druze, founder mutations in monogenic disorders were expected and, indeed, reported: two recurring ATM gene mutations in Druze individuals residing in communities of Jordan, Lebanon, and Syria;6 a single mutation in the beta globin gene;7 and a unique non-sense mutation in the LDL receptor gene in Druze familial hypercholesterolemia pedigrees.8 Furthermore, analyses in the Israeli Druze population of several clinically relevant sequence alterations showed that allele distribution and variability were limited, and different from those of other Jewish and non-Jewish populations in Israel (eg, Muslims and Bedouins). For example, compared with other Israeli populations, Druze individuals have a unique distribution of allelic variants in the TMPT gene (associated with Azathiprine metabolism),9 a lower frequency of the Gluthatione S Transferase (GSTT1) null mutation,10 and unique allelic variants of the CYP29C gene.11 Moreover, there are multifactorial disorders that are more common in the Druze community than in any other Israeli ethnic group, primarily attributable to genetic factors: for example, Behcet disease was reported at a rate of 50–185:100 000 in the Druze population, compared with the often quoted rate of less than 1:100 000 individuals of other ethnicities.12
A number of previous studies have investigated the genetic structure of the Druze. Shlush et al13, 14 used mitochondrial DNA and Y chromosome markers to find particularly high diversity of uniparental haplogroups in Galilee Druze. Zalloua et al13 and more recently, Haber et al15 studied the genetic structure of Lebanon and showed that Druze are genetically distinct from other Lebanese religious groups (Muslims and Christians). The Human Genome Diversity Project15 as well as Behar et al16, 17 used genome-wide SNP arrays in a large sample of world-wide populations to demonstrate close genetic relations between Druze and other Middle-Eastern populations, such as Bedouins, Palestinians, Syrians, Lebanese, and Jews. Haber et al17 as well as Moorjani and coworkers18 found evidence for an African admixture event, replacing ≈4% of the population about ≈1000 years ago. However, no previous genome-wide study has aimed to adequately represent the various ancestral origins of the Druze population.
In this study, we describe genotyping of 40 Druze trios from the Galilee and the Golan Druze communities, selected to maximize representation of different Druze ancestries. Data analysis confirmed the Middle-Eastern origins of the Druze and their distinction from other regional populations. Taking advantage of trio phasing, we detected, to high resolution, a large number of identical-by-descent (IBD) segments, revealing a recent bottleneck. We then merged our data with previously published genotypes of Druze from the Carmel region in Israel and from Lebanon, and found that all four Druze communities were genetically distinct. Furthermore, the four Druze groups showed different levels of IBD sharing and heterozygosity, suggesting complex demographic history.
MATERIALS AND METHODS
Recruitment and genotyping of Druze participants
Overall, 120 participants (40 trios) were recruited from the Druze communities in Beit Jan (located in the Upper Galilee in Israel; 20 trios) and in the Golan Heights (primarily the village of Majdal Shams; 20 trios). Each trio was selected based on the presumed origin of the extended family (hamulla; based on family names and verbally transmitted, trans-generational information), to try and evenly represent the ancestral geographical origins of the Druze population.4 In all cases, subjects were included only if all four grandparents came from the same community (mostly originating in Syria or Lebanon) and there were no known first- or second-degree relatives among the other participants. Recruitment occurred at the Ziv Medical Center and its affiliate in the Golan for Development Medical Center in Majdal Shams. The study's protocol and informed consent were approved by the local ethics committee and the Israeli Ministry of Health Genetics Ethics Review Board, and each participant signed a written informed consent in Arabic (the Druze native language). Genotyping was performed using the Affymetrix Genome-Wide human SNP Array 6.0 at the genomic facility at Albert Einstein College of Medicine, Bronx, NY. The genotypes are publicly available at the European Genome-phenome Archive (EGA, http://www.ebi.ac.uk/ega/), which is hosted by the EBI, under accession number EGAB00000000809.
Comparison populations
To examine the genetic structure of the Druze population in the context of other populations, we combined the Druze data set generated in the present study (parents only) with an additional Lebanese Druze data set (25 individuals from Haber et al17) and with data from the Human Genome Diversity Project (HGDP15, 19, 20), containing Illumina 650k genotypes of 967 unrelated individuals from 56 world-wide populations, including Druze from the Carmel region in Israel.19, 20 After removing SNPs not existing on all platforms and SNPs with low call rate, a total of 183 381 SNPs were analyzed in 1043 unrelated individuals. To remove unintentionally sampled relatives, we ran Plink21 (after merging) with the – genome command and removed (arbitrarily) one individual of each pair with relatedness coefficient 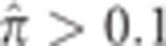 (leaving 51 Druze individuals). We phased the combined data set using SHAPEIT.22
(leaving 51 Druze individuals). We phased the combined data set using SHAPEIT.22
Principal component and ADMIXTURE analyses
Principal Component Analysis (PCA) was performed using JMP Genomics (version 6, Cary, NC, USA). With the exception of an initial run, all analyses were performed without removal of outliers. To improve resolution at the Middle-East region, we ran PCA only on West-Eurasian (and Mozabite) populations (Figure 1). To test for the significance in the differences between population pairs, we performed ANOVA for each subgroup's PC average (Supplementary Table 1).
Figure 1.

Principal component analysis (PCA) of West-Eurasian (and Mozabite) populations from HGDP along with Druze samples from this study and from Lebanon (Haber et al17). The plot places all Druze sub-populations between Middle-Eastern and European populations, but distinct from any other population.
The ADMIXTURE program23 takes as an input a number of hypothetical ancestral populations (K) and provides a maximum likelihood estimate of allele frequencies in each ancestral population and the admixture proportions of each individual. We run ADMIXTURE on all 1043 unrelated individuals, using K between 2 and 7.
IBD detection
The merged Druze-HGDP data set was processed as follows. First, the genotypes were statistically phased using SHAPEIT22 without using the trios (to guarantee a fair comparison between genotypes coming from different platforms). We then assigned genetic map distances using the HapMap2 genetic map24 followed by linear interpolation at the remaining sites. We then ran Germline25 with a window size (bits) of 100, one homozygous (err_hom) and one heterozygous (err_het) error per window, ‘genotype extension' mode (where only double homozygous SNPs are being matched), and a minimal segment length (min_m) of 3 cM. We then filtered segments using HaploScore,26 which ranks segments based on the number of genotype errors and phase switches needed to explain a segment that is truly IBD. HaploScore values were occasionally high (indicating many false positives), and we removed all segments with score >2. We also removed segments with >20% overlap with any of the gaps in the reference genome (UCSC Table Browser27).
After computing the average amount of sharing between all pairs of populations, the significance of differences between different population pairs was evaluated by jackknifing. For each of 104 iterations, we removed a random 10% of all individuals, and recomputed the average level of sharing between all populations. Differences between population pairs were either very significant (P<10−4) or insignificant (P>0.1).
The Galilee and Golan genotypes were then reprocessed as follows. First, we re-phased those genotypes, again using SHAPEIT22 and now using the trio information (followed by removing all children, but without merging with the other data sets). We then ran Germline25 with window size (bits) of 50, one allowed heterozygous mismatch in a window (err_het), no homozygous mismatches (err_hom), ‘haplotype extension' mode (h_extend), and a minimal segment length (min_m) of 0.5 cM. Those parameters seemed reasonable for the trio-phased data,28 in which we expect Germline to be accurate even for very short segments. We then again removed segments with HaploScore >2.26 Indeed, manual inspection showed that most scores were very low (high accuracy). To confidently guarantee that relatives were removed, we eliminated one individual of each pair sharing more than overall 300 cM (which roughly corresponds to individuals who are second cousins) as well as four Galilee individuals who seemed to have been misclassified based on a multidimensional scaling plot. We also removed 14 individuals who contained particularly long runs of homozygosity (>70 MB; roughly corresponding to children of first cousins; detected using Plink's – homozyg command).
Genetic distances
Genetic distances between populations were measured using the pairwise Fst statistic (Table 1), computed using GENEPOP. Fst ranges were calculated by bootstrap re-sampling, with 500 replications (Supplementary Table 2).
Table 1. F st distances between HGDP and Druze populations.

Demographic inference
Our method is based on the approach of Palamara et al29 that matches the decay of IBD sharing at different segment lengths to the theoretical expectation for a given demographic model.
For the trio-phased, Druze-only data, we attempted to infer the parameters of the bottleneck and exponential expansion model shown in Figure 2a. On average, a pair of Golan Druze shared (in segments >0.5 cM) 111 cM, a pair of Galilee Druze shared 179 cM, and a pair of Galilee-Golan individuals shared 81 cM. Despite the somewhat lower rate of sharing between compared to within communities, inter-community sharing was of sufficient magnitude for the two groups to be considered as a single panmictic population for our demographic modeling.
Figure 2.

A demographic model for the recent Druze history. (a) A suggested bottleneck and expansion model. The inferred values of the parameters are given in Table 2 under ‘Druze-only model'. The horizontal arrows correspond to effective population sizes. (b) After trio phasing the Galilee and Golan genotypes, we detected IBD segments of length >0.5 cM and plotted the fraction of the genome shared vs the segment length (after logarithmic binning; circles are at mid-bins). We then identified the demographic model that best fits the observed curve (line).
To create the IBD decay curve (Figure 2b), we binned the lengths of segments into 30 equally (log−) spaced bins between [0.5,15] cM. For each bin, we summed the total length (in cM) of segments shared between all pairs of individuals and divided by the total genome size (3546 cM24) and by the total number of (haplotype) pairs. We then used a grid search to find the demographic model that best fitted the decay curve. Similarity between curves was measured as a sum over all bins of the square of the log of the ratio between the model and observed data points. The theoretical curve was computed essentially as in Eq. (6) in Palamara et al29 with the following (haploid) effective population sizes at each generation g in the past (corresponding to the model in Figure 2a):
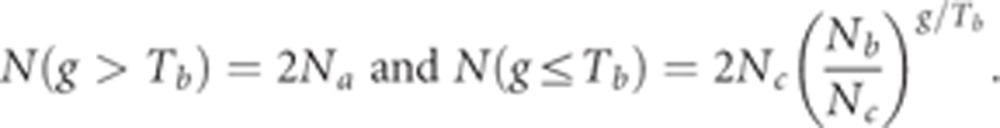 |
In these equations, Na is the ancestral (diploid, effective) population size, Tb is the bottleneck time (in generations), Nb is the bottleneck population size, and Nc is the current population size.
To infer the split time between Druze and Middle-Eastern (non-Druze) populations (Figure 3b), we used the merged Druze and non-Druze data set and a slightly modified theoretical framework. We considered only segments shared between Druze and non-Druze and computed a decay curve as above, but for just two bins: [3,4]cM and >4 cM. The theoretical expectation was computed similarly to Eq. (4) in Palamara et al29 using the fact that the fraction of the genome shared at lengths >mcM is
Figure 3.

Sharing between Druze and non-Druze, Middle-Eastern populations. (a) Identical-by-descent (IBD) segmental sharing between Druze and other Middle-Eastern samples (Palestinians and Bedouins). Each bar corresponds to the average sharing (in cM, over all pairs of individuals) between any pair of populations or within each population. D-GA: Druze-Galilee, D-GO: Druze-Golan, D-C: Druze-Carmel, D-L: Druze-Lebanon, PAL: Palestinians, BED: Bedouins. Using jackknifing, the differences in overall sharing between each of the 12 left-most pairs of populations in the figure were significant at P<10−4, except for PAL:PAL vs D-GO:D-GO, D-GA:D-C vs D-GO:D-C, and D-GA:D-L vs D-L:D-L (for which the differences were not significant). (b) A suggested demographic model for the Druze-Middle-East split. The parameters of the model were inferred using segments shared between any Druze to any non-Druze at two different length bins (see Materials and methods), and are given in Table 2 under ‘Middle-East model'. The horizontal arrow corresponds to an effective population size.
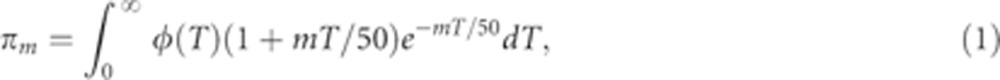 |
where φ(T) is the probability density function (PDF) of the time to the most recent common ancestor (the coalescence time). Equation (1) is the average of the probability of an SNP to lie in a segment longer than mcM, (1+mT/50)e−mT/50,29, 30 over all possible coalescence times, T. According to the model in Figure 3b, a Druze and a non-Druze lineage can coalesce no more recently than Ts generations in the past (their split time), but beyond that, the coalescence times are as in the standard coalescent with population size 2N0 (the ancestral population size). Therefore,31
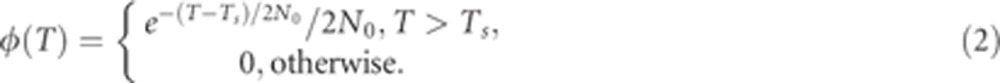 |
Combining Equations 1 and 2, we have
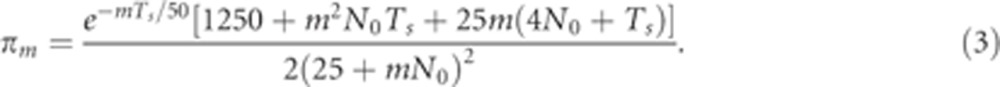 |
The expected fraction of the genome shared in segments of lengths between [3,4]cM is π3–π4, and in segments >4 cM is π4. As these quantities depend on two unknowns, N0 and Ts, matching them to the observed data points yields two equations in two variables, which we solved numerically using Matlab's fsolve. We verified that a grid search converged to the same solution.
In the population split model, we did not consider segments shared within Druze or within the other Middle-Eastern populations; therefore, the results are unaffected by and are not informative on population size fluctuations more recently than the split. The illustration, in Figure 3b, of each population having a constant size history was made for simplicity. The post-split isolation assumption is justified due to the prohibition of conversion in and out of the Druze religion. The assumption that the population had been panmictic before the split is crude but necessary, since the IBD analysis would not have been powered enough to estimate the parameters of a more detailed model. Inferring a unified model for both the Druze-specific recent history and the regional, more ancient history will require more data and more elaborate methods, and is left for future work.
To generate confidence intervals for our estimated parameters, we used jackknifing. For the trio-based, Druze-only model, we ran 100 iterations, where in each iteration we randomly removed five individuals. For the Druze-HGDP model, we ran 1000 iterations, where in each we randomly removed 10% of the individuals (Druze or non-Druze). For each parameter θ, we computed the 95% confidence interval, assuming a normal distribution of the estimated parameter  , as
, as 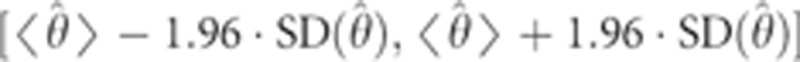 , where
, where 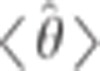 and
and 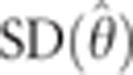 were the mean and standard deviation of
were the mean and standard deviation of  , respectively, over all iterations.
, respectively, over all iterations.
RESULTS
Druze form distinct Middle-Eastern clusters in PC and ADMIXTURE analyses
We genotyped 40 trios of Druze individuals from two sub-populations (Galilee and Golan, in Israel) on the genome-wide Affymetrix 6.0 platform. After strict filtering for hidden relatives (which are abundant in this population), we were left with 51 unrelated individuals. To examine the genetic structure of the Druze in the global and regional contexts, we merged SNP data generated in the present study with genotypes from the Human Genome Diversity Project (HGDP,15, 19, 20 which includes Druze samples from the Carmel region in Israel) and from 25 Druze from Lebanon 17 (see Materials and methods). PCA of West-Eurasian populations confirmed that Druze individuals cluster close to other Middle-Eastern groups (Figure 1). Nevertheless, the Druze cluster (containing four sub-populations: Galilee and Golan (this study), Carmel, and Lebanon) was distinct compared with other Middle-Eastern populations (Bedouins and Palestinians), suggesting that genetic stratification in the region is due to religious, rather than geographic barriers.17 The Druze populations clustered closer to the European samples compared with Palestinians and Bedouins, and closer to Palestinians than to Bedouins, likely due to different levels of African ancestry.18 Subsequent PC analysis of the four Druze groups alone (Figure 4) revealed that all four groups are genetically distinct. Interestingly, Lebanese Druze were tightly clustered in the center of the PC space and were less diverse than the other groups.
Figure 4.

A PCA plot of four Druze sub-populations studied here: Galilee, Golan, Carmel, and Lebanon.
The relative positions of the population clusters assigned by PCA were further confirmed by an ADMIXTURE analysis (Figure 5). The populations with composition most similar to the two Druze groups studied herein (Golan and Galilee) were the other Druze groups (from Lebanon the Carmel region), followed by Palestinians and Bedouins (who had slightly more African ancestry). As expected, Fst distances between the Druze groups and the other HGDP populations were smallest for Middle-Eastern, European, and Central/South-Asians population (Table 1).
Figure 5.

An ADMIXTURE analysis of the Druze and HGDP world-wide populations. The plot demonstrates similarity between Druze and Palestinians and among Druze sub-populations, with the genetic composition being mostly Middle-Eastern with minor European and Central/South-Asia-related components.
IBD sharing within and between the Druze and Middle-Eastern populations
To gain further insight into the interrelation between the Druze and other Middle-Eastern populations, we searched for long IBD segments (>3 cM; using Germline25 and HaploScore;26 see Materials and methods) in the merged Druze-HGDP data, limiting ourselves to the four Druze communities, Palestinians, and Bedouins. The average amount of sharing (in cM) within and between populations is shown in Figure 3a. As expected, IBD sharing within populations was the most abundant (≈10–80 cM per pair), particularly in the Galilee and Carmel communities, with the exception of Lebanese Druze (≈1 cM per pair). Sharing between Druze communities was also abundant, with, again, the least amount of sharing between Lebanese other Druze. Sharing between Druze and non-Druze was much less frequent, yet non-zero (≈0.05 cM per pair).
The lower levels of IBD sharing in Lebanese Druze could be due to the more widespread geographic distribution of those samples or due to less consanguinity. Heterozygosity is indeed higher in Lebanese Druze compared with the Carmel, Galilee, and Golan Druze (31.3, vs 29.7, 30.0, and 30.3%, respectively, P<10−4), supporting lower levels of consanguinity.
Reconstruction of the Druze demographic history using shared segments
Recently, it was demonstrated that the lengths of IBD segments are highly informative on recent demographic history.29, 30, 32, 33, 34 To date the split between the Druze and the non-Druze Middle-Eastern populations, we pooled all Druze populations and all non-Druze populations and considered two data points: the mean fraction of the genome covered by segments shared between Druze and non-Druze at lengths between [3,4]cM and above 4 cM. Assuming no gene flow between Druze and other Middle-Eastern populations since the split between them, we are left with just two parameters to infer (Figure 3b), which we did using a simple theoretical extension of the model of Palamara et al29 (see Materials and methods). The results (Table 2) suggest that the split occurred ≈47–66 generations ago (≈4th–9th centuries CE), at about the time of the formation of Islam.
Table 2. The parameters of our inferred demographic models.
| Parameter | Na | Nb | Nc | Tb |
|---|---|---|---|---|
| Druze-only model (trios) | 41 000 (30 700–50 900) | 3000 (39–6170) | 6100 (0–24 700) | 36 (22–47) |
| Parameter | N0 | Ts | ||
| Middle-East model (unrelateds) | 510 000 (269 000–759 000) | 56 (47–66) |
The ‘Druze-only' model corresponds to Figure 2a, and the ‘Middle-East' model to Figure 3b. Population sizes (Ns) are in diploid effective sizes and times (Ts) are in generations. The top number in each cell is the best fit using the entire data; confidence intervals (95%), obtained using jackknifing, are shown in parentheses (see Materials and Methods).
The large amount of segments shared within and between the Israeli Druze communities suggested a recent bottleneck in their history. To infer the parameters of such a bottleneck, we took advantage of our trio genotypes for the Galilee and Golan communities and re-phased the genomes of these trios, improving the phasing accuracy and the resolution of IBD segment detection. Then, using all segments longer than 0.5 cM, we inferred the parameters of the model shown in Figure 2a, of a sharp population size reduction followed by gradual expansion up to the present size. The model's parameters were inferred by fitting the fraction of the genome shared at each segment length bin29 (Figure 2b), followed by jackknifing (see Materials and methods). The results, presented in Table 2, suggest an ≈15-fold reduction in the Druze population size, occurring ≈22–47 generations ago. The timing of the bottleneck, corresponding to the 9th–15th centuries, is around the documented time of the foundation of the Druze religion at the 11th century. The post-bottleneck increase in the population size was modest (in fact, in some jackknife iterations the population size declined), perhaps due to the organization of the Druze population in clans. Interestingly, the bottleneck is estimated to post-date the split from the non-Druze populations (see the Discussion for possible historical interpretations).
DISCUSSION
We presented the first population genetic study of the Druze to employ careful sample selection from the Galilee and Golan communities, along with genome-wide trio genotyping. Our data confirm previous findings, namely, that Druze are genetically close to other Middle-Eastern ethnic groups (ie, Palestinians and Bedouins), and, to lesser extent, to Europeans and Central/South-Asians. This supports the inclusion of Druze individuals (along with Palestinians and Bedouins) in Middle-Eastern reference panels in previous population genetic studies.35, 36, 37, 38 The data also show, perhaps surprisingly, that Druze communities from different locales are closer to each other than to other Middle-Eastern groups, consistently with the Druze's strict ban on intermarriage. Yet, each of the four communities studied (Galilee, Golan, Carmel, and Lebanon) was genetically distinct, even the more geographically proximate Israeli communities.
Our historical reconstruction using IBD segments suggested a split between Druze and (Middle-Eastern) non-Druze around 47–66 generations ago, and an ≈15-fold reduction in population size around 22–47 generations ago. A plausible historical interpretation is that the ancestral population leading to the Druze has split from other Middle-Eastern groups at about the time of the formation of Islam. According to our results, that split did not involve any dramatic population crash, which happened only ≈500 years later, at or just after the foundation of the Druze religion. We note that care must be taken in interpreting these results and the proposed historical connections due to a number of factors, such as imperfect detection of IBD segments, misspecification (or oversimplification) of the historical models (in particular, the assumption of a panmictic ancestral Middle-Eastern population), the small sample size, and the large degree of relatedness within the sample. On the other hand, we note that our estimate is free from the issues associated with the uncertainty in the human mutation rate.39
Our study did not establish whether the Druze arose as a single founder group (and only then split to communities), or alternatively, whether each community descended from a unique ancestral population (by accepting the Druze religion), followed by homogenization by gene flow. A combination of these two scenarios is also possible, namely, a single ancestral group followed by admixture with local populations. Currently, we cannot rule out either option, since the Druze communities we studied are genetically closer to each other than to other Middle-Eastern populations, but at the same time are genetically distinct. We also did not identify the ancestral founder population (in case such a single population existed) or which of the four communities studied is closest to that founder population. Evidence from this study, in particular with respect to Lebanese Druze, is ambiguous: Lebanese Druze clustered more closely on PCA but shared significantly less IBD segments. These patterns are not necessarily contradicting (ie, they could represent different time scales; see also McVean40 for interpretation of PCA results), but are nevertheless not decidedly informative regarding Druze origins. Since the Druze population was only recently established (≈1000 years ago by historical accounts), we expect that with more data (eg, a finer mapping of Middle-Eastern populations as well as larger sample sizes) and improved modeling, questions of Druze origins will be further clarified. Additionally, sampling of more Druze communities (eg, from Syria) will be necessary to validate the conclusions reached in this study. Finally, due to the consanguineous nature of this population and hence its importance in medical genetics, we believe that the trio-based data set that we generated will be indispensable in mapping and investigating medically-relevant haplotypes.
Acknowledgments
This study was in part sponsored by a grant from the Binational Science Foundation (BSF) to EF and Harry Ostrer and generous donations from private donors to the Golan for Development organization and the Oncology Institute at the Ziv medical center. SC acknowledges financial support from the Human Frontier Science Program and GA NIH (1R01AG042188).
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary Material
References
- Hitti PK: Origins of the Druze People and Religion: with Extracts from Their Sacred Writings, Columbia University Oriental Studies 28 (new ed.). London: Saqi, 2007 [1924], pp 13–14
- Swayd S: The Druzes: An Annotated Bibliography. Kirkland, WA, USA: ISES Publications, 1998. [Google Scholar]
- Cohen N, Czamanski D, Hefetz A: Internal migration of Ethno-national Minorities: the case of Arabs in Israel. Int Migr 2012, e-pub ahead of print 29 November 2012. doi:10.1111/imig.12014
- Hitti PK: The Origins of the Druze People and Religion. Netlancers Inc, 2014 [Google Scholar]
- Vardi-Saliternik R, Friedlander Y, Cohen T: Consanguinity in a population sample of Israeli Muslim Arabs, Christian Arabs and Druze. Ann Hum Biol 2002; 29: 422–431. [DOI] [PubMed] [Google Scholar]
- Fares F, Axelord Ran S, David M et al: Identification of two mutations for ataxia telangiectasia among the Druze community. Prenat Diagn 2004; 24: 358–362. [DOI] [PubMed] [Google Scholar]
- Filon D, Oron V, Krichevski S et al: Diversity of beta-globin mutations in Israeli ethnic groups reflects recent historic events. Am J Hum Genet 1994; 54: 836–843. [PMC free article] [PubMed] [Google Scholar]
- Landsberger D, Meiner V, Reshef A et al: A nonsense mutation in the LDL receptor gene leads to familial hypercholesterolemia in the Druze sect. Am J Hum Genet 1992; 50: 427–433. [PMC free article] [PubMed] [Google Scholar]
- Ronen O, Cohen SB, Rund D: Evaluating frequencies of thiopurine S-methyl transferase (TPMT) variant alleles in Israeli ethnic subpopulations using DNA analysis. Isr Med Assoc J 2010; 12: 721–725. [PubMed] [Google Scholar]
- Karban A, Krivoy N, Elkin H et al: Non-Jewish Israeli IBD patients have significantly higher glutathione S-transferase GSTT1-null frequency. Dig Dis Sci 2011; 56: 2081–2087. [DOI] [PubMed] [Google Scholar]
- Efrati E, Elkin H, Sprecher E, Krivoy N: Distribution of CYP2C9 and VKORC1 risk alleles for warfarin sensitivity and resistance in the Israeli population. Curr Drug Saf 2010; 5: 190–193. [DOI] [PubMed] [Google Scholar]
- Klein P, Weinberger A, Altmann VJ, Halabi S, Fachereldeen S, Krause I: Prevalence of Behcet's disease among adult patients consulting three major clinics in a Druze town in Israel. Clin Rheumatol 2010; 29: 1163–1166. [DOI] [PubMed] [Google Scholar]
- Shlush LI, Behar DM, Yudkovsky G et al: The Druze: a population genetic refugium of the Near East. PLoS One 2008; 3: e2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalloua PA, Xue Y, Khalife J et al: Y-chromosomal diversity in Lebanon is structured by recent historical events. Am J Hum Genet 2008; 82: 873–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JZ, Absher DM, Tang H et al: Worldwide human relationships inferred from genome-wide patterns of variation. Science 2008; 319: 1100–1104. [DOI] [PubMed] [Google Scholar]
- Behar DM, Yunusbayev B, Metspalu M et al: The genome-wide structure of the Jewish people. Nature 2010; 466: 238–242. [DOI] [PubMed] [Google Scholar]
- Haber M, Gauguier D, Youhanna S et al: Genome-wide diversity in the levant reveals recent structuring by culture. PLoS Genet 2013; 9: e1003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorjani P, Patterson N, Hirschhorn JN et al: The history of African gene flow into Southern Europeans, Levantines, and Jews. PLoS Genet 2011; 7: e1001373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann HM, de Toma C, Cazes L et al: A human genome diversity cell line panel. Science 2002; 296: 261–262 [DOI] [PubMed] [Google Scholar]
- Rosenberg NA: Standardized subsets of the HGDP-CEPH Human Genome Diversity Cell Line Panel, accounting for atypical and duplicated samples and pairs of close relatives. Ann Hum Genet 2006; 70: 841–847. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K et al: PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81: 559–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O, Marchini J, Zagury JF: A linear complexity phasing method for thousands of genomes. Nat Methods 2012; 9: 179–181. [DOI] [PubMed] [Google Scholar]
- Alexander DH, Novembre J, Lange K: Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009; 19: 1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium, Frazer KA, Ballinger DG et al: A second generation human haplotype map of over 3.1 million SNPs. Nature 2007; 449: 851–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Lowe JK, Stoffel M et al: Whole population, genome-wide mapping of hidden relatedness. Genome Res 2009; 19: 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand EY, Eriksson N, McLean CY: Reducing pervasive false positive identical-by-descent segments detected by large-scale pedigree analysis. Mol Biol Evol 2014; 31: 2212–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer LR, Zweig AS, Hinrichs AS et al: The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res 2013; 41: D64–D69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Palamara PF, Aponte G et al: The architecture of long-range haplotypes shared within and across populations. Mol Biol Evol 2012; 29: 473–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palamara PF, Lencz T, Darvasi A, Pe'er I: Length distributions of identity by descent reveal fine-scale demographic history. Am J Hum Genet 2012; 91: 809–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmi S, Palamara PF, Vacic V, Lencz T, Darvasi A, Pe'er I: The variance of identity-by-descent sharing in the Wright-Fisher Model. Genetics 2013; 193: 911–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeley J: Coalescent Theory: An Introduction. Greenwood Village, Colorado, USA: Roberts & Company Publishers, 2009. [Google Scholar]
- Browning SR, Browning BL: Identity by descent between distant relatives: detection and applications. Annu Rev Genet 2012; 46: 617–633. [DOI] [PubMed] [Google Scholar]
- Ralph P, Coop G: The geography of recent genetic ancestry across Europe. PLoS Biol 2013; 11: e1001555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atzmon G, Hao L, Pe'er I et al: Abraham's children in the genome era: major Jewish diaspora populations comprise distinct genetic clusters with shared Middle Eastern Ancestry. Am J Hum Genet 2010; 86: 850–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhaes TR, Casey JP, Conroy J et al: HGDP and HapMap analysis by Ancestry Mapper reveals local and global population relationships. PLoS One 2012; 7: e49438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopelman NM, Stone L, Wang C et al: Genomic microsatellites identify shared Jewish ancestry intermediate between Middle Eastern and European populations. BMC Genet 2009; 10: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Li Y, Singleton AB et al: Genotype-imputation accuracy across worldwide human populations. Am J Hum Genet 2009; 84: 235–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodoglugil U, Mahley RW: Turkish population structure and genetic ancestry reveal relatedness among Eurasian populations. Ann Hum Genet 2012; 76: 128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scally A, Durbin R: Revising the human mutation rate: implications for understanding human evolution. Nat Rev Genet 2012; 13: 745–753. [DOI] [PubMed] [Google Scholar]
- McVean G: A genealogical interpretation of principal components analysis. PLoS Genet 2009; 5: e1000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.