

SUMMARY
We report that the protein kinase Cι (PKCι) oncogene controls expression of NOTCH3, a key driver of stemness, in KRAS-mediated lung adenocarcinoma (LADC). PKCι activates NOTCH3 expression by phosphorylating the ELF3 transcription factor and driving ELF3 occupancy on the NOTCH3 promoter. PKCι-ELF3-NOTCH3 signaling controls the tumor-initiating cell (TIC) phenotype by regulating asymmetric cell division, a process necessary for tumor initiation and maintenance. Primary LADC tumors exhibit PKCι-ELF3-NOTCH3 signaling, and combined pharmacologic blockade of PKCι and NOTCH synergistically inhibits tumorigenic behavior in vitro and LADC growth in vivo demonstrating the therapeutic potential of PKCι-ELF3-NOTCH3 signal inhibition to more effectively treat KRAS LADC.
Keywords: Protein Kinase Cι, ELF3, NOTCH Signaling, KRAS-driven lung adenocarcinoma, therapeutic intervention
eTOC Blurb
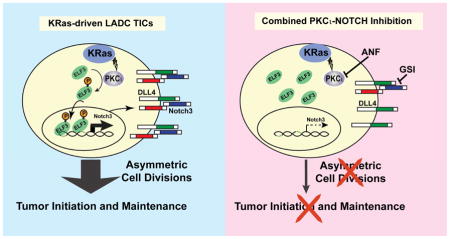
Ali et al. show that in KRAS-mediated lung adenocarcinoma, PKCι controls NOTCH3 expression by phosphorylating ELF3 and driving occupancy at the NOTCH3 promoter. PKCι-ELF3-NOTCH3 signaling controls the TIC phenotype and combined blockade of PKCι and NOTCH has a synergistic anti-tumor effect in vitro and in vivo.
INTRODUCTION
Lung cancer is the major cause of cancer death worldwide (Jemal et al., 2011). LADC is the most prevalent form of lung cancer accounting for ~40% of cases. Major oncogenic drivers of LADC include activating mutations in epidermal growth factor receptor (EGFR), chromosomal translocations that generate oncogenic EML4-ALK fusions, and activating mutations in KRAS. KRAS mutations, the most prevalent oncogenic driver in human LADC, are present in ~30% of cases. Highly selective and potent EGFR and ALK kinase inhibitors are promising targeted therapies for mutant EGFR and EML4-ALK LADCs, respectively (Herbst, 2002; Koivunen et al., 2008; Nakajima et al., 2010; Pao et al., 2004), and represent a new paradigm of individualized therapy to treat cancers harboring specific driver mutations. Despite intensive efforts however, targeted treatment options for KRAS LADC remain elusive. KRAS has proven to be an intractable target, leading to efforts to target critical KRAS effectors that are more amenable to therapeutic intervention (Vasan et al., 2014). There remains a need to better understand the molecular mechanisms that drive KRAS LADC and translate this knowledge into new intervention strategies.
We previously identified PRKCI as an oncogene in non-small cell lung cancer (NSCLC) (Regala et al., 2005b). PKCι is overexpressed in KRAS LADC and PKCι expression predicts poor outcome (Regala et al., 2005b). Genetic silencing of PRKCI inhibits transformed growth and invasion of KRAS LADC cells in vitro, and tumor development in vivo (Regala et al., 2005a; Regala et al., 2005b). Furthermore, lung-specific genetic disruption of Prkci in the LSL-KrasG12D mouse LADC model blocks tumor initiation by inhibiting clonal expansion of putative lung cancer stem cells (Regala et al., 2009). A synthetic lethality screen identified a small molecule oncrasin that selectively inhibits oncogenic KRAS LADC in a PKCι-dependent manner (Guo et al., 2008). These studies establish PRKCI as a critical oncogenic effector of KRAS in LADC.
KRAS LADC tumors consist of a hierarchy of cells of differing tumorigenic potential. Atop this cellular hierarchy are highly malignant TICs exhibiting potent tumor-initiating activity and the ability to recapitulate KRAS LADC in vivo (Hassan et al., 2013; Sullivan et al., 2010; Zheng et al., 2013). Functional characterization of LADC TICs revealed a requirement for NOTCH (Hassan et al., 2013; Sullivan et al., 2010; Zheng et al., 2013). Here, we define a major molecular mechanism by which PRKCI maintains a highly tumorigenic TIC phenotype in KRAS LADC cells in vitro and drives tumorigenesis in vivo. Our results provide molecular insights into KRAS LADC TIC biology and inform a targeted therapeutic approach that may improve treatment outcomes for KRAS LADC patients.
RESULTS
Characterization of human KRAS LADC tumor-initiating cells
TICs function to drive tumor initiation, maintenance and progression (Clarke et al., 2006; Kreso and Dick, 2014). Cell surface markers such as CD133 can mark LADC TICs, however considerable heterogeneity in expression of these markers across LADC cell lines makes them problematic for TIC identification (Hassan et al., 2013). Therefore, we took an unbiased approach to enrich for TICs by culturing three human oncogenic KRAS LADC cell lines (A549, H358 and H23) under low adherence conditions in defined stem cell media (Eramo et al., 2008; Hassan et al., 2013; Justilien et al., 2012). These cells successfully grow as large masses termed oncospheres in non-adherent stem culture (Fig. 1A, middle panels). Oncosphere cells re-differentiate and acquire morphology comparable to parental cells when returned to adherent culture (Fig. 1A, compare upper and lower panels). Oncosphere cells exhibit enhanced anchorage-independent growth (Fig. 1B) and clonal expansion efficiency (>65–80%) when compared to parental or re-differentiated oncosphere cells (Fig. 1C). Quantitative PCR (QPCR) reveals that oncosphere cells express elevated levels of stem-related genes, including BMI, NANOG, OCT3/4, ALDH1A1 and CD133, which are decreased upon re-differentiation (Fig. S1A). Consistent with their stem-like behavior, oncosphere cells exhibit enhanced tumor initiation when injected orthotopically into the lungs of immunodeficient mice (Fig. 1D–F). As few as 100 oncosphere cells result in efficient tumor take (5/5) whereas 1×106 parental cells achieve only partial tumor take (3/5) (Fig. 1D, see also Fig. S1B). Extreme limiting dilution analysis (ELDA) revealed a highly significant (p=1.9×10−49) enrichment in TIC frequency in oncosphere cultures (1/TIC frequency ~42 cells) when compared to parental cells (1/TIC frequency ~1.4×106 cells) (Fig. S1C). 10,000 oncosphere cells generate robust multifocal lung tumors, whereas 10,000 parental cells fail to propagate tumors (Fig. 1E, a–d; Fig. 1F, e and f). Histology reveals that oncosphere-derived and parental cell tumors (from 1 × 106 parental cells) exhibit similar morphology (Fig. 1F, g and h). Serial xenotransplantation demonstrates that oncosphere cells propagate histologically similar tumors through five successive generations (Fig. S1D). Thus, oncosphere cultures are highly enriched in TICs exhibiting cancer stem-like behavior in vitro and in vivo.
Figure 1.

LADC oncospheres exhibit tumor initiating cell properties. (A) Micrographs of parental cells (top, parental), oncospheres in low adherence (middle, oncosph) and re-differentiated oncosphere cells returned to adherent culture (bottom, Re. oncosph). Scale bars 50 μm. (B) Soft agar growth expressed as fold parental ± SEM, n=6, *p≤0.05 compared to parental, **p≤0.05 compared to oncospheres. (C) Single cells were assessed for clonal expansion. Plotted as % expanded +/−SEM; Cells expanded/total cells scored: A549: Parental (11/91), oncosph. (71/97), re. oncosph. (10/87); H358: parental (7/81), oncosph. (75/95), re. oncosph. (9/86); H23: parental (9/81), oncosph. (61/87), re. oncosph. (12/86). *p<0.0001 compared to parental; **p<0.0001 compared to oncospheres using Fisher’s exact test. Scale bars 50 μm. (D) Extreme Limited Dilution Analysis (ELDA) of A549 parental (black) and oncosphere (red) cells for orthotopic tumor engraftment. Results plotted as log fraction not responding (no engraftment) vs. cells inoculated. (E) Bioluminescent images of representative tumor bearing-mice at weeks 1 and 5 post-injection (a–d) and (F) corresponding images of lung tissue upon dissection (e, f). H&E staining reveals similar histology of oncosphere- and parental cell-derived tumors (g, h). Scale bars 50 μm. See also Figure S1.
PKCι is required for LADC oncosphere behavior
QPCR revealed no significant change in PKCι expression, but a consistent increase in the PKCι target gene MMP10 (Frederick et al., 2008) in LADC oncosphere cells (Fig. 2A). Immunoblot analysis demonstrated elevated levels of pT410 PKCι, a marker of PKCι activity (Le Good et al., 1998; Standaert et al., 1999), in oncosphere cells compared to parental cells, consistent with enhanced PKCι activation (Fig. S2A). To assess involvement of PKCι in oncosphere behavior, we used two shRNA lentiviruses targeting PKCι (KD1 and KD2) to achieve stable KD (Fig. 2B; and Fig. S2B and C). PKCι KD significantly decreased oncosphere growth (Fig. 2C and D), clonal expansion efficiency (Fig. 2E) and anchorage-independent growth (Fig. 2F) compared to oncosphere cells expressing non-target (NT) control shRNA. Two independent PKCι KD constructs induced similar phenotypes indicating the observed effects are due to PKCι loss, a conclusion validated by expressing exogenous PKCι in PKCι KD1 A549 oncosphere cells as described previously (Frederick et al., 2008; Justilien and Fields, 2009). Expression of exogenous PKCι (Fig. 2G) significantly restored oncosphere growth (Fig. 2H and I) and anchorage-independent growth (Fig. 2J). Similar results were obtained in H358 and H23 oncosphere cells demonstrating the generalizability of these findings to other KRAS LADC cells (Fig. S2D–G).
Figure 2.

Role of PKCι in the LADC TIC phenotype. (A) QPCR of parental and oncosphere cells for PKCι and MMP10. Results expressed as fold parental +/−SEM. n=3. *p<0.05 compared to parental. (B) PKCι knock down (KD) in A549 oncospheres using two RNAi constructs. Immunoblot (top), and QPCR (bottom). QPCR results expressed as fold parental cells +/−SEM, n=3. *p≤ 0.05 compared to parental. (C) Representative micrographs showing effect of PKCι KD on oncosphere growth. Scale bars 50 μm. (D) Oncosphere size expressed as mean diameter (μm) +/−SEM. Oncospheres assessed: A549: NT (63), KD1 (63), KD2 (58); H358: NT (44), KD1 (40), KD2 (39); H23: NT (41), KD1 (33), KD2 (37). *p≤0.05. (E) Single oncosphere cells were assessed for clonal expansion plotted as % expanded +/−SEM. Cells expanded/total cells: A549: NT (55/63), KD1 (7/63), KD2 (7/58); H358: NT (37/44), KD1 (5/40), KD2 (4/39); H23: NT (35/41), KD1 (4/33), KD2 (4/37). *p≤0.0001 compared to NT by Fisher’s exact test. (F) Soft agar growth expressed as colony number +/−SEM, n=6. *p≤ 0.05 compared to NT. (G) Immunoblot of A549 oncosphere cells stably transduced with NT or PKCι RNAi and stably transfected with either empty vector (EV) or vector encoding PKCι (+PKCι). (H) Representative micrographs of PKCι reconstitution on A549 oncosphere growth. Scale bars 50 μm. (I) Oncosphere size expressed as mean diameter (μm) +/−SEM. Number assessed: NT + EV (39), KD + EV (34), KD + PKCι (35). *p≤ 0.05 compared to NT+EV, **p≤0.05 compared to PKCι KD+EV. (J) Soft agar growth plotted as colony number +/−SEM, n=6. *p≤ 0.05 compared to NT+EV, **p≤0.05 compared to PKCι KD+EV. See also Figure S2.
PKCι drives expression of NOTCH3 in LADC oncospheres
NOTCH3 plays a major role in maintenance of KRAS LADC TICs (Sullivan et al., 2010; Zheng et al., 2013). Interestingly, we observed significantly elevated NOTCH3 expression in LADC oncospheres that was decreased by PKCι KD, suggesting PKCι may regulate NOTCH3 expression (Fig. 3A). PKCι selectively affected NOTCH3 without changing NOTCH1 or NOTCH2 levels (Fig. S3A); NOTCH4 was below the detection limit of QPCR in these cells. PKCι KD had no significant effect on expression of NOTCH ligands JAG1, JAG2, DLL1, DLL3 or DLL4 (Fig. S3B), indicating that PKCι selectively regulates NOTCH3 and not NOTCH ligand expression.
Figure 3.

Role of NOTCH3 in the LADC TIC phenotype. (A) QPCR for NOTCH3. Results expressed as fold parental +/−SEM, n=5. *p≤0.05 compared with parental; **p≤0.05 compared with NT oncospheres. (B) NOTCH3 KD in A549 oncospheres using two RNAi constructs. Immunoblot (top) and QPCR (bottom). QPCR results expressed as fold parental +/−SEM, n=3, *p<0.05. (C) QPCR for NOTCH1 and NOTCH2. Results expressed as fold parental +/−SEM, n=3. *p<0.05. (D) Growth of A549 oncospheres expressed as mean cell number +/−SEM, n=3, *p≤0.05. Scale bars 50 μm. (E) Effect of NOTCH3 KD on A549 oncosphere growth. Scale bars 50 μm. (F) A549 oncosphere size expressed as mean diameter (μm) +/−SEM. Number assessed: NT (40), KD1 (35), KD2 (33); *p≤0.05. (G) Single oncosphere cells were assessed for clonal expansion plotted as % expanded. Cells expanded/total cells scored: NT (35/40), KD1 (5/35), KD2 (4/33); *p<0.0001 by Fisher’s exact test. (H) A549 TIC viability plotted as fold NT +/− SEM, n=3, *p<0.05. (I) Soft agar growth plotted as colony number +/−SEM, n=6 and *p<0.05. (J) Representative micrographs showing NOTCH3 KD and reconstitution of A549 oncosphere growth. Scale bars 50 μm. (K) QPCR for NOTCH3. Results expressed as fold NT +/−SEM, n=5. *p<0.05 compared to NT; **p<0.05 compared to NOTCH3 KD. (L) Oncosphere size expressed as mean diameter (μm) +/−SEM. Number assessed: NT + EV (25), KD + EV (25), KD + PKCι (25). *p≤ 0.05 compared to NT+EV, **p≤0.05 compared to PKCι KD+EV. (M) Single oncosphere cells were assessed for clonal expansion plotted as % expanded +/−SEM. Cells expanded/total cells scored: NT (22/25), NOTCH3 KD (4/25), NOTCH3 KD+NOTCH3 (19/25). *p≤0.0001 compared to NT and **p<0.0001 compared to NOTCH3 KD by Fisher’s exact test. (N) Oncosphere viability (MTT reduction) plotted as %NT +/−SEM, n=3, *p<0.05 compared to NT; **p<0.05 compared to NOTCH3 KD. For (K, L, M and N) open bars, NT; black bars, Notch3 KD; gray bars, Notch3 KD+ Notch 3. See also Figure S3.
To assess NOTCH3 function we knocked down NOTCH3 using two lentiviral shRNAs (Fig. 3B). NOTCH3 KD was specific since no change in NOTCH1 or NOTCH2 was observed (Fig. 3C). NOTCH3 KD decreased oncosphere growth (Fig. 3D–F), clonal expansion (Fig. 3G), cell viability (Fig. 3H), and soft agar growth (Fig. 3I) when compared to NT oncospheres. Similar results were observed in H358 and H23 oncospheres (Fig. S3C–J). The cellular effects of NOTCH3 KD were rescued by expressing exogenous NOTCH3 (Fig. 3K–N), demonstrating these effects are specific to NOTCH3 loss. Knock down of NOTCH ligands revealed that DLL4 KD significantly inhibited oncosphere growth, whereas JAG1, JAG2, DLL1 and DLL3 KD had either no significant effect, or only a modest inhibitory effect (Fig. S3K). Thus, DLL4 may preferentially mediate NOTCH3 signaling in KRAS LADC oncosphere cells.
We previously demonstrated that PKCι maintains a LSCC TIC phenotype (Justilien et al., 2014). Thus, we assessed NOTCH3 levels in oncospheres from four human LSCC cell lines harboring PRKCI gene copy gains (Chago, H1299, H1703 and H520 cells). Interestingly, NOTCH3 was not induced (Fig. S3L), and PKCι KD (Fig. S3M) did not regulate NOTCH3 in LSCC oncospheres (Fig. S3N). Similarly, NOTCH3 was not significantly induced, and PKCι KD did not significantly affect NOTCH3 expression, in oncospheres from two LADC cell lines (H661 and H1437) harboring wild-type KRAS (Fig. S3O). Interestingly, expression of an oncogenic KrasG12V allele led to significant induction of NOTCH3 in H661 and H1437 oncospheres that is inhibited by PKCι KD (Fig. S3P). Thus, PKCι regulates NOTCH3 in oncogenic KRAS oncosphere cells but not in LSCC or LADC oncospheres expressing wild-type KRAS.
PKCι recruits ELF3 to the NOTCH3 promoter
PKCι maintains a LSCC TIC phenotype by regulating the transcriptional activity of the stemness factor SOX2 (Justilien et al., 2014). However, SOX2 is a LSCC-specific stem factor that is not highly expressed in LADC (Tatsumori et al., 2014; Yuan et al., 2010) suggesting that another transcription factor(s) may be targeted by PKCι in KRAS LADC oncospheres. In a meta-analysis, we identified four genes whose expression correlates with PKCι in multiple LADC gene expression datasets (Erdogan et al., 2009). Among these genes was ELF3, an ETS family transcription factor implicated in lung epithelial stem cell maintenance (Oliver et al., 2011). Interestingly, QPCR demonstrates that ELF3 is elevated in LADC oncospheres (Fig 4A), and co-immunoprecipitation indicates that PKCι and ELF3 interact in LADC oncospheres (Fig. S4A). To assess the role of ELF3 in TIC behavior we knocked down ELF3 (Fig. 4B). Similar to PKCι or NOTCH3 KD, ELF3 KD decreased oncosphere growth (Fig. 4C and D), clonal expansion (Fig. 4E), cell viability (Fig. 4F) and soft agar growth (Fig. 4G).
Figure 4.

Effect of PKCι on ELF3 occupancy at the NOTCH3 promoter and NOTCH3 expression. (A) QPCR for ELF3 presented as fold parental +/−SEM, n=3. *p≤ 0.05. (B) Immunoblot (top) and QPCR (bottom) of ELF3 KD. QPCR expressed as fold NT +/−SEM, n=3. *p≤ 0.05 compared to NT. (C) Representative micrographs of ELF3 KD on oncosphere growth Scale bars 50 μm. (D) Oncosphere size expressed as mean diameter (μm) +/−SEM. Oncospheres assessed: A549: NT (37), KD (37); H358: NT (34), KD (34); H23: NT (34), KD (38). *p≤0.05 compared to NT. (E) Single oncosphere cells were assessed for clonal expansion plotted as %expanded. Cells expanded/total cells scored: A549: NT (33/37), KD (5/37); H358: NT (26/34), KD (4/34); H23: NT (26/34), KD (5/38). *p≤0.001 based on Fisher’s exact test. (F) TIC viability plotted as fold NT +/−SEM, n=3, *p≤0.05 compared to NT. (G) Soft agar growth plotted as colony number +/−SEM, n=6. *p<0.05 compared to NT. (H) ChIP analysis of ELF3 occupancy of the NOTCH3 promoter in A549 TICs. Inset depicts the NOTCH3 promoter; ChIP primer-probes used are indicated as A and B. Consensus ELF3 binding sites indicated by vertical slashes. Data presented as % Input +/−SEM, n=3. *p<0.05; data representative of two independent experiments. immunoglobulin G, (IgG). (I) QPCR of NOTCH3 expressed as fold NT +/−SEM, n=3. *p≤ 0.05 compared to NT. (J) NOTCH3 promoter reporter (inset) activity plotted as fold NT +/−SEM, n=5, *p<0.05 compared to NT. (K) NOTCH3 promoter occupancy. Data presented as % Input +/−SEM, n = 3; *p<0.05. (L) QPCR for NOTCH3 expressed as fold NT +/−SEM, n=3. *p<0.05 compared to NT, **p<0.05 compared to PKCι KD. See also Figure S4.
The proximal NOTCH3 promoter contains multiple ELF3 sites 5′ to the transcriptional start site (Fig. 4H, inset). Chromatin immunoprecipitation (ChIP) using two primer/probe sets spanning two clusters of putative ELF3 binding sites (A and B) revealed enhanced ELF3 binding to the NOTCH3 promoter in LADC oncospheres compared to parental cells (Fig 4H). Moreover, ELF3 KD significantly decreased NOTCH3 expression (Fig 4I), without affecting NOTCH1 or NOTCH2 expression (Fig. S4B). Thus, ELF3 occupies the NOTCH3 promoter and selectively regulates NOTCH3 expression in LADC oncospheres.
To assess whether PKCι and ELF3 regulate NOTCH3 promoter activity NT, PKCι KD and ELF3 KD oncospheres were transfected with a ~1 kb NOTCH3 promoter reporter construct (pGL4-NOTCH3-luc, Fig. 4J, inset) and assessed for promoter-dependent luciferase activity (Fig. 4J). NOTCH3 promoter activity was decreased in PKCι and ELF3 KD oncospheres compared to NT oncospheres (Fig. 4J), consistent with the loss of NOTCH3 expression in PKCι KD (Fig. 3A) and ELF3 KD oncospheres (Fig. 4I). Expression of exogenous PKCι in PKCι KD oncospheres restored ELF3 NOTCH3 promoter occupancy (Fig. 4K) and NOTCH3 expression (Fig. 4L), validating PKCι-dependent ELF3 occupancy at the NOTCH3 promoter and NOTCH3 expression.
PKCι phosphorylates ELF3 to drive NOTCH3 expression and the TIC phenotype
Since PKCι and ELF3 interact in LADC oncosphere cells (Fig. S4A), we determined whether PKCι can phosphorylate ELF3. Recombinant PKCι and Flag tagged-ELF3 were combined in a PKCι kinase reaction and bands corresponding to ELF3 (Fig. 5A) subjected to mass spectrometry. Analysis identified a phosphopeptide corresponding to phosphorylation at Serine 68 (pS68) (Fig. 5A). S68 resides within the Pointed (PNT) domain of ELF3 that is implicated in binding to other ETS proteins (Mackereth et al., 2004). To assess the functional significance of S68 phosphorylation, we generated non-phosphorylatable (S68A) and phosphomimetic (S68D) ELF3 mutants (Fig. 5A, lower panel). ELF3 KD oncospheres transfected with ELF3 mutants (Fig. 5B, top panel) were assessed for NOTCH3-luc promoter activity (Fig. 5B, lower panel). ELF3 KD led to decreased NOTCH3-luc promoter activity that was restored by WT and S68D ELF3 but not S68A ELF3 (Fig. 5B). ChIP revealed that both WT and S68D ELF3 occupy the endogenous NOTCH3 promoter and induce NOTCH3 expression whereas S68A ELF3 does not (Fig. 5C and D; Fig. S5A–B). WT and S68D ELF3 specifically induced NOTCH3 but not NOTCH1 or NOTCH2 expression (Fig. S5C). Since S68A ELF3 did not significantly bind and activate the NOTCH3 promoter, we assessed its intracellular localization relative to WT ELF3 and S68D ELF3. Both WT ELF3 and S68D ELF3 localize predominantly to the nucleus, whereas S68A ELF3 exhibited diminished nuclear localization (Fig. 5E and F), suggesting that S68 phosphorylation participates in nuclear import, accumulation, and/or retention of ELF3. PKCι-mediated ELF3 phosphorylation is functionally significant since expression of WT ELF3 or S68D ELF3, but not empty vector or S68A ELF3, significantly restored growth of ELF3 KD oncospheres (Fig. 5G and H). Similar results were obtained in H23 oncospheres (Fig. S5B–D).
Figure 5.

Role of PKCι-mediated ELF3 phosphorylation on NOTCH3 expression and oncosphere growth. (A) Recombinant human ELF3 incubated in the absence or presence of recombinant PKCι. ELF3 was subjected to mass spectrometric analysis. Schematic of ELF3 domain structure indicating the PKCι phosphorylation site at S68, and the S68A and S68D mutants generated for subsequent experiments (inset). (B) ELF3 expression (top) and NOTCH3 promoter reporter activity (bottom) assayed in NT or ELF3 KD cells stably transduced with empty vector (EV), or ELF3 KD cells transduced with WT-ELF3 (WT), S68A-ELF3 (S68A), or S68D-ELF3 (S68D). Activity is plotted as fold NT +/−SEM, n=5, *p<0.05 compared to NT+EV, **p<0.05 compared to ELF3-KD+EV. (C) ChIP analysis of ELF3 occupancy at the NOTCH3 promoter. Data presented as % input +/−SEM, n=3; *p<0.05 compared to EV. Data are representative of two independent experiments. (D) QPCR of NOTCH3 expressed as fold EV +/−SEM, n=3. *p<0.05 compared to EV. (E) Representative micrographs of immunofluorescence localization of exogenous WT-ELF3, S68A-ELF3 and S68D-ELF3 mutants in the nucleus (dotted line) and cytoplasm in ELF3-KD cells. Scale bars 10 μm. (F) Cells (300–350/ELF3 construct) were assessed for intracellular localization of ELF3 (nucleus vs. cytoplasm) as described in Experimental Procedures. (G) Representative micrographs showing the effect of ELF-WT and ELF3 mutants on oncosphere growth. Scale bars 50 μm. (H) Oncosphere size expressed as mean diameter (μm) +/−SEM. Oncospheres assessed: A549: NT+EV (27), KD+EV (29), KD+WT ELF3 (28), KD+S68A ELF3 (30), KD+S68D (29); H358: NT+EV (31), KD+EV (27), KD+WT ELF3 (29), KD+S68A ELF3 (28), KD+S68D ELF3 (30). *p<0.05 compared to NT, **p<0.05 compared to EV. See also Figure S5.
PKCι-ELF3-NOTCH3 signaling regulates asymmetric cell divisions
A key defining characteristic of TICs is the ability to propagate cancer stem-like cells and generate differentiated, transiently-amplifying cells that populate the bulk tumor (Lathia et al., 2011). This feat is accomplished through a balance of symmetric and asymmetric cell divisions (Lathia et al., 2011; Pine et al., 2010). PKCι and NOTCH3 have both been implicated in polarity, cell fate, and maintenance of lung cancer stem-like phenotypes (Justilien et al., 2014; Zheng et al., 2013). Thus, we hypothesized that PKCι, ELF3 and NOTCH3 KD may alter the balance between symmetric and asymmetric cell divisions. CD133 is a cell surface antigen that exhibits asymmetric distribution during asymmetric cell divisions in LADC oncosphere cells (Pine et al., 2010). Immunofluorescence of interphase LADC oncosphere cells revealed polar distribution of cell surface CD133 (Fig. 6A, panels a–c), consistent with previous reports (Lathia et al., 2011; Pine et al., 2010). Mitotic LADC oncosphere cells undergo both symmetric cell divisions to generate two CD133+ daughter cells (Fig. 6A, symmetric, panels d–f), and asymmetric cell divisions to generate one CD133+ and one CD133− daughter cell (Fig. 6A, asymmetric, panels g–i). As expected, PKCι, ELF3 and NOTCH3 KD oncosphere cells exhibit a decreased mitotic index (Fig. 6B) consistent with inhibition of oncosphere growth. Interestingly, NT oncosphere cells undergo approximately equal numbers of symmetric and asymmetric cell divisions whereas PKCι KD, ELF3 KD and NOTCH3 KD oncosphere cells exhibit a significant and selective decrease in asymmetric cell divisions compared to NT cells (Fig. 6C), indicating that PKCι-ELF3-NOTCH3 signaling regulates cell fate by driving asymmetric cell divisions.
Figure 6.

Effect of the PKCι-ELF3-NOTCH3 axis on asymmetric cell division and tumor initiation. (A) Immunofluorescence of A549 oncosphere cells for CD133 (red) and DAPI (blue) during interphase (a–c) and mitosis (d–i) demonstrating symmetric (d–f) and asymmetric (g–i) cell divisions. Scale bars 10 μm. (B) PKCι, ELF3 and NOTCH3 KD inhibit mitotic index in LADC oncosphere cells. Data expressed as mitotic index (% mitotic cells) +/−SEM. n=3. *p<0.05 compared to NT. (C) Effect of PKCι, ELF3 and NOTCH3 KD on asymmetric cell division. Results expressed as % asymmetric cell divisions (>300 mitoses evaluated/cell line). *p≤0.0001 compared to NT using Fisher exact test. (D) Bioluminescent images of representative tumor bearing-mice 5 weeks after injection of 50,000 NT, PKCι KD, ELF3 KD or NOTCH3 KD A549 oncosphere cells. (E) Tumor size expressed as bioluminescence flux +/−SEM, n=10. *p<0.05 compared to NT control. (F) Representative bioluminescence images of lung tumors ex vivo. (G) Kaplan-Meier survival analysis of mice injected with the indicated A549 oncosphere cells (50,000 cells). N=10. *p<0.05 compared to NT. (H) QPCR for ELF3, NOTCH3 and CD133. Results are expressed as fold parental +/−SEM, n=10, *p<0.05 compared to NT parental, **p<0.05 compared to injected NT oncosphere cells.
Asymmetric cell divisions are necessary for LADC TICs to propagate a tumor (Pine et al., 2010) suggesting that loss of PKCι-ELF3-NOTCH3 signaling may impair tumor initiation in vivo. To test this hypothesis, we injected 50,000 NT, PKCι KD, ELF3 KD and NOTCH3 KD oncosphere cells expressing firefly luciferase orthotopically into the lungs of immunodeficient mice. 50,000 cells are sufficient for oncosphere, but not parental, cultures to engraft (Fig. 1D, Fig. S1A), ensuring that we are monitoring TIC behavior and allowing us to assess effects of PKCι, ELF3 and NOTCH3 KD on TIC engraftment. NT oncosphere cells produce large tumors whereas PKCι KD, ELF3 KD and NOTCH3 KD oncosphere cells fail to generate tumors (no or only weak residual bioluminescence at the injection site) (Fig. 6D). PKCι, ELF3 and NOTCH3 KD oncosphere cells exhibited a >90% inhibition in tumor size compared to NT oncosphere cells (Fig. 6E). Ex vivo imaging at sacrifice confirmed the presence of large, locally-invasive multi-focal tumors in NT oncosphere cell-injected mice, and a lack of tumors in PKCι, ELF3 and NOTCH3 KD mice (Fig. 6F). Kaplan-Meier analysis revealed a statistically significant increase in survival (>80% survival rate) in PKCι, ELF3 and NOTCH3 KD oncosphere cell-injected mice compared to NT mice (22% survival) six weeks after injection (Fig. 6G). QPCR revealed that NT oncosphere-derived tumor cells express ELF3, NOTCH3 and CD133 levels comparable to parental cells (Fig. 6H) indicating that injected NT oncosphere cells differentiate in vivo. Blockade of PKCι-ELF3-NOTCH3 signaling inhibits tumor growth in vitro and in vivo
The anti-rheumatoid gold salts aurothiomalate (ATM) and auranofin (ANF) are selective PKCι inhibitors that block NSCLC growth in vitro and in vivo (Erdogan et al., 2006; Stallings-Mann et al., 2006). Thus, we assessed whether PKCι-ELF3-NOTCH3 signaling can be pharmacologically targeted with ANF. Treatment of oncospheres with ANF inhibited NOTCH3 expression (Fig. S6A). The inhibitory effect of ANF was dependent upon ELF3 since ELF3 KD inhibited NOTCH3 expression and abolished ANF-mediated inhibition of NOTCH3 (Fig. S6A). Re-expression of wild-type ELF3 reconstituted NOTCH3 expression and response to ANF, whereas S68A ELF3 did not (Fig. S6A). However, re-expression of S68D ELF3 restored NOTCH3 expression and conferred resistance to ANF-mediated inhibition of NOTCH3 (Fig. S6A), indicating that ANF selectively inhibits PKCι-ELF3-NOTCH3 signaling in LADC oncospheres. We next assessed the therapeutic potential of ANF alone and in combination with γ-secretase inhibitor (GSI). GSIs are effective inhibitors of NOTCH signaling that exhibit antitumor effects in NSCLC (Fan et al., 2010; Konishi et al., 2007; Sullivan et al., 2010). Combination index analysis (Chou and Talalay, 1984) revealed that GSI and ANF exhibit synergistic activity against oncosphere growth in vitro (Fig. 7A). Furthermore, both GSI and ANF inhibit oncosphere growth (Fig. 7B–C), cell viability (Fig. 7D) and soft agar growth (Fig. 7E). In each case, combined GSI and ANF treatment led to enhanced inhibition compared to either drug alone (Fig. 7C–E). Furthermore, ANF and GSI treatment led to a selective loss of asymmetric cell divisions in LADC oncosphere cells which was reversed upon withdrawal of the drugs (Fig. S6B and C).
Figure 7.

Effect of Auranofin (ANF) and γ-secretase inhibitor (GSI) on oncosphere formation and tumor growth. (A) Combination index analysis of ANF and GSI. (B) Representative micrographs of ANF and GSI effects on A549 and H358 oncosphere growth. Scale bars 50 μm. (C) Effect of ANF, GSI and the combination on oncosphere size expressed as mean diameter (μm) +/−SEM. Oncospheres assessed: A549: DMSO (54), GSI (45) ANF (49), GSI/ANF (55); H358: DMSO (57), GSI (49), ANF (51), GSI/ANF (55). *p<0.05. (D) Cell viability expressed as MTT reduction. n=3. *p<0.05. (E) Soft agar growth expressed as colonies +/−SEM, n=6. *p<0.05 compared to DMSO. **p<0.05 compared to ANF or GSI alone. (F) Tumor volume plotted +/−SEM. N=20. *p<0.05 compared to DMSO; **p<0.05 compared to ANF or GSI alone. (G) Tumor burden assessed at time of sacrifice (30 days). Results expressed as mean +/−95% confidence intervals. n=10. *p<0.05 compared to DMSO. **p<0.05 compared to ANF or GSI alone. (H) Association analysis for expression of the indicated genes in primary LADC tumors (see Experimental Procedures). See also Figure S6.
We next determined the effect of these compounds on the growth of LADC oncosphere cell-derived tumors in vivo. Established A549 oncosphere cell-derived subcutaneous tumors were treated with ANF, GSI, ANF+GSI or diluent. Both GSI and ANF significantly inhibited tumor growth compared to diluent, and combined GSI+ANF treatment led to a significantly larger inhibitory effect than either compound alone (Fig. 7F and G).
We next assessed whether the PKCι-ELF3-NOTCH3 signaling axis is operative in primary LADC tumors. We first interrogated a TCGA LADC tumor dataset for associations between expression of KRAS, pathway components PRKCI and NOTCH3, and the major downstream NOTCH gene target HES1. Analysis revealed a positive correlation of KRAS with PRKCI, NOTCH3, and HES1 (Fig. 7H). Furthermore, significant correlations of PRKCI with NOTCH3 and HES1, and of NOTCH3 with HES1 were also observed (Fig. 7H). We next validated these associations in an independent primary LADC dataset (Kalari et al., 2012). Significant correlations were again observed between KRAS and PRKCI, NOTCH3 and HES1, between PRKCI and NOTCH3 and HES1, and between NOTCH3 and HES1 (Fig. S6D). The correlations of KRAS and PRKCI with NOTCH3 were specific since no associations were observed between KRAS or PRKCI with NOTCH1, NOTCH2 or NOTCH4 (Fig. S6E). Furthermore, analysis of a TCGA LSCC dataset revealed no significant correlations between these pathway components (Fig. S6F). These data provide compelling evidence for active PKCι-ELF3-NOTCH3 signaling in primary human KRAS LADC tumors but not LSCC tumors.
DISCUSSION
Despite introduction of new targeted therapies into clinical practice, median survival for NSCLC patients remains <18 months. One reason for the recalcitrance of NSCLC tumors to therapeutic intervention is the existence of highly malignant, chemoresistant stem-like cells, alternatively termed tumor initiating cells (TICs), tumor propagating cells (TPCs) or cancer stem cells (CSCs) that survive therapy and drive relapse. We recently showed that PKCι is required for Kras LADC tumorigenesis in the mouse (Regala et al., 2009), suggesting a role for PKCι in KRAS LADC TICs. Here, we identify and molecularly characterize a PKCι-ELF3-NOTCH3 signaling axis through which PKCι drives a KRAS LADC TIC phenotype. Our results are consistent with recent reports implicating NOTCH3 in KRAS LADC TIC behaviors (Sullivan et al., 2010; Zheng et al., 2013), and provide significant mechanistic insight into NOTCH3 regulation in KRAS LADC TICs. Moreover, our data provide molecular insight into how PKCι and NOTCH3 drive the tumorigenic behavior of KRAS LADC cells. Specifically, we find that PKCι-mediated ELF3 phosphorylation regulates NOTCH3 by driving ELF3 occupancy on the NOTCH3 promoter. We recently demonstrated that PKCι also maintains a TIC phenotype in human LSCC harboring PRKCI gene amplification (Justilien et al., 2014). Thus, PKCι is a critical oncogenic kinase in at least two major forms of lung cancer.
Although PKCι drives a TIC phenotype in both LSCC and LADC, it does so through distinct, lineage-specific mechanisms. In KRAS LADC, PKCι drives PKCι-ELF3-NOTCH3 signaling whereas in LSCC PKCι activates Hedgehog (Hh) signaling by inducing SOX2-dependent expression of HHAT, the rate limiting step in Hh ligand production (Justilien et al., 2014). Interestingly, both biochemical and bioinformatics studies indicate that these two PKCι-dependent pathways are lineage-restricted and relevant to primary KRAS LADC and LSCC, respectively. Thus, PKCι emerges as a master regulator of LSCC and KRAS LADC TIC phenotypes through control of distinct, lineage-restricted cell fate pathways. PKCι was recently implicated in maintenance of normal pluripotent stem cells through a NOTCH1-dependent pathway (Mah et al., 2015) suggesting that PKCι may be even more widely involved in stem cell maintenance in different contexts, including normal development.
Though distinct PKCι signaling mechanisms drive KRAS LADC and LSCC tumors respectively, some commonalities emerge. First, in both KRAS LADC and LSCC, PKCι establishes and maintains a highly malignant stem-like phenotype that drives tumor initiation and maintenance in vivo. Second, PKCι drives TIC behavior by regulating the transcriptional activity of key, stem-related transcription factors through direct phosphorylation; ELF3 in KRAS LADC and SOX2 in LSCC (Justilien et al., 2014). Interestingly, PKCι can phosphorylate and activate GLI1 in basal cell carcinoma cells (Atwood et al., 2013) indicating that PKCι may act as a key transcriptional regulator of stemness in many cancer types.
A hallmark of TICs is their capacity to self-renew and give rise to differentiated bulk tumor cells through symmetric and asymmetric cell divisions (Morrison and Kimble, 2006). In lower eukaryotes, disruption of the balance between symmetric and symmetric cell divisions can drive tumor development (Morrison and Kimble, 2006; Neumuller and Knoblich, 2009). Whereas symmetric cell divisions of stem-like cells can drive tumor formation in some model systems (Neumuller and Knoblich, 2009), recent evidence reveals that asymmetric cell divisions are critical for KRAS LADC TICs to initiate and propagate tumors in vivo (Pine et al., 2010). However, the specific mechanism(s) controlling these critical cell fate decisions is still poorly understood. Here we show that PKCι-ELF3-NOTCH3 signaling is responsible for controlling asymmetric cell division and tumor-initiating activity of KRAS LADC oncosphere cells. Our results are consistent with the well-established role of atypical PKCs in cell polarity (Lin et al., 2000; Ohno, 2001) and cell fate (Vorhagen and Niessen, 2014). We now implicate PKCι in cell fate decisions in human LADC cells, and directly and mechanistically link PKCι-mediated control of cell polarity, cell fate and oncogenesis in human cancer.
Asymmetrically dividing LADC oncosphere cells can be identified using CD133, whose asymmetric segregation during mitosis has been implicated in both LADC (Pine et al., 2010) and glioma cell fate decisions (Lathia et al., 2011). Though the exact function of CD133 in TIC biology is unclear, CD133 localization to cholesterol-based lipid micro-domains in the apical membrane (Roper et al., 2000; Weigmann et al., 1997) and asymmetric segregation during TIC differentiation (Pine et al., 2010) suggest the intriguing possibility that CD133 is functionally linked to the PKCι-ELF3-NOTCH3 cell fate pathway. Further studies will be required to assess if a direct functional link exists between PKCι and CD133 in determining cell fate.
TICs have emerged as critical therapeutic targets for cancer therapy since they often exhibit intrinsic drug resistance and may mediate therapeutic failure. NOTCH signaling participates in TIC fate in gliomas (Fan et al., 2006) and lung cancer (Sullivan et al., 2010; Zheng et al., 2013). GSI inhibitors of NOTCH exhibit potent anti-tumor activity in glioma (Fan et al., 2010; Fan et al., 2006), lung (Konishi et al., 2007), and ovarian cancers (Groeneweg et al., 2014a; Groeneweg et al., 2014b) and the potent GSI PF-03084014 has shown clinical promise in advanced cancer patients (Messersmith et al., 2015). The anti-rheumatoid gold salts ATM and ANF selectively inhibit PKCι signaling and block growth of lung (Erdogan et al., 2006; Fields et al., 2007; Regala et al., 2008; Stallings-Mann et al., 2006), pancreatic (Butler et al., 2015; Scotti et al., 2012) and ovarian (Wang et al., 2013) tumors in vitro and in vivo, and two clinical studies have demonstrated proof-of-principle for PKCι inhibitor-based therapy with ATM (Mansfield et al., 2013) and ANF (Jatoi et al., 2014). Our finding that GSI and ANF exhibit highly synergistic anti-tumor activity suggests a novel therapeutic approach to treat KRAS LADC, a tumor sub-type for which there is a dire need for effective targeted therapeutics. Our findings demonstrate the efficacy of “vertical blockade” of a key signaling pathway to achieve a desired therapeutic effect; a concept perhaps best documented by combined BRAF and MEK inhibition for treatment of NRAS and BRAF-mutant melanomas (Flaherty et al., 2012; Kwong et al., 2012). The NOTCH ligand DLL4, which may mediate NOTCH3 action, represents another attractive therapeutic target in KRAS LADC. In this regard, the humanized DLL4 antibody, Enoticumab, has shown preliminary clinical activity in NSCLC and other solid malignancies (Chiorean et al., 2015). We recently demonstrated that combined PKCι and Hh inhibition exhibits synergistic growth inhibitory effects in LSCC TICs (Justilien and Fields, 2015), consistent with the role of PKCι-Hh signaling in LSCC TIC behavior (Justilien et al., 2014). Thus, combined PKCι and Hh inhibition represents a promising therapeutic approach for LSCC tumors harboring PRKCI amplification. Our current findings suggest an individualized therapeutic approach to treating both KRAS LADC and LSCC in which ANF is strategically combined with a second agent that provides vertical blockade of the lineage-restricted oncogenic PKCι signaling driving that tumor type. Clinical trials are currently being designed to evaluate the clinical utility of this personalized approach for both PRKCI-amplified LSCC and KRAS LADC.
Experimental Procedures
Antibodies and Plasmids. Antibodies
αTubulin, GAPDH and Phospho-T410-PKCι (Cell Signaling), PKCλ/ι (Santa Cruz Biotechnology), ELF3 and NOTCH3 (Abcam), PKCι (BD Pharmingen), CD133/1 (Miltenyi Biotech). The human PKCι cDNA plasmid was described previously (Justilien et al., 2014); human NOTCH3 plasmid (Myc-NOTCH3/cCMV6; cat.# RC224711) was from OriGene Technologies. PKCι and NOTCH3 KD reconstitutions were performed as described previously (Justilien et al., 2014).
Cell lines, enrichment of tumor-initiating cells, anchorage-independent growth and clonal expansion
Human lung adenocarcinoma (LADC) and squamous carcinoma (LSCC) cell lines (A549, H358, H23, H661, H1437, H1703, H1299, H520 and ChagoK1) were obtained from American Type Culture Collection. Cell lines and oncospheres were cultured, and assayed for anchorage-independent growth, clonal expansion and redifferentiation as described previously (Justilien et al., 2014). Details provided in Supplemental Experimental Procedures.
Quantitative real-time polymerase chain reaction (QPCR)
QPCR was performed using a ViiA7 thermal cycler and associated reagents (Applied Biosystems) or custom designed reagents (Invitrogen) (primers/probes listed in Supplemental Experimental Procedures). Efficiency of target protein knock down was monitored by immunoblot as described previously (Justilien et al., 2014). Experimental details in Supplemental Experimental Procedures.
In vivo tumor formation
Lung orthotropic injections and tumor assessments of A549 cells expressing firefly luciferase were performed as described previously (Justilien et al., 2014). All animal procedures were approved by the Mayo Clinic Institutional Animal Care and Use Committee. Details provided in Supplemental Experimental Procedures.
Lentiviral RNAi constructs and transfections
Lentiviral vectors containing short hairpin RNAi against human PKCι, NOTCH3 and ELF3 (Sigma-Aldrich) were packaged into recombinant lentivirus as described previously (Frederick et al., 2008). Lentiviral target sequences are listed in Supplemental Experimental Procedures. A non-target (NT) control vector that does not recognize any mouse or human genes was used as a negative control. Details provided in Supplemental Experimental Procedures.
Chromatin Immunoprecipitation
CHiP assays were performed to assess ELF3 occupancy of the NOTCH3 promoter as described in Supplemental Experimental Procedures.
NOTCH3 promoter Luciferase assays
A ~1kB fragment of the NOTCH3 promoter was PCR cloned into pGL4.14 [luc2/Hygro] (PromegaI) using primers listed in Supplemental Experimental Procedures. Luciferase assays were performed as described in Supplemental Experimental Procedures.
In vitro PKCι kinase assays, MS analysis and ELF3 mutants
PKCι kinase assays were performed using recombinant ELF3 and PKCι as described previously (Justilien et al., 2011) and phosphorylation site analysis performed by the Mayo Clinic Cancer Center Protein Chemistry and Proteomics Shared Resource as described previously (Justilien et al., 2011; Justilien et al., 2014). ELF3 S68A and ELF3 S68D mutants were generated by site-directed mutagenesis using pIRES-puro-ELF3 (Addgene) as template as described previously (Justilien et al., 2014). Resulting plasmids were sequenced to ensure sequence fidelity. Primers and details provided in Supplemental Experimental Procedures.
Analysis of Gene Expression Data
TCGA gene expression datasets for LADC and LSCC were analyzed using cBioPortal for Cancer Genomics (http://www.cbioportal.org/public-portal/) software (Cerami et al., 2012; Gao et al., 2013). Associations between expression of KRAS, PRKCI, NOTCH3, ELF3 and HES1 mRNAs were determined using cBioPortal software, mRNA expression z-scores (microarray) and a z-score threshold of +/− 1.0. Validation was performed on an independent dataset of mutant KRAS LADC described previously (Kalari et al., 2012) using Kendall Tau rank order analysis.
Supplementary Material
HIGHLIGHTS.
PKCι maintains a tumor initiating cell (TIC) phenotype in KRAS LADC
PKCι phosphorylates and recruits ELF3 to the NOTCH3 promoter
PKCι-ELF3-NOTCH3 signaling drives LADC tumor formation
Combined PKCι and NOTCH blockade synergistically inhibits KRAS LADC growth in vivo
SIGNIFICANCE.
Lung cancer is the leading cause of cancer deaths worldwide. Activating mutations in KRAS are the oncogenic driver in ~30% of cases of LADC, the most prevalent form of lung cancer. KRAS LADC is characterized by poor therapeutic response and a high relapse rate, underscoring the need for new therapeutic options. Here we define a PKCι-ELF3-NOTCH3 signaling axis that drives a TIC phenotype in vitro and KRAS LADC tumorigenesis in vivo. Combined pharmacologic blockade of PKCι and NOTCH produces synergistic anti-tumor activity against KRAS LADC in vitro and in vivo. Our findings support the use of combined PKCι and NOTCH inhibition in the treatment of KRAS LADC.
Acknowledgments
We thank Drs. R. Bergen and B. Madden, Mayo Clinic Proteomics Research Center, for MS analysis of ELF3 phosphorylation, Dr. E.A. Thompson for gene expression analysis, Ms. C. Weems for technical assistance, and Ms. B. Edenfield for tumor tissue processing. This work was supported by grants from the National Institutes of Health (R01 CA081436-18 and R21 CA151250-02 to APF; R01 CA14090-05 to NRM); the James and Esther King Biomedical Research Program (1KG-05-33971) and the Mayo Clinic Center for Individualized Medicine to APF; and a National Institutes of Health Research Supplement to Promote Diversity in Health-related Research Award from the National Cancer Institute to VJ. APF is the Monica Flynn Jacoby Professor of Cancer Research. SAA is the recipient of the Edward C. Kendall Fellowship in Biochemistry from the Mayo Clinic Graduate School.
Footnotes
Author Contributions
SAA performed most experiments with technical assistance, input and advice from LJ and VJ. The experiments were conceived by SAA, NRM and APF, and the work was supervised by NRM and APF. The manuscript was written by SAA, NRM and APF with input from LJ and VJ.
The authors declare that they have no conflicts of interest to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atwood SX, Li M, Lee A, Tang JY, Oro AE. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature. 2013;494:484–488. doi: 10.1038/nature11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AM, Scotti Buzhardt ML, Erdogan E, Li S, Inman KS, Fields AP, Murray NR. A small molecule inhibitor of atypical protein kinase C signaling inhibits pancreatic cancer cell transformed growth and invasion. Oncotarget. 2015;6:15297–15310. doi: 10.18632/oncotarget.3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiorean EG, LoRusso P, Strother RM, Diamond JR, Younger A, Messersmith WA, Adriaens L, Liu L, Kao RJ, DiCioccio AT, et al. A Phase I First-in-Human Study of Enoticumab (REGN421), a Fully Human Delta-like Ligand 4 (Dll4) Monoclonal Antibody in Patients with Advanced Solid Tumors. Clin Cancer Res. 2015;21:2695–2703. doi: 10.1158/1078-0432.CCR-14-2797. [DOI] [PubMed] [Google Scholar]
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells--perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, Conticello C, Ruco L, Peschle C, De Maria R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–514. doi: 10.1038/sj.cdd.4402283. [DOI] [PubMed] [Google Scholar]
- Erdogan E, Klee EW, Thompson EA, Fields AP. Meta-analysis of oncogenic protein kinase Ciota signaling in lung adenocarcinoma. Clin Cancer Res. 2009;15:1527–1533. doi: 10.1158/1078-0432.CCR-08-2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdogan E, Lamark T, Stallings-Mann M, Lee J, Pellecchia M, Thompson EA, Johansen T, Fields AP. Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Ciota. J Biol Chem. 2006;281:28450–28459. doi: 10.1074/jbc.M606054200. [DOI] [PubMed] [Google Scholar]
- Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J, et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells. 2010;28:5–16. doi: 10.1002/stem.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li YM, Eberhart CG. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66:7445–7452. doi: 10.1158/0008-5472.CAN-06-0858. [DOI] [PubMed] [Google Scholar]
- Fields AP, Frederick LA, Regala RP. Targeting the oncogenic protein kinase Ciota signalling pathway for the treatment of cancer. Biochem Soc Trans. 2007;35:996–1000. doi: 10.1042/BST0350996. [DOI] [PubMed] [Google Scholar]
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick LA, Matthews JA, Jamieson L, Justilien V, Thompson EA, Radisky DC, Fields AP. Matrix metalloproteinase-10 is a critical effector of protein kinase Ciota-Par6alpha-mediated lung cancer. Oncogene. 2008;27:4841–4853. doi: 10.1038/onc.2008.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groeneweg JW, DiGloria CM, Yuan J, Richardson WS, Growdon WB, Sathyanarayanan S, Foster R, Rueda BR. Inhibition of notch signaling in combination with Paclitaxel reduces platinum-resistant ovarian tumor growth. Front Oncol. 2014a;4:171. doi: 10.3389/fonc.2014.00171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groeneweg JW, Foster R, Growdon WB, Verheijen R, Rueda BR. Notch signaling in serous ovarian cancer. J Ovarian Res. 2014b;7:95. doi: 10.1186/s13048-014-0095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Wu S, Liu J, Fang B. Identification of a small molecule with synthetic lethality for K-ras and protein kinase C iota. Cancer Res. 2008;68:7403–7408. doi: 10.1158/0008-5472.CAN-08-1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan KA, Wang L, Korkaya H, Chen G, Maillard I, Beer DG, Kalemkerian GP, Wicha MS. Notch pathway activity identifies cells with cancer stem cell-like properties and correlates with worse survival in lung adenocarcinoma. Clin Cancer Res. 2013;19:1972–1980. doi: 10.1158/1078-0432.CCR-12-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS. Targeted therapy using novel agents in the treatment of non-small-cell lung cancer. Clin Lung Cancer. 2002;3(Suppl 1):S30–38. doi: 10.3816/clc.2002.s.006. [DOI] [PubMed] [Google Scholar]
- Jatoi A, Radecki Breitkopf C, Foster NR, Block MS, Grudem M, Wahner Hendrickson A, Carlson RE, Barrette B, Karlin N, Fields AP. A Mixed-Methods Feasibility Trial of Protein Kinase C Iota Inhibition with Auranofin in Asymptomatic Ovarian Cancer Patients. Oncology. 2014;88:208–213. doi: 10.1159/000369257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- Justilien V, Fields AP. Ect2 links the PKCiota-Par6alpha complex to Rac1 activation and cellular transformation. Oncogene. 2009;28:3597–3607. doi: 10.1038/onc.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justilien V, Fields AP. Molecular pathways: novel approaches for improved therapeutic targeting of hedgehog signaling in cancer stem cells. Clin Cancer Res. 2015;21:505–513. doi: 10.1158/1078-0432.CCR-14-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justilien V, Jameison L, Der CJ, Rossman KL, Fields AP. Oncogenic activity of Ect2 is regulated through protein kinase C iota-mediated phosphorylation. J Biol Chem. 2011;286:8149–8157. doi: 10.1074/jbc.M110.196113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justilien V, Regala RP, Tseng IC, Walsh MP, Batra J, Radisky ES, Murray NR, Fields AP. Matrix metalloproteinase-10 is required for lung cancer stem cell maintenance, tumor initiation and metastatic potential. PLoS One. 2012;7:e35040. doi: 10.1371/journal.pone.0035040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justilien V, Walsh MP, Ali SA, Thompson EA, Murray NR, Fields AP. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell. 2014;25:139–151. doi: 10.1016/j.ccr.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalari KR, Rossell D, Necela BM, Asmann YW, Nair A, Baheti S, Kachergus JM, Younkin CS, Baker T, Carr JM, et al. Deep Sequence Analysis of Non-Small Cell Lung Cancer: Integrated Analysis of Gene Expression, Alternative Splicing, and Single Nucleotide Variations in Lung Adenocarcinomas with and without Oncogenic KRAS Mutations. Front Oncol. 2012;2:12. doi: 10.3389/fonc.2012.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen JP, Mermel C, Zejnullahu K, Murphy C, Lifshits E, Holmes AJ, Choi HG, Kim J, Chiang D, Thomas R, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14:4275–4283. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi J, Kawaguchi KS, Vo H, Haruki N, Gonzalez A, Carbone DP, Dang TP. Gamma-secretase inhibitor prevents Notch3 activation and reduces proliferation in human lung cancers. Cancer Res. 2007;67:8051–8057. doi: 10.1158/0008-5472.CAN-07-1022. [DOI] [PubMed] [Google Scholar]
- Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. 2014;14:275–291. doi: 10.1016/j.stem.2014.02.006. [DOI] [PubMed] [Google Scholar]
- Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, Jakubosky D, Genovese G, Muller FL, Jeong JH, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18:1503–1510. doi: 10.1038/nm.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Hitomi M, Gallagher J, Gadani SP, Adkins J, Vasanji A, Liu L, Eyler CE, Heddleston JM, Wu Q, et al. Distribution of CD133 reveals glioma stem cells self-renew through symmetric and asymmetric cell divisions. Cell Death Dis. 2011;2:e200. doi: 10.1038/cddis.2011.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Good JA, Ziegler WH, Parekh DB, Alessi DR, Cohen P, Parker PJ. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science. 1998;281:2042–2045. doi: 10.1126/science.281.5385.2042. [DOI] [PubMed] [Google Scholar]
- Lin D, Edwards AS, Fawcett JP, Mbamalu G, Scott JD, Pawson T. A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat Cell Biol. 2000;2:540–547. doi: 10.1038/35019582. [DOI] [PubMed] [Google Scholar]
- Mackereth CD, Scharpf M, Gentile LN, MacIntosh SE, Slupsky CM, McIntosh LP. Diversity in structure and function of the Ets family PNT domains. J Mol Biol. 2004;342:1249–1264. doi: 10.1016/j.jmb.2004.07.094. [DOI] [PubMed] [Google Scholar]
- Mah IK, Soloff R, Hedrick SM, Mariani FV. Atypical PKC-iota Controls Stem Cell Expansion via Regulation of the Notch Pathway. Stem Cell Reports. 2015;5:866–880. doi: 10.1016/j.stemcr.2015.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield AS, Fields AP, Jatoi A, Qi Y, Adjei AA, Erlichman C, Molina JR. Phase I dose escalation study of the PKCiota inhibitor aurothiomalate for advanced non-small-cell lung cancer, ovarian cancer, and pancreatic cancer. Anticancer Drugs. 2013;24:1079–1083. doi: 10.1097/CAD.0000000000000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messersmith WA, Shapiro GI, Cleary JM, Jimeno A, Dasari A, Huang B, Shaik MN, Cesari R, Zheng X, Reynolds JM, et al. A Phase I, Dose-Finding Study in Patients with Advanced Solid Malignancies of the Oral gamma-Secretase Inhibitor PF-03084014. Clin Cancer Res. 2015;21:60–67. doi: 10.1158/1078-0432.CCR-14-0607. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441:1068–1074. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Kimura H, Takeuchi K, Soda M, Mano H, Yasufuku K, Iizasa T. Treatment of lung cancer with an ALK inhibitor after EML4-ALK fusion gene detection using endobronchial ultrasound-guided transbronchial needle aspiration. J Thorac Oncol. 2010;5:2041–2043. doi: 10.1097/JTO.0b013e3181f77b58. [DOI] [PubMed] [Google Scholar]
- Neumuller RA, Knoblich JA. Dividing cellular asymmetry: asymmetric cell division and its implications for stem cells and cancer. Genes Dev. 2009;23:2675–2699. doi: 10.1101/gad.1850809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno S. Intercellular junctions and cellular polarity: the PAR-aPKC complex, a conserved core cassette playing fundamental roles in cell polarity. Curr Opin Cell Biol. 2001;13:641–648. doi: 10.1016/s0955-0674(00)00264-7. [DOI] [PubMed] [Google Scholar]
- Oliver JR, Kushwah R, Wu J, Pan J, Cutz E, Yeger H, Waddell TK, Hu J. Elf3 plays a role in regulating bronchiolar epithelial repair kinetics following Clara cell-specific injury. Lab Invest. 2011;91:1514–1529. doi: 10.1038/labinvest.2011.100. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, Singh B, Heelan R, Rusch V, Fulton L, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pine SR, Ryan BM, Varticovski L, Robles AI, Harris CC. Microenvironmental modulation of asymmetric cell division in human lung cancer cells. Proc Natl Acad Sci U S A. 2010;107:2195–2200. doi: 10.1073/pnas.0909390107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regala RP, Davis RK, Kunz A, Khoor A, Leitges M, Fields AP. Atypical protein kinase C{iota} is required for bronchioalveolar stem cell expansion and lung tumorigenesis. Cancer Res. 2009;69:7603–7611. doi: 10.1158/0008-5472.CAN-09-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regala RP, Thompson EA, Fields AP. Atypical protein kinase C iota expression and aurothiomalate sensitivity in human lung cancer cells. Cancer Res. 2008;68:5888–5895. doi: 10.1158/0008-5472.CAN-08-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regala RP, Weems C, Jamieson L, Copland JA, Thompson EA, Fields AP. Atypical protein kinase Ciota plays a critical role in human lung cancer cell growth and tumorigenicity. J Biol Chem. 2005a;280:31109–31115. doi: 10.1074/jbc.M505402200. [DOI] [PubMed] [Google Scholar]
- Regala RP, Weems C, Jamieson L, Khoor A, Edell ES, Lohse CM, Fields AP. Atypical protein kinase C iota is an oncogene in human non-small cell lung cancer. Cancer Res. 2005b;65:8905–8911. doi: 10.1158/0008-5472.CAN-05-2372. [DOI] [PubMed] [Google Scholar]
- Roper K, Corbeil D, Huttner WB. Retention of prominin in microvilli reveals distinct cholesterol-based lipid micro-domains in the apical plasma membrane. Nat Cell Biol. 2000;2:582–592. doi: 10.1038/35023524. [DOI] [PubMed] [Google Scholar]
- Scotti ML, Smith KE, Butler AM, Calcagno SR, Crawford HC, Leitges M, Fields AP, Murray NR. Protein kinase C iota regulates pancreatic acinar-to-ductal metaplasia. PLoS One. 2012;7:e30509. doi: 10.1371/journal.pone.0030509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallings-Mann M, Jamieson L, Regala RP, Weems C, Murray NR, Fields AP. A novel small-molecule inhibitor of protein kinase Ciota blocks transformed growth of non-small-cell lung cancer cells. Cancer Res. 2006;66:1767–1774. doi: 10.1158/0008-5472.CAN-05-3405. [DOI] [PubMed] [Google Scholar]
- Standaert ML, Bandyopadhyay G, Perez L, Price D, Galloway L, Poklepovic A, Sajan MP, Cenni V, Sirri A, Moscat J, et al. Insulin activates protein kinases C-zeta and C-lambda by an autophosphorylation-dependent mechanism and stimulates their translocation to GLUT4 vesicles and other membrane fractions in rat adipocytes. J Biol Chem. 1999;274:25308–25316. doi: 10.1074/jbc.274.36.25308. [DOI] [PubMed] [Google Scholar]
- Sullivan JP, Spinola M, Dodge M, Raso MG, Behrens C, Gao B, Schuster K, Shao C, Larsen JE, Sullivan LA, et al. Aldehyde dehydrogenase activity selects for lung adenocarcinoma stem cells dependent on notch signaling. Cancer Res. 2010;70:9937–9948. doi: 10.1158/0008-5472.CAN-10-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsumori T, Tsuta K, Masai K, Kinno T, Taniyama T, Yoshida A, Suzuki K, Tsuda H. p40 is the best marker for diagnosing pulmonary squamous cell carcinoma: comparison with p63, cytokeratin 5/6, desmocollin-3, and sox2. Appl Immunohistochem Mol Morphol. 2014;22:377–382. doi: 10.1097/PAI.0b013e3182980544. [DOI] [PubMed] [Google Scholar]
- Vasan N, Boyer JL, Herbst RS. A RAS renaissance: emerging targeted therapies for KRAS-mutated non-small cell lung cancer. Clin Cancer Res. 2014;20:3921–3930. doi: 10.1158/1078-0432.CCR-13-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorhagen S, Niessen CM. Mammalian aPKC/Par polarity complex mediated regulation of epithelial division orientation and cell fate. Exp Cell Res. 2014;328:296–302. doi: 10.1016/j.yexcr.2014.08.008. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hill KS, Fields AP. Protein Kinase Ciota maintains a tumor-initiating cell phenotype that is required for ovarian tumorigenesis. Mol Cancer Res. 2013 doi: 10.1158/1541-7786.MCR-13-0371-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigmann A, Corbeil D, Hellwig A, Huttner WB. Prominin, a novel microvilli-specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non-epithelial cells. Proc Natl Acad Sci U S A. 1997;94:12425–12430. doi: 10.1073/pnas.94.23.12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan P, Kadara H, Behrens C, Tang X, Woods D, Solis LM, Huang J, Spinola M, Dong W, Yin G, et al. Sex determining region Y-Box 2 (SOX2) is a potential cell-lineage gene highly expressed in the pathogenesis of squamous cell carcinomas of the lung. PLoS One. 2010;5:e9112. doi: 10.1371/journal.pone.0009112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, de la Cruz CC, Sayles LC, Alleyne-Chin C, Vaka D, Knaak TD, Bigos M, Xu Y, Hoang CD, Shrager JB, et al. A rare population of CD24(+)ITGB4(+)Notch(hi) cells drives tumor propagation in NSCLC and requires Notch3 for self-renewal. Cancer Cell. 2013;24:59–74. doi: 10.1016/j.ccr.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.