

Abstract
Mycobacterium tuberculosis is intrinsically resistant to most β-lactam antibiotics because of the constitutive expression of the blaC-encoded β-lactamase. This enzyme has extremely high activity against penicillins and cephalosporins, but weaker activity against carbapenems. The enzyme can be inhibited by clavulanate, avibactam, and boronic acids. In this study, we investigated the ability of 6-methylidene β-lactams to inhibit BlaC. One such compound, penem 2, inhibited BlaC more than 70 times more efficiently than clavulanate. The compound forms a covalent complex with BlaC as shown by mass spectrometry. Crystallization of the complex revealed that the bound inhibitor was covalently attached via the Ser70 active site residue and that the covalently, acylated form of the inhibitor had undergone additional chemistry yielding a 4,7-thiazepine ring in place of the β-lactam and a thiazapyroline ring generated as a result of β-lactam ring opening. The stereochemistry of the product of the 7-endo-trig cyclization was the opposite of that observed previously for class A and D β-lactamases. Addition of penem 2 greatly synergized the antibacterial properties of both ampicillin and meropenem against a growing culture of M. tuberculosis. Strikingly, penem 2 alone showed significant growth inhibition, suggesting that in addition to its capability of efficiently inhibiting BlaC, it also inhibited the peptidoglycan cross-linking transpeptidases.
Graphical abstract
The emergence, and rapid molecular evolution, of bacterial β-lactamases threatens the clinical utility of β-lactam antibacterials.1,2 These are the largest class of antibacterial agents and are widely prescribed for the treatment of both Gram-negative and Gram-positive infections. There are four classes of β-lactamases, originally classified on the basis of primary amino acid sequence,3 termed Ambler classes A–D.3 Classes A, C, and D enzymes contain an active site serine nucleophile, while the class B enzymes are metalloenzymes containing zinc at the active site. The class A and C enzymes are the most common and are believed to have arisen from unique ancestral penicillin binding proteins (PBPs), which are the targets of β-lactams and share the initial serine acylation mechanism, and subsequent evolution of β-lactam hydrolyzing activity (reviewed in ref 1). A more recent attempt to classify β-lactamses according to substrate and inhibitor specificity has been proposed.4 With the appearance of broad (extended) spectrum β-lactamases (ESBLs), which catalyze the hydrolysis of both penicillins and cephalosporins, the development and combinatorial use of β-lactamase inhibitors with β-lactams was adopted. There are presently only four Food and Drug Administration-approved and clinically used β-lactamase inhibitors: clavulanic acid, the two penicillanic acid sulfones (tazobactam and sulbactam), and most recently avibactam. However, there has been the emergence of resistance even to these combinations among formerly susceptible bacteria.5–7 Further, the class C β-lactamases have weak affinity for these inhibitors and are only poorly inhibited by them, while organisms expressing metallo-β-lactamases or class D β-lactamases (where the SXN motif is replaced with a YXN motif) are not susceptible to any of the β-lactamase inhibitors2 and can hydrolyze carbapenems.8
To address these concerns, several new classes of β-lactamase inhibitors have recently been approved or are in development.9 These include the non-lactam diazabicyclooctanes that are powerful inhibitors of several classes of β-lactamases10 and include avibactam (formerly NXL104) whose covalently bound structure to a class A β-lactamase has recently been reported.11 A second class under development is the 6-methylidene penems that have a broad spectrum of activity against all the non-metallo-β-lactamases.12–14 The β-lactam rings of the penems are nucleophilically attacked and ring opened to form acylated penem–β-lactamase complexes but then undergo a rearrangement and cyclization to generate a covalently bound 1,4-thiazepine seven-membered ring system, first suggested in 199512 and confirmed crystallographically in 2003.15 Since that time, a number of crystal structures of various 6-methylidene penem derivatives have been reported in complex with a number of β-lactamases.16–19
Mycobacterium tuberculosis was shown to constitutively express a “penicillinase” in 1949.20 The publication of the genome revealed a single, chromosomally encoded β-lactamase whose sequence allowed its classification as a class A β-lactamase.21 A subsequent genetic study revealed that the blaC gene product (BlaC) was responsible for the lack of susceptibility of mycobacterial species, including M. tuberculosis, to β-lactam antibacterials.22 An expression construct in which the N-terminal 40-residue “membrane-anchoring” sequence was truncated allowed for the production of a soluble, active enzyme that was crystallized and structurally characterized.23 A subsequent detailed kinetic analysis of the substrate specificity of the enzyme revealed that BlaC exhibited a broad spectrum of activity against penicillins, cephalosporins, and the carbapenem, imipenem.24 That study demonstrated that both tazobactam and sulbactam only transiently inhibited the enzyme, while clavulanate irreversibly inhibited the enzyme, and the structure of the BlaC–clavulanate covalent complex was reported soon after.25 This provided biochemical confirmation of the earlier report that amoxicillin in combination with clavulanate exhibited in vitro activity against laboratory strains of M. tuberculosis26 and a resolution to the paradoxical lack of activity of β-lactams against mycobacteria known to harbor essential penicillin-sensitive targets (e.g., D,D-transpeptidases27). Finally, the combination of the single effective β-lactamase inhibitor, clavulanate, with the extremely poor BlaC substrate, meropenem, was shown to exhibit bactericidal activity against both rapidly replicating and dormant cultures of both laboratory and extensively drug-resistant (XDR) strains of M. tuberculosis.28This same combination has been used successfully to treat patients infected with XDR-TB strains.29
This study was performed to assess the interaction of 6-methylidene penems as potential inhibitors of BlaC, structurally characterize the covalent complex formed between the penem and BlaC, and determine the efficacy and potential synergies of the inhibitor with selected β-lactam antibiotics.
MATERIALS AND METHODS
Materials
Penem 1 and penem 2 (Scheme 1) were kindly provided by Pfizer (Groton, CT). Buffer reagents for crystallography were purchased from Hampton Research (Aliso Viejo, CA). Unless noted, other chemicals were from Sigma-Aldrich (St. Louis, MO).
Scheme 1.
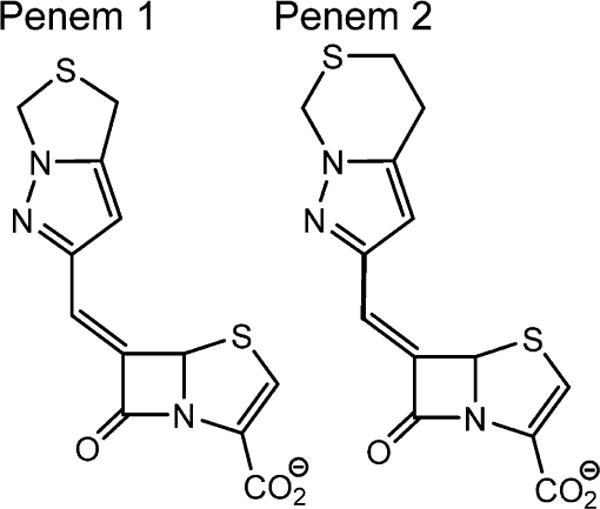
Structures of Penems 1 and 2
Cloning and Purification of Wild-Type (WT) BlaC
The blaC gene was amplified from genomic M. tuberculosis H37Rv DNA and cloned into pET28 using NdeI and HindIII. BlaC was expressed as an N-terminally truncated form, lacking the first 40 amino acids, as previously described.24 After the construct was confirmed by sequencing, the plasmid expressing His6-tagged wild-type BlaC was transformed into Escherichia coli BL21/DE3 cells and cultured in LB broth at 37 °C. When the culture OD600 reached 0.6, the culture was cooled, and protein expression was induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). After incubation for 18 h at 16 °C, cells were harvested, resuspended in 25 mM Tris-HCl containing 300 mM NaCl (pH 7.5), and disrupted by sonication. After centrifugation, the soluble extract was loaded onto a Ni-NTA agarose column (Qiagen) and eluted with 200 mM imidazole, in 25 mM Tris-HCl containing 300 mM NaCl (pH 7.5). The eluted fractions were dialyzed against the same buffer without imidazole. To remove the His tag, the eluted protein was incubated with thrombin (Novagen, Madison, WI) overnight at 4 °C (1.6 units/mg of protein). The cleaved protein was separated from the His6-tagged peptide by size-exclusion chromatography using a HiLoad 26/60 Superdex 200 column (GE Healthcare Life Science, Uppsala, Sweden).
Inactivation Kinetics of BlaC with Clavulanate and Penems 1 and 2
Steady state kinetics were performed on an Agilent (Palo Alto, CA) 8453 diode array spectrophotometer in sodium phosphate buffer (50 mM, pH 7.2) and a 1 cm path length cuvette. Inhibitor kinetics were performed using nitrocefin (NCF) as a reporter substrate (Δε482 = 17400 M−1 cm−1). The kinetic behavior of mechanism-based inhibitors was characterized using a two-phase model of time-dependent inactivation with kinact being the maximal rate of inactivation or the rate limit at infinite inhibitor concentration, and KI reflecting the concentration that results in a half-maximal rate of inhibition. KI and kinact were determined as follows. Product formation from NCF hydrolysis at an initial concentration of 100 μM in the presence of 0.05 μg/mL BlaC was assessed at 482 nm over a period of 300 s and plotted as a function of time t. The data points were fitted to eq 1 using Origin8.0 (Origin Corp., Northampton, MA) to obtain the individual apparent first-order rate constant, kobs, for the interconversion of initial velocity v0 to final velocity vf for a given inhibitor concentration:
| (1) |
Serial measurements using increasing inhibitor concentrations, [I], were performed, and the resulting individual rate constants (kobs) were plotted versus [I]. Fitting to eq 2 allowed determination of kinact and KI:
| (2) |
KI values were corrected for the affinity of NCF according to eq 3.
| (3) |
using the determined KM value for NCF of 58 μM.
Turnover numbers for penem 2 were obtained as follows. Penem 2 and BlaC were co-incubated at molar ratios (I:E) of 5:1, 10:1, 50:1, and 100:1 over 20 h, and residual activities were determined using NCF. No residual activity was detected at any I:E ratio, indicating a turnover number of <5. Then, after time-dependent inactivation, equimolar inhibitor:enzyme ratios were obtained at 5, 10, 30, and 60 min. No residual activity was detected after 5 min, indicating stoichiometric inactivation. Assays were performed in triplicate, including control reactions.
Mass Spectrometry of the BlaC–Penem 2 Covalent Adduct
To test if penem 2 forms a stable, covalent complex with BlaC, the sizes of native BlaC and BlaC co-incubated with penem 2 were determined using electrospray ionization (ESI) mass spectrometry (MS). Spectra were generated on an Applied Biosystems (Foster City, CA) QStar Elite quadrupole time-of-flight mass spectrometer equipped with a TurboIon spray source. BlaC was preincubated with a 10-fold excess of penem 2 for 15 min. Samples were desalted using a C18ZipTip (Millipore, Billerica, MA) following the manufacturer’s protocol. Proteins were diluted with 50% acetonitrile and 0.2% formic acid. The samples were infused at a rate of 0.5 μL/min, and data were collected for 2 min. Spectra were deconvoluted using Analyst from Applied Biosystems (Framingham, MA).
Growth Inhibition of M. tuberculosis
M. tuberculosis strain H37Rv was inoculated into 7H9 liquid medium, grown at 37 °C to an OD600 of 1.0, and then diluted to a starting OD600 of 0.18. Fractions (5 mL) were inoculated in duplicate with the following compounds each at a concentration of 10 μg/mL: ampicillin, clavulanate, meropenem, penem 2, ampicillin and clavulanate, and meropenem and clavulanate. Growth was assessed over 9 days by measuring OD600 and compared to that of a control culture.
Crystallization of the BlaC–Penem 2 Adduct
The hanging drop, vapor diffusion method was used for the crystallization of BlaC. The composition of the well consisted of 0.1 M HEPES (pH 7.5) and 2 M NH4H2PO4, which makes the final pH of the well solution 4.1. Protein at a concentration of 12 mg/mL was mixed 1:1 with the well solution and incubated at 10 °C. Initial crystals grew within 4–5 days but were very small needles. Repeated microseeding was performed, which resulted in efficient large crystal growth with improved morphology producing diffraction quality crystals of the active enzyme.
Data Collection and Refinement
Crystals were soaked with ~100 mM penem 2 in mother liquor. The solution was incubated for 2, 4, 8, 12, 24, and 48 h. Before the crystals were frozen, mineral oil was added to the solution as a cryoprotectant. Data were collected at the RAXIS IV+ home source on crystals frozen after 2, 4, 8, and 24 h soaks with penem 2. The data from the 8 h soak were processed using HKL2000.30 Our previous structure of tebipenem bound covalently to M. tuberculosis BlaC31 [Protein Data Bank (PDB) entry 4Q8I] was used to phase the data, using the CCP4 software suite.32 Multiple rounds of structural refinement and model building were performed in Refmac5,33,34 Phenix,35 and Coot.36 Table 1 lists the data collection statistics for the structure as well as the final refinement statistics. Structure figures were generated using PyMOL. Atomic coordinates and experimental structure factors have been deposited as PDB entry 4QHC.
Table 1.
Kinetics of BlaC Inhibition
| KI (μM) | kinact(s−1) | kinact/KI | |
|---|---|---|---|
| clavulanate | 6.3 | 0.02 | 0.004 |
| penem 1 | 0.8 | 0.06 | 0.07 |
| penem 2 | 0.2 | 0.07 | 0.30 |
RESULTS AND DISCUSSION
The kinetics of inactivation of BlaC by clavulanate, penem 1, and penem 2 were determined. Clavulanate has previously been shown to be a rapid, irreversible inactivator of BlaC.24 Both penem 1 and penem 2 were shown to inactivate BlaC, with rates of inactivation (kinact) that were higher (0.06 and 0.07 s−1, respectively) and KI values that were lower (0.8 and 0.2 μM, respectively) than those of clavulanate (Table 1). The ratio of these kinetic constants (kinact/KI) is a commonly used measure of the effectiveness of an inhibitor. Upon comparison of this ratio for penem 2 and clavulanate, penem 2 is 75 times more effective as an inhibitor. Thus, we sought to examine the nature of the binding of penem 2 to BlaC.
Using BlaC alone, or BlaC that had been incubated with a 10-fold molar excess of penem 2, the mass spectra shown in Figure 1 were obtained. On the left is the spectrum of BlaC alone, while on the right (shown with a gray background) is the spectrum of BlaC in the presence of penem 2. The BlaC peak completely disappears and is replaced by a peak that is mass shifted by 322 ± 3 Da. The predicted mass shift for the formation of the covalent complex is 321 ± 3 Da, suggesting that penem 2 reacts with Ser70 to form a stable, covalent acylated complex. This has been observed previously with other β-lactamases, including the class A enzyme, SHV-1,18 and the class D enzyme, OXA-1.14
Figure 1.
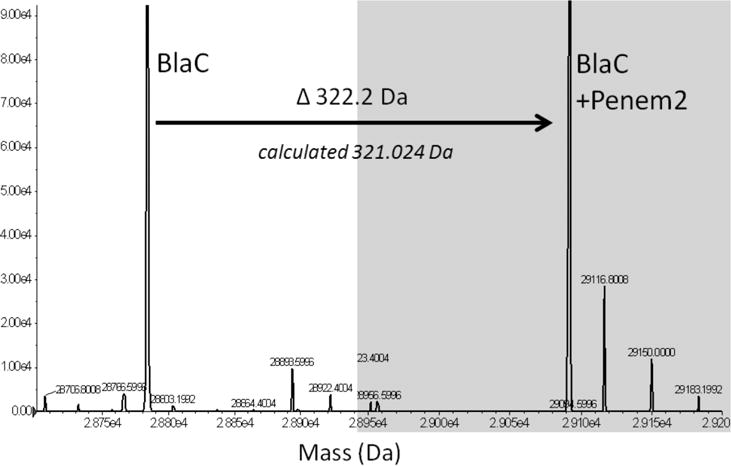
Overlay of the mass spectra of apoenzyme (mass determined, 28784.2 Da) and the BlaC–penem 2 covalent complex (mass determined, 29406.4). The mass increase of 322.2 Da corresponds to the molecular weight of penem 2 of 321.02 Da. No fragments were observed.
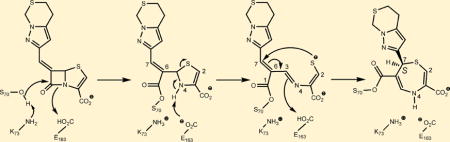
M. tuberculosis BlaC has been crystallized in a number of complexes with the β-lactamase inhibitors clavulanate25 and avibactam11 as well as several carbapenems.28,31,37 These structures have revealed that after acylation at Ser70, that additional chemical reactions occur at the active site, causing fragmentation of the bound molecules. Previous structural studies of 6-methylidene penems and penem sulfones bound to class A (Klebsiella pneumoniae SHV-1) and C (Enterobacter cloacae GC1) β-lactamases have revealed that after β-lactam ring opening of these molecules, the thiazoline ring of the covalently attached inhibitor is cleaved to generate a C6 thiolate intermediate (Scheme 2) much like the oxazole ring of clavulanate is opened. The thiolate can now attack the C6–C7 ester conjugated, unsaturated bond to form an enzyme-bound 1,4-thiazepine species. This has been shown to occur stereospecifically, with the unique formation of the C7 R stereoisomer.15 We thus sought to determine whether similar chemistry and stereochemistry apply to the BlaC β-lactamase.
Scheme 2.
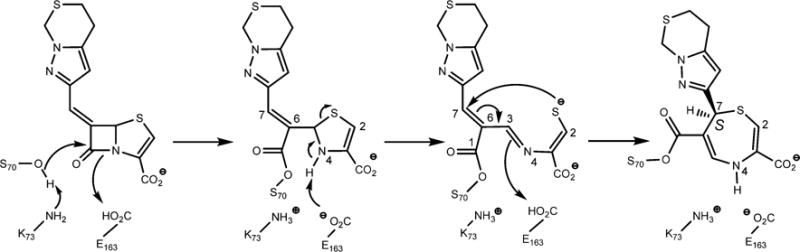
Chemical Steps Leading to the Final 1,4-Thiazepine Covalent Adduct
When crystals of BlaC were soaked for varying lengths of time with penem 2, the data sets were indistinguishable, suggesting that penem 2 bound and completely reacted with BlaC within 2 h. The data set from the 8 h soak was refined to 1.9 Å resolution using the BlaC–tebipenem structure as a model with excellent statistics (Table 2). There was clear electron density in the active site that was continuous with the side chain of Ser70. The bound species was modeled as the 1,4-thiazepine product previously observed in other β-lactamase structures (Figure 2B). The two ring systems could be unambiguously positioned because of the carboxyl group located on the 1,4-thiazepine ring and the sulfur anomalous signal of the sulfur atoms in both the thiazepine and C7 heterocyclic substituent.
Table 2.
Data Collection and Refinement Statistics for the BlaC–Penem 2 Complex
| Data Collection | |
| X-ray source | rotating anode |
| wavelength (Å) | 1.5418 (Cu anode) |
| temperature (K) | 100 |
| resolution range (Å) | 27.00–1.89 |
| no. of reflections | 21290 |
| completeness (%) | 96.89 |
| redundancy | 4.1 |
| space group | P21P21P21 |
| unit cell dimensions (Å) | |
| a | 49.70 |
| b | 68.33 |
| c | 75.25 |
| α = β = γ | 90.00 |
| no. of molecules per asymmetric unit | 1 |
| Refinement | |
| Rwork (%) | 17.52 |
| Rfree (%) | 21.09 |
| no. of atoms | |
| protein (chain A) | 2027 |
| phosphate (chain B) | 20 |
| penem 2 (chain T) | 21 |
| water (chain W) | 209 |
| root-mean-square deviation | |
| bond lengths (Å) | 0.007 |
| bond angles (deg) | 1.148 |
| average B factor (Å2) | |
| protein main chain | 12.43 |
| protein side chain | 14.84 |
| protein whole chain | 13.56 |
| phosphate | 37.94 |
| penem 2 | 17.44 |
| water | 21.34 |
| PDB entry | 4QHC |
Figure 2.
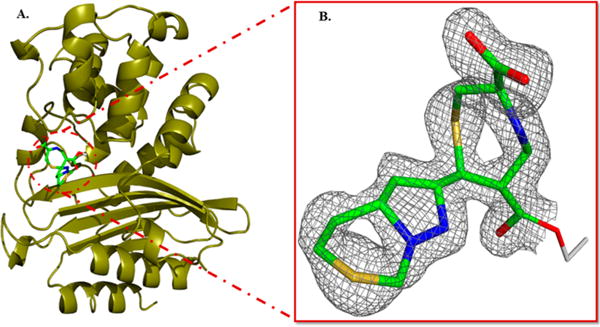
Structure of the BlaC–penem 2 covalent adduct. (A) The BlaC backbone is shown as gold ribbons, while the 1,4-thiazepine covalent adduct is colored by atom type. (B) The 2Fo – Fc electron density into which the inhibitor was modeled.
The reactions leading to the formation of the thiazepine ring are shown in Scheme 2. After binding to BlaC, Ser70, assisted by the general base Lys73, nucleophilically attacks the β-lactam ring of penem 2. Ring opening leads to the covalently bound inhibitor with the thiazapyroline ring intact. Deprotonation of the ring nitrogen leads to cleavage of the thiazapyroline ring and formation of the extensively conjugated open chain thiolate. The stereochemistry at C7 is clearly S in the experimental structure, in contrast to the R configuration observed in the penem complexes of the class A (SHV-1)18 and class D (OXA-1)14 β-lactamases. In the class C GC1 penem structure,16 the stereochemistry is mixed, consisting of approximately 30% R and 70% S isomers. The final product of this 7-endo-trig cyclization requires that the thiolate attack the C6–C7 double bond on the re face of C7 to generate the seven-membered thiazepine ring. We draw the final ring as the 4,7-dihydro tautomer primarily on the basis of the bond angles in the structure. The measured S1–C2–C3 and C2–C3–N4 angles of 118.5° and 120.4°, respectively, support the presence of a C2–C3 double bond. Similarly, the C5–C6–CO bond angle of 119.9° supports the predominant enzyme-bound tautomer as being the 4,7-dihydro form. Figure 3 shows the overlay of the covalent complexes of the BlaC–penem covalent complex (carbons colored green) with that of the K. pneumoniae SHV-1–penem complex (carbons colored orange). The orientation of the thiazepine ring is opposite in the two complexes because the stereochemistry at C7 of the two final products is opposite, thus causing the sulfur of the ring to be oriented toward the solvent in the SHV structure but inward in the BlaC complex.
Figure 3.
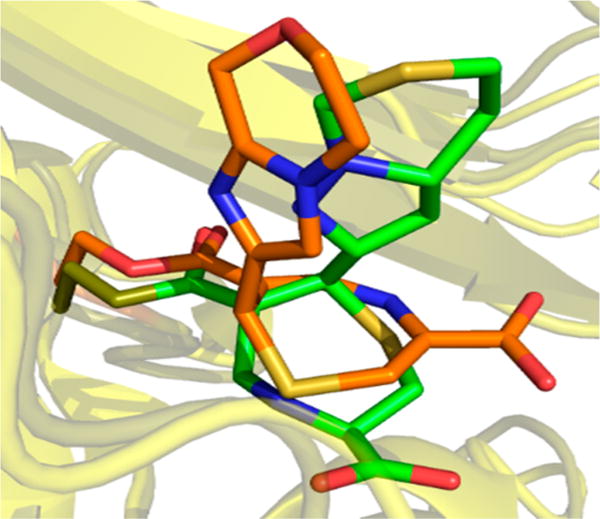
Comparison of the binding modes of the BlaC–penem 2 complex of M. tuberculosis (green) and the K. pneumoniae BlaC–penem 2 complex (orange).
As discussed by others, while all substrates and inhibitors of β-lactamases interact with their shared carboxylic acid and the “carboxylate binding pocket” composed of arginine and threonine residues, the structures of the β-lactamse–thiazepine complexes reveal no interaction in this complex. That is true for the BlaC–thiazepine complex here, as well. The only interaction of the carboxylate is with a backbone amide of Ile105 and a water molecule (Figure 4A,B). This same water molecule interacts with the ring nitrogen atom of the thiazepine ring, as well. The carbonyl of the ester remains in the “oxyanion hole” composed of the backbone amide nitrogens of Thr239 and Ser70. The only other electrostatic interaction of the bound inhibitor is an interaction between one of the ring nitrogens of the thiazepine heterocycle and the conserved “deacylation” water molecule and the bound phosphate anion observed in many of the BlaC complexes as a result of the high concentration (2 M) of phosphate used in the crystallization medium.
Figure 4.
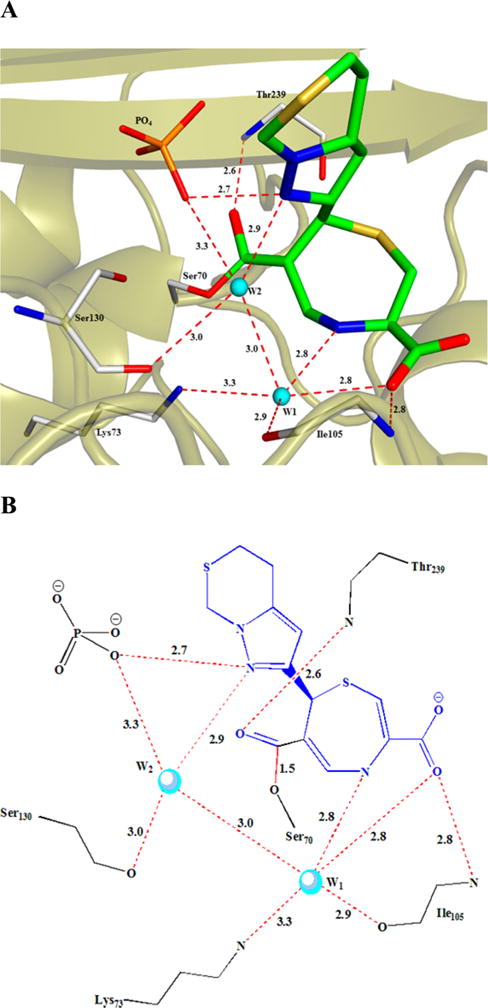
Interactions between BlaC active site residues and the covalently bound inhibitor.
To determine whether the biochemical demonstration of inhibition of BlaC was relevant to inhibition of the enzyme in the pathogen, we performed growth experiments with the virulent H37Rv strain of M. tuberculosis. Robust growth was observed in control cultures, as well in the presence of either ampicillin or clavulanate added individually (Figure 5). When ampicillin and clavulanate were tested together, the synergistic effect of the inhibition of BlaC by clavulanate24 on the activity of ampicillin was clear. Meropenem, a carbapenem, inhibited growth when added alone to approximately the same extent as the combination of ampicillin and clavulanate. When clavulanate was added to meropenem, this combination yielded a nearly complete inhibition of growth. This combination has been shown to have excellent bactericidal activity against H37Rv and extensively drug-resistant (XDR) clinical strains of M. tuberculosis28 and recently shown to have activity in patients with MDR and XDR strains of M. tuberculosis.29 Penem 2 when added with ampicillin had a more synergizing effect than clavulanate, suggesting that this compound can enter the periplasmic space and inhibit BlaC.
Figure 5.
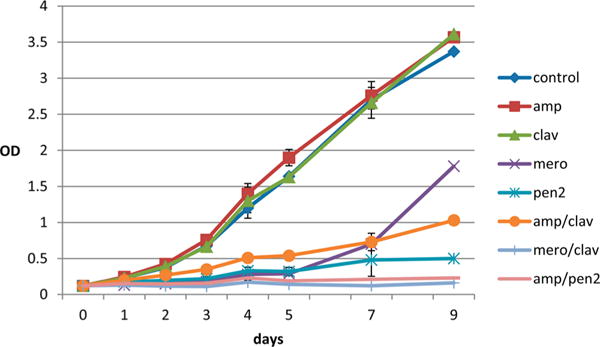
Growth of a M. tuberculosis strain H37Rv fluid culture in duplicate. All compounds were added at a concentration of 10 μg/mL, and the OD600 was measured over 9 days.
The most surprising finding to come from these studies was the ability of penem 2 alone to inhibit the growth of M. tuberculosis H37Rv. This suggests that in addition to its demonstrated ability to potently inhibit the BlaC β-lactamase, it also has antibacterial properties. The results suggest that penem 2 may be able to bind to, and inhibit, the D,D- and L,D-transpeptidases that are responsible for peptidoglycan cross-linking. Mycobacteria are unusual in having a large percentage of 3–3 cross-links generated by the L,D-transpeptidases compared to the more common 3–4 cross-links generated by the D,D-transpeptidases. These L,D-transpeptidases are poorly inhibited by penicillins, such as ampicillin, while they, and the D,D-carboxypeptidases that generate the truncated substrates for the transpeptidases, have been shown to be inhibited by the carbapenems, including meropenem.38,39 The long-lasting inhibition of growth by penem 2 also suggests that once the covalent, cyclized thiazepine generated at the BlaC active site is formed, it remains stable to subsequent hydrolysis. In contrast, meropenem and other carbapenems are extremely poor substrates for BlaC (kcat = 0.08 min−1)28 but are eventually hydrolyzed from the enzyme. In summary, we have shown that penem 2 inhibits BlaC more effectively than any currently available β-lactamase inhibitor. We have also shown by mass spectrometry and crystallography that the inhibitor is not fragmented and forms a stable covalent complex. Most importantly, we show that novel stereochemistry is formed by this inhibitor and that inhibitor alone significantly retards the growth of mycobacteria in culture.
Acknowledgments
Funding
This work was supported by National Institutes of Health Grants AI060899 (to J.S.B.) and AI100560 and AI063517 (to R.A.B). S.H. is currently Assistant Professor at the Indian Institute of Technology, Roorke. S.G.K. is currently Assistant Professor at Tufts University School of Medicine.
Footnotes
Accession Codes
The Protein Data Bank entry for the BlaC–penem 2 adduct is 4QHC.
The authors declare no competing financial interest.
References
- 1.Massova I, Mobashery S. Kinship and Diversification of Bacterial Penicillin-Binding Proteins and β-Lactamases. Antimicrob Agents Chemother. 1998;42:1–17. doi: 10.1128/aac.42.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Helfand MS, Bonomo RA. β-Lactamases: A Survey of Protein Diversity. Curr Drug Targets: Infect Disord. 2003;3:9–23. doi: 10.2174/1568005033342181. [DOI] [PubMed] [Google Scholar]
- 3.Ambler RP. The structure of β-lactamases. Philos Trans R Soc B. 1980;289:321–331. doi: 10.1098/rstb.1980.0049. [DOI] [PubMed] [Google Scholar]
- 4.Bush K, Jacoby GA. Updated functional classification of β-lactamses. Antimicrob Agents Chemother. 2010;54:969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaye KS, Gold HA, Schwaber MJ, Venkataraman L, Qi Y, De Girolami PC, Samore MH, Anderson G, Rasheed JK, Tenover FC. Variety of β-lactamases produced by amoxicillin-clavulanate-resistant Escherichia coli isolated in the northeastern United States. Antimicrob Agents Chemother. 2004;48:1520–1525. doi: 10.1128/AAC.48.5.1520-1525.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stapleton P, Wu PJ, King A, Shannon K, French G, Phillips I. Incidence and mechanism of resistance to the combination of amoxicillin and clavulanic acid in Escherichia coli. Antimicrob Agents Chemother. 1995;39:2478–2483. doi: 10.1128/aac.39.11.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lister PD. Beta-lactamase inhibitor combinations with extended-spectrum penicillins: factors influencing antibacterial activity against enterobacteriaceae and Pseudomonas aeruginosa. Pharmacotherpy. 2000;20:213S–218S. doi: 10.1592/phco.20.14.213s.35045. [DOI] [PubMed] [Google Scholar]
- 8.Afzal-Shah MN, Woodford N, Livermore DM. Characterization of OXA-25, OXA-26 and OXA-27, molecular Class D β-lactamses associated with carbapenem resistance in clinical isolates of Acinetobacter baumannii. Antimicrob Agents Chemother. 2001;45:583–588. doi: 10.1128/AAC.45.2.583-588.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watkins RR, Papp-Wallace KM, Drawz SM, Bonomo RA. Novel β-lactamase inhibitors: a therapeutic hope against the scourge of multidrug resistance. Front Microbiol. 2013;4:1–8. doi: 10.3389/fmicb.2013.00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coleman K. Diazabicyclooctanes (DBO’s): a potent new class of non-β-lactam β-lactamase inhibitors. Curr Opin Microbiol. 2011;14:550–555. doi: 10.1016/j.mib.2011.07.026. [DOI] [PubMed] [Google Scholar]
- 11.Xu H, Hazra S, Blanchard JS. NXL104 Irreversibly Inhibits the β-Lactamase from Mycobacterium tuberculosis. Biochemistry. 2012;51:4551–4557. doi: 10.1021/bi300508r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bulychev A, Massova I, Lerner SA, Mobashery S. Penem BRL 42715: An Effective Inactivator for β-Lactamases. J Am Chem Soc. 1995;117:4797–4801. [Google Scholar]
- 13.Weiss WJ, Petersen PJ, Murphy TM, Tardio L, Yang Y, et al. In vitro and in vivo activities of novel 6-methylidene penems as beta-lactamase inhibitors. Antimicrob Agents Chemother. 2004;48:4589–4596. doi: 10.1128/AAC.48.12.4589-4596.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Venkatesan AM, Agarwal MA, Abe T, Ushirogochi H, Yamamura I, Ado M, Tsuyoshi T, Dos Sanos O, et al. Structure-activity relationship of 6-methylidene penems bearing 6,5 bicyclic heterocycles as broad-spectrum β-lactamase inhibitors: evidence for 1,4-thiazepine intermediates with C7R stereochemistry by computational methods. J Med Chem. 2006;49:4623–4637. doi: 10.1021/jm060021p. [DOI] [PubMed] [Google Scholar]
- 15.Nukaga M, Abe T, Venkatesan AM, Mansour TS, Bonomo RA, Knox JR. Inhibition of Class A and Class C β-lactamases by Penems: crystallographic structures of a novel 1,4-thiazepine intermediate. Biochemistry. 2003;42:13152–12159. doi: 10.1021/bi034986b. [DOI] [PubMed] [Google Scholar]
- 16.Venkatesan AM, Gu Y, Dos Santos O, Abe T, Agarwal A, Yang Y, Petersen PJ, Weiss WJ, Mansour TS, Nukaga M, Hujer AM, Bonomo RA, Knox JR. Structure-activity relationship of 6-methylidene penems bearing tricyclic heterocycles as broad-spectrum β-lactamase inhibitors: crystallographic structure showing unexpected binding of 1,4-thiazepinee intermediates. J Med Chem. 2004;47:6556–6568. doi: 10.1021/jm049680x. [DOI] [PubMed] [Google Scholar]
- 17.Bou G, Santillana E, Sheri A, Beceiro A, Smapson, et al. Design, synthesis and crystal structures of 6-alkylidene-2′-substituted penicillanic sulfones as potent inhibitors of Acinetobacter baumannii OXA-24 carbapenemase. J Am Chem Soc. 2010;132:13320–13331. doi: 10.1021/ja104092z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ke W, Pattanaik P, Bethel CR, Sheri A, Buynak JD, Bonomo RA, van den Akker F. Structures of SHV-1 β-lactamase with penem and penam sulfone inhibitors that form cyclic intermediates stabilizied by carbonyl conjugation. PLoS One. 2012;7:e49035. doi: 10.1371/journal.pone.0049035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodkey EA, Winkler ML, Bethel CR, Pagadala SR, Buynak JD, Bonomo RA, van den Akker F. Penam sulfones and β-lactamase inhibition: SA2–13 and the importance of the C2 side chain length and composition. PLoS One. 2014;9:e85892. doi: 10.1371/journal.pone.0085892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iland CN, Baines S. The effect of penicillin on the tubercle bacillus: tubercle penicillinase. J Pathol Bacteriol. 1949;61:329–335. [Google Scholar]
- 21.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 22.Flores AR, Parsons LM, Pavelka MS., Jr Genetic analysis of the β-lactamases of Mycobacterium tuberculosis and Mycobacterium smegmatis and susceptibility to β-lactam antibiotics. Microbiology. 2005;151:521–532. doi: 10.1099/mic.0.27629-0. [DOI] [PubMed] [Google Scholar]
- 23.Wang F, Cassidy C, Sacchettini JC. Crystal structure and activity studies of the Mycobacterium tuberculosis β-lactamase reveal its critical role in resistance to β-lactam antibiotics. Antimicrob Agents Chemother. 2006;50:2762–2771. doi: 10.1128/AAC.00320-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hugonnet JE, Blanchard JS. Irreversible Inhibition of the Mycobacterium tuberculosis β-lactamase by Clavulanate. Biochemistry. 2007;46:11998–12004. doi: 10.1021/bi701506h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tremblay L, Hugonnet JE, Blanchard JS. Structure of the Covalent Adduct formed between Mycobacterium tuberculosis β-Lactamase and Clavulanate. Biochemistry. 2008;47:5312–5316. doi: 10.1021/bi8001055. [DOI] [PubMed] [Google Scholar]
- 26.Cynamon MH, Palmer GS. In vitro activity of amoxicillin in combination with clavulanic acid against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1983;24:429–431. doi: 10.1128/aac.24.3.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goffin C, Ghuysen JM. Biochemistry and acomparative genomics of SxxK superfamily acyltranferases offer a clue to the mycobacterial paradox: presence of penicillin-suseptible targets proteins versus lack of efficiency of penicillin as a therapeutic agent. Microbiol Mol Biol Rev. 2002;66:702–738. doi: 10.1128/MMBR.66.4.702-738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hugonnet JE, Tremblay LW, Boshoff HI, Barry CE, 3rd, Blanchard JS. Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science. 2009;323:1215–1218. doi: 10.1126/science.1167498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Payen MC, De Wit S, Martin C, Sergysels R, Muylle I, Van Laethem Y, Clumeck N. Clinical use of the Meropenem-Clavunate combination for extensively drug-resistant tuberculosis. Int J Tuberc Lung Dis. 2012;16:558–560. doi: 10.5588/ijtld.11.0414. [DOI] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.Hazra S, Xu H, Blanchard JB. Tebipenem, a New Carbapenem Antibiotic is a Slow Substrate that Inhibits the β-Lactamase from Mycobacterium tuberculosis. Biochemistry. 2014;53:3671–4678. doi: 10.1021/bi500339j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Potterton E, Briggs P, Turkenburg M, Dodson E. A graphical user interface to the CCP4 program suite. Acta Crystallogr Sect D: Biol Crystallogr. 2003;59:1131–1137. doi: 10.1107/s0907444903008126. [DOI] [PubMed] [Google Scholar]
- 33.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 34.Pannu NS, Murshudov GN, Dodson EJ, Read RJ. Incorporation of prior phase information strengthens maximum-likelihood structure refinement. Acta Crystallogr Sect D: Biol Crystallogr. 1998;54:1285–1294. doi: 10.1107/s0907444998004119. [DOI] [PubMed] [Google Scholar]
- 35.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D: Biol Crystallogr. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 37.Tremblay LW, Fan F, Blanchard JS. Biochemical and Structural Characterization of Mycobacterium tuberculosis β-Lactamase (BlaC) with the Carbapenems Ertapenem and Doripenem. Biochemistry. 2010;49:3766–3773. doi: 10.1021/bi100232q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar P, Arora K, Lloyd JR, Lee IY, Nair V, Fischer E, Boshoff HI, Barry CE., 3rd Meropenem Inhibits the D,D-carboxypeptidase activity in Mycobacterium tuberculosis. Mol Microbiol. 2012;86:367–381. doi: 10.1111/j.1365-2958.2012.08199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cordillot M, Dubee V, Triboulet S, Dubost L, Marie A, Hugonnet JE, Arthur M, Mainardi JL. In vitro Cross-linking of Mycobacerium tuberculosis peptidoglycan by L,D-transpeptidases and Inactivation of these Enzymes by Meropenem. Antimicrob Agents Chemother. 2013;57:5940–5945. doi: 10.1128/AAC.01663-13. [DOI] [PMC free article] [PubMed] [Google Scholar]