

This CUGC intends to give guidance regarding molecular genetic testing in patients with thoracic aortic aneurysm and dissection (for definition see Loeys et al.1). It includes genes associated with non-syndromic and syndromic conditions. In cases of a strong clinical suspicion for a particular syndrome, it recommends testing the respective associated genes first. In cases without such suspicion, it recommends either a stepwise approach by Sanger sequencing or, if available, NGS-based procedures according to the ESHG recommendations (Matthijs et al.2) based on ‘core genes' and ‘additional genes'. The recommendations suggest that core gene lists are to be established by consensus among experts in the field. The ‘list must result in a ‘substantial contribution' to the quality of life of a patient, and hence the genes must be chosen with care; a two tier system would be acceptable, whereby some genes are scrutinized more in detail (in other words: with a more complete coverage) than others; the list must not inflict with the efficiency of a service, that is, overzealous testing is not helpful; the use of core gene panels must lead to better diagnosis of the group of disorders, if not it lacks clinical utility.'2 Additional genes (those with a lower disease contribution) are optional components of the panel.
1. Disease characteristics
1.1 Name of the disease (synonyms)
See Table 1 column 1—‘Disease'.
Table 1. Overview of diseases and genes associated with thoracic aortic aneurysm.
| Disease | OMIM# of disease | Orpha number of disease | Associated gene(s) | OMIM# of associated gene(s) |
|---|---|---|---|---|
| Aortic aneurysm, familial thoracic (AAT); Aneurysm, thoracic aortic; aortic dissection, familial; thoracic aortic aneurysm and dissection, familial | 615436 (AAT8) | 91387 | PRKG1 | 176894 |
| 613780 (AAT7) | 91387 | MYLK | 600922 | |
| 611788 (AAT6) | 91387 | ACTA2 | 102620 | |
| 132900 (AAT4) | 91387 | MYH11 | 160745 | |
| Alport syndrome, X-linked (ATSa) | 301050 | 88917 | COL4A5 | 303630 |
| Aortic valve disease 1 (AOVD1); bicuspid aortic valve | 109730 | 402075 | NOTCH1 | 190198 |
| Arterial tortuosity syndrome (ATSa) | 208050 | 3342 | SLC2A10 | 606145 |
| Contractural arachnodactyly, congenital, Beals syndrome (CCA) | 121050 | 115 | FBN2 | 612570 |
| Cutis laxa, autosomal dominant 1 (ADCL1) | 123700 | 90348 | ELN | 130160 |
| Cutis laxa, autosomal recessive, type 1B (ARCL1B) | 614437 | 90349 | EFEMP2 | 604633 |
| Ehlers–Danlos syndrome, classical type/type I (EDS I) | 130000 | 90309 | COL1A1 | 120150 |
| COL5A1 | 120215 | |||
| COL5A2 | 120190 | |||
| Ehlers–Danlos syndrome, classical type/type II (EDS II) | 130010 | 90318 | COL5A1 | 120215 |
| 130010 | 90318 | COL5A2 | 120190 | |
| Ehlers–Danlos syndrome, vascular type/type IV (EDS IV) | 130050 | 286 | COL3A1 | 120180 |
| Ehlers–Danlos syndrome, kyphoskoliotic type/type VI (EDS VI) | 225400 | 1900 | PLOD1 | 153454 |
| Ehlers–Danlos syndrome, arthrochalasic type/type VIIA (EDS VIIA) | 130060 | 1899 | COL1A1 | 120150 |
| Familial thoracic aortic aneurysm and aortic dissection | 91387 | TGFB2 | 190220 | |
| Heterotopia, periventricular, Ehlers–Danlos variant (PVNH4) | 300537 | 82004 | FLNA | 300017 |
| Juvenile polyposis/hereditary hemorrhagic telangiectasia syndrome (JPHT) | 175050 | 2929 | SMAD4 | 600993 |
| Loeys–Dietz syndrome type 1, Furlong syndrome (LDS1/FS/AAT5) | 609192 | 60030 | TGFBR1 | 190181 |
| Loeys–Dietz syndrome type 2, Marfan syndrome type 2 (LDS2/MFS2/AAT3) | 610168 | 60030 | TGFBR2 | 190182 |
| Loeys–Dietz syndrome type 3, Aneurysm osteoarthritis syndrome (LDS3/AOS) | 613795 | 284984 | SMAD3 | 603109 |
| Loeys–Dietz syndrome type 4 (LDS4) | 614816 | TGFB2 | 190220 | |
| Marfan syndrome (MFS) | 154700 | 558 | FBN1 | 134797 |
| Moyamoya disease 5 (MYMY5) | 614042 | 2573 | ACTA2 | 102620 |
| Shprintzen–Goldberg craniosynostosis syndrome (SGS) | 182212 | 2462 | SKI | 164780 |
Note: the nomenclature of diseases associated with thoracic aortic aneurysm is in part controversial.15, 16 This CUGC does not attempt to resolve these controversies. In this table, both OMIM and Orpha numbers are given together with their alternative disease designations in the first column.
ATS is used in OMIM as a symbol for both conditions.
1.2 OMIM# of the disease
See Table 1 column 2—‘OMIM# of Disease', and Table 2.
Table 2. Genes associated with thoracic aortic aneurysm not yet given an OMIM or Orpha number.
1.3 Name of the analysed genes or DNA/chromosome segments:
1.3.1 Core genes (irrespective if being tested by Sanger sequencing or next-generation sequencing)
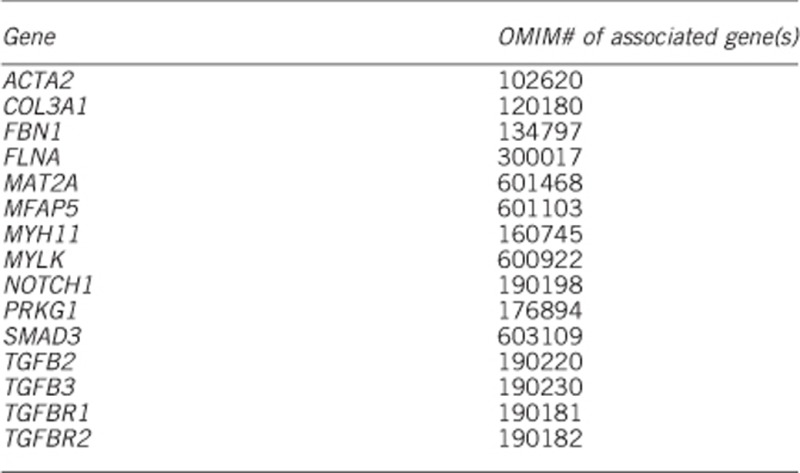
1.3.2 Additional genes (if tested by next-generation sequencing, including whole-exome/genome sequencing and panel sequencing)
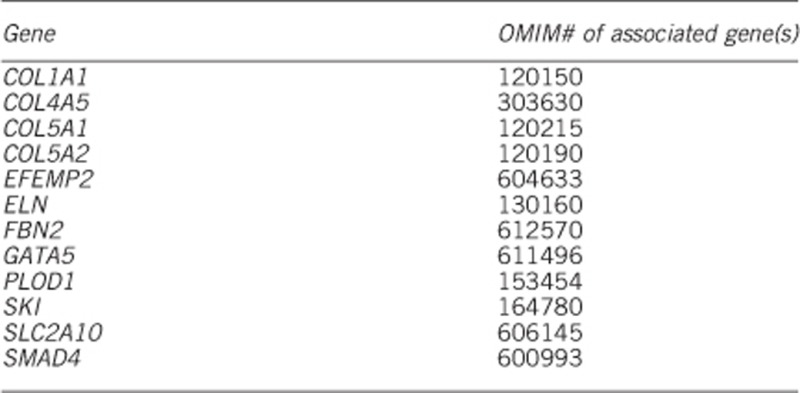
1.4 Mutational spectrum:
(Please note the EJHG instructions for authors http://mts-ejhg.nature.com/cgi-bin/main.plex?form_type=display_auth_instructions regarding sequence variants and genetic databases.)
All types of variants have been reported3 (missense, nonsense, splice site, small and large deletions/insertions). Many variants are listed in the Human Gene Mutation Database (http://www.hgmd.org/) and in ClinVar.4 The ‘Locus Specific Mutation Database' from HGVS gives an overview of gene-specific mutation databases, for example, FBN1, FBN2, TGFBR1, TGFBR2, ACTA2, SMAD3, MYH11 and MYLK variants are also registered in The Universal Mutation Database (www.umd.be). In general, there are no frequent disease-causing mutations or hot spots for disease-causing mutations in the vast majority of the genes. Causative mutations are distributed throughout the genes. However, some trends are observed for disease-causing mutations in specific genes (for example, exons encoding intracellular domains of TGFBR1 and TGFBR2, and recurrent mutations in PRKG1).
SNPs or rare variants are listed in the dbSNP Database (http://www.ncbi.nlm.nih.gov/snp/), the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu/EVS/) and in the Exome Aggregation Consortium (http://exac.broadinstitute.org/). Please note that the above-mentioned databases include pathogenic mutations.
1.5 Analytical validation
Sequencing of both DNA strands. Disease-causing mutations should be confirmed using genomic DNA from a new extraction. Causative mutations found with next-generation sequencing should be verified using Sanger sequencing or other specific molecular methods (eg, PCR digest); for further details, see the Eurogentest Guideline.2
1.6 Estimated frequency of the disease (Incidence at birth (‘birth prevalence') or population prevalence)
If known to be variable between ethnic groups, please report):
Estimated population prevalence ranges between 1:5000 and 1:4 000 000 in adults depending on the occurrence of an isolated thoracic aortic aneurysm or as a symptom of a syndromic disorder, excluding non-genetic causes, for example, atherosclerosis.
1.7 Diagnostic setting:

Comment: panel diagnostic or WES/WGS filtering should be preferred if clinical signs of a specific syndrome are missing, for example, in young patients with an emerging syndrome. But also in older persons, a specific syndrome can have a widely variable expression.
Time constraints, for example, in pregnancy, is another reason to choose panel diagnostic, if there is a request for prenatal diagnosis (rarely) or if the modus of delivery is dependent on a specific condition of the child.
2. Test characteristics
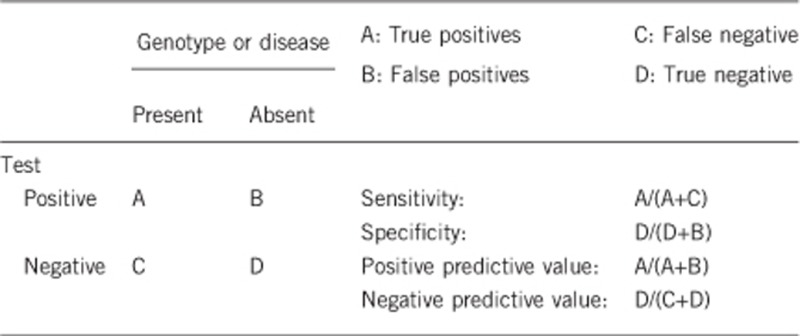
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present in the analyte)
2.1.1 if tested by conventional Sanger sequencing
Less than 100%.
The proportion is likely <100%, because primers may be localized on sequences containing SNPs or rare variants, which results in a preferential amplification of one allele (allele drop out). A supplementary deletion/duplication diagnostic test should be performed for genes with a known proportion of large genomic deletions/duplications.
2.1.2 if tested by next-generation sequencing
Less than 100%.
The proportion is likely <100%, because there might be disease-causing mutations in regions that could not be enriched and/or sequenced by NGS due to suboptimal coverage of some regions of interest with this technology but depending on NGS strategy. If amplicon-based enrichment strategies are being used, primers may be localized on SNPs or rare variants, which results in a preferential amplification of one allele. In patients with a highly suggestive phenotype in whom initial testing for specific gene alterations proves negative, a supplementary deletion/duplication diagnostic test should be performed for genes with a known proportion of large genomic deletions/duplications.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
2.2.1 if tested by conventional Sanger sequencing
Nearly 100%. False positives may at the most arise due to misinterpretation of rare polymorphic variants.
2.2.2 if tested by next-generation sequencing
Less than 100%. The risk of false positives due to misinterpretation of rare polymorphic variants may even be higher compared with Sanger sequencing because of the greater number of analysed genes.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases, a general statement should be given, even if a quantification can only be made case by case.
2.3.1 if tested by conventional Sanger sequencing
In about 20% of patients presenting with familial AAT, a disease-causing mutation is found5 (eg, ACTA2: 4–14%, TGFBR2: 4%, SMAD3: 2%, TGFBR1: 1%, MYH11: 1%, MYLK: 1%, TGFB2, MAT2A, PRKG1, MFAP5).
In syndromic forms of heritable thoracic aortic aneurysm clinical sensitivity is highly dependent on fulfillment of specific clinical criteria for a given entity.
2.3.2 if tested by next-generation sequencing
See 2.3.1.
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases, a general statement should be given, even if a quantification can only be made case by case.
2.4.1 if tested by conventional Sanger sequencing
Unknown.
2.4.2 if tested by Next-generation sequencing
See 2.4.1.
2.5 Positive clinical predictive value (life time risk to develop the disease if the test is positive)
Dependent on clinical subtype, typically >50%.
2.6 Negative clinical predictive value (Probability not to develop the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity must need to be considered.
Index case in that family had been tested and a causative mutation identified:
Nearly 100%. If the non-affected relative is not carrier of an identified disease-causing mutation, he or she has no increased risk, except a small risk related to the prevalence of the disease in the general population.
Index case in that family had not been tested or no causative mutation identified:
Up to 19% of patients with TAAD without a known genetic syndrome have a first-degree relative with TAAD.6 In syndromic forms of heritable thoracic aortic aneurysm, the negative clinical predictive value corresponds to the detection rate in the known genes mutated in the different diseases.7
3. Clinical utility
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.8 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
A clinically affected person has to be regularly examined by echocardiogram, CT or MR imaging.8
Alternative diagnostic methods might not capture early detection of none cardiovascular symptoms in syndromic cases.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
No data available.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive Setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.8 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe), see 3.1.4.
If the test result is negative (please describe), if the causative mutation is identified in the index case and not in the clinically unaffected proband, regular examinations are not necessary unless otherwise indicated.
Follow-up is recommended if the disease-causing mutation could not be identified. In contrast, follow-up is dispensable in a family member, if a familial mutation has been excluded.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
That person should avoid sport activity/professional activity with a high static burden, competitive sports and body contact sports.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.8 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes, if a causative mutation could be identified in the index patient.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
If a disease-causing mutation is identified in the index patient, family members can be tested (cascade screening). Test negative family members can be released from otherwise indicated diagnostic monitoring.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes.
3.4 Prenatal diagnosis
(To be answered if in 1.8 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes.
4. If applicable, further consequences of testing
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe).
The genetic diagnostics contributes substantially to the classification of a heritable thoracic aortic aneurysm to a syndromic or non-syndromic entity.14 Genetic testing gives insight to inheritance pattern and allows reasonable genetic counseling. If a causative mutation is identified in a gene also responsible for a syndromic form of TAAD, further clinical investigations regarding symptoms of this specific syndrome should be performed. In some cases it might be justified to start medical treatment at an earlier stage.
Acknowledgments
CUGCs were developed within a work package of EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469). Currently, the initiative is supported by the European Society of Human Genetics.
The authors declare no conflict of interest.
References
- Loeys BL, Dietz HC, Braverman AC et al: The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010; 47: 476–485. [DOI] [PubMed] [Google Scholar]
- Matthijs G, Souche E, Alders M et al: Guidelines for diagnostic next generation sequencing. Eur J Hum Genet 2016; 24: 2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner-Ellis JP, Aldubayan SH, Hernandez AL et al: The spectrum of FBN1, TGFβR1, TGFβR2 and ACTA2 variants in 594 individuals with suspected Marfan Syndrome, Loeys-Dietz Syndrome or Thoracic Aortic Aneurysms and Dissections (TAAD). Mol Genet Metab 2014; 112: 171–176. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR et al: ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014; 42: D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milewicz DM, Regalado E. Thoracic aortic aneurysms and aortic dissections. In: Pagon RA, Adam MP, Ardinger HH (eds): Gene Reviews. Seattle, WA, USA: University of Washington, 1993–2014. [Google Scholar]
- Albornoz G, Coady MA, Roberts M et al: Familial thoracic aortic aneurysms and dissections—incidence, modes of inheritance, and phenotypic patterns. Ann Thorac Surg 2006; 82: 1400–1405. [DOI] [PubMed] [Google Scholar]
- Malfait F, De Paepe A: The Ehlers-Danlos syndrome. Adv Exp Med Biol 2014; 802: 129–143. [DOI] [PubMed] [Google Scholar]
- Chu LC, Johnson PT, Dietz HC, Fishman EK: CT angiographic evaluation of genetic vascular disease: role in detection, staging, and management of complex vascular pathologic conditions. AJR Am J Roentgenol 2014; 202: 1120–1129. [DOI] [PubMed] [Google Scholar]
- Hiratzka LF, Bakris GL, Beckman JA et al: 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010; 121: e266–e369. [DOI] [PubMed] [Google Scholar]
- Erbel R, Aboyans V, Boileau C et al: 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: Document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The Task Force for the Diagnosis and Treatment of Aortic Diseases of the European Society of Cardiology (ESC). Eur Heart J 2014; 35: 2873–2926. [DOI] [PubMed] [Google Scholar]
- Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH: Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet Med 2014; 16: 881–888. [DOI] [PubMed] [Google Scholar]
- Paterick TE, Humphries JA, Ammar KA et al: Aortopathies: etiologies, genetics, differential diagnosis, prognosis and management. Am J Med 2013; 126: 670–678. [DOI] [PubMed] [Google Scholar]
- Schoenhoff F, Schmidli J, Czerny M, Carrel TP: Management of aortic aneurysms in patients with connective tissue disease. J Cardiovasc Surg (Torino) 2013; 54 (1 Suppl 1): 125–134. [PubMed] [Google Scholar]
- Pyeritz RE: Heritable thoracic aortic disorders. Curr Opin Cardiol 2014; 29: 97–102. [DOI] [PubMed] [Google Scholar]
- Pyeritz R, Jondeau G, Moran R et al: Loeys-Dietz syndrome is a specific phenotype and not a concomitant of any mutation in a gene involved in TGF-β signaling. Genet Med 2014; 16: 641–642. [DOI] [PubMed] [Google Scholar]
- MacCarrick G, Loeys B, Dietz HC 3rd: Response to Pyeritz et al. Genet Med 2014; 16: 642–644. [DOI] [PubMed] [Google Scholar]
- Padang R, Bagnall RD, Richmond DR, Bannon PG, Semsarian C: Rare non-synonymous variations in the transcriptional activation domains of GATA5 in bicuspid aortic valve disease. J Mol Cell Cardiol 2012; 53: 277–281. [DOI] [PubMed] [Google Scholar]
- Guo DC, Gong L, Regalado ES et al: MAT2A mutations predispose individuals to thoracic aortic aneurysms. Am J Hum Genet 2015; 96: 170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbier M, Gross MS, Aubart M et al: mfap5 loss-of-function mutations underscore the involvement of matrix alteration in the pathogenesis of familial thoracic aortic aneurysms and dissections. Am J Hum Genet 2014; 95: 736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoli-Avella AM, Gillis E, Morisaki H et al: Mutations in a TGFβ ligand, TGFB3, cause syndromic aortic aneurysms and dissections. JACC 2015; 65: 1324–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]