

Abstract
Osteoclasts are required for bone resorption. A new study in mice indicates that osteoclast differentiation is stabilized by DNA methylation at Irf8 (encoding interferon regulatory factor 8) mediated by DNA methyltransferase 3a (Dnmt3a), which suppresses Irf8 gene expression. The activity of Dnmt3 in osteoclasts requires elevated oxidative metabolism.
Bone resorption is the osteoclast-mediated degradation and removal of mineralized bone matrix, and it is required for physiological bone remodeling. Excessive bone resorption that is not balanced by bone formation results in bone loss in diseases such as osteoporosis, leading to weak bones and increased fractures. Osteoclasts are cells that are derived from myeloid bone marrow precursor cells that migrate from the bone marrow into specialized bone-resorbing niches. In these niches, osteoclast precursors respond to the stromal cell–expressed cytokine RANKL and bone-derived signals to differentiate into multi-nucleated osteoclasts that are cells efficient in bone resportion1. RANKL signaling is carried out via MAPK-AP-1, NF-κB, and calcium-regulated pathways that converge to induce and activate NFATc1, a transcription factor that induces the expression of genes important for cell fusion and bone resorption, and that coordinates the osteoclast differentiation program1. More recently, the importance of RANKL-induced downregulation of transcription factors that repress osteoclast differentiation, such as IRF8, has become better understood2,3.
In the context of cell differentiation, epigenetics refers to developmentally or environmentally induced modifications to DNA or chromatin that regulate gene expression in a tissue- and context-specific manner. Epigenetic processes such as DNA methylation can be stable and are known to be involved in regulation of differentiation in order to maintain gene expression patterns. Furthermore, regulation of epigenetic mechanisms by cellular metabolic processes is an emerging theme in biological research4. Very little is known about epigenetic mechanisms that regulate commitment to the osteoclast- differentiation pathway, although RANKL-induced changes in histone modifications at the Nfatc1 locus that may stabilize Nfatc1 expression have been reported5,6. The role of RANKL-induced increases in cellular oxidative metabolism has also not been clarified. In this issue of Nature Medicine, Nishikawa et al.7 identify an intriguing link between osteoclast oxidative metabolism and epigenetic regulation by DNA methylation.
To investigate the potential role of epigenetic regulators of osteoclast differentiation, the authors carried out an in vitro analysis of the transcriptome of mouse osteoclast precursors stimulated with RANKL7. Expression of the de novo DNA methyltransferase Dnmt3a was induced by RANKL during osteoclast differentiation. Dnmt3a is known to be expressed during developmental processes, and in this manner it can stably silence gene expression. To test the role of this epigenetic regulator in osteoclast differentiation, Nishikawa et al.7 inhibited this enzyme with a panel of pharmacological inhibitors, which suggested that de novo DNA methylation is indeed necessary for this process. This conclusion was supported by cell culture experiments that the authors carried out in which Dnmt3a methyltransferase domain mutants were unable to promote osteoclast differentiation7. An important role for Dnmt3a in osteoclast differentiation was also shown by reducing levels of Dnmt3a in vitro with RNAi7. Conditional osteoclast lineage–specific gene deletion in vivo further indicated that Dnmt3a is important in osteoclast differentiation and bone resorption under homeostatic conditions. Notably, in a mouse model of osteoporosis induced by ovariectomy (OVX), the authors showed that Dnmt3a has a role in pathological bone loss7.
To identify genes that are repressed by Dnmt3a-mediated DNA methylation, Nishikawa et al. 7 then carried out a genome-wide approach using transcriptomics coupled with methyl-CpG binding domain (MBD) sequencing—a technique analyzing genome-wide DNA methylation patterns—to identify genes whose expression was repressed and that were methylated after RANKL stimulation in cell culture. This approach indicated that Irf8, a negative regulator of osteoclastogenesis2, has increased methylation at CpG sites downstream of the gene body and concomitant altered gene expression (Fig. 1).
Figure 1.
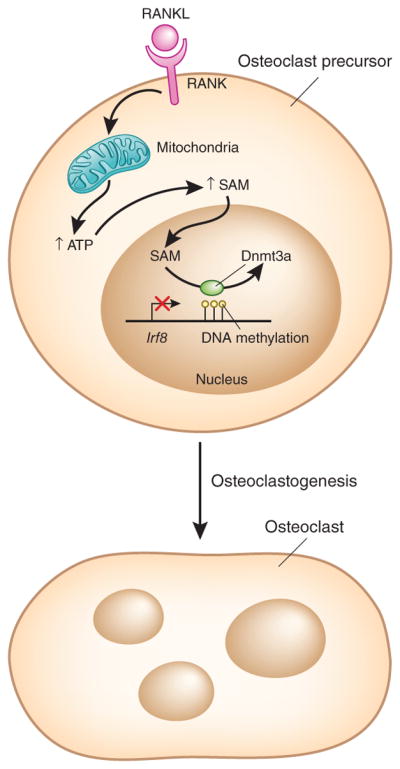
The role of DNA methylation in osteoclast differentiation. RANKL is a regulator of osteoclastogenesis. Nishikawa et al.7 show in mouse osteoclast precursor cells that it induces the expression of the de novo DNA methyltransferase Dnmt3a and increases oxidative metabolism to provide the SAM substrate for Dnmt3a. After RANKL-induced signaling, the expression of Irf8, a negative regulator of osteoclastogenesis is repressed. Dnmt3a maintains this repression by methylating CpG motifs in putative regulatory elements downstream of the Irf8 gene body, thereby promoting osteoclast differentiation and bone resorption. RANK, receptor activator of NF-κB.
The epigenetic silencing of a negative regulator to stabilize osteoclast differentiation introduces a new concept into the myeloid differentiation field. Importantly, DNA methylation occurred after Irf8 expression had already been downregulated, approximately 12 h after RANKL stimulation. Thus, it is likely that Dnmt3a stabilized Irf8 expression at a low level at an early phase of osteoclast precursor differentiation into osteoclasts, which typically requires 3–4 d of RANKL stimulation in cell culture. Signals that initially downregulate Irf8 expression after RANKL stimulation remain to be identified but are probably related to Notch-RBP-J signaling8 which also regulates osteoclastogenesis. Downregulation of Irf8 expression may also be mediated by chromatin- based mechanisms, as changes in histone methylation in Irf8 that can alter gene expression and potentially recruit Dnmt3a were observed in this study7. This work further supports the notion that IRF8 is a key negative regulator of osteoclast phenotype that needs to be epigenetically silenced for osteoclastogenesis to proceed.
Another striking finding of the study, on the basis of Dnmt3a overexpression in osteoclast precursors, was that Dnmt3a expression alone was not sufficient to suppress Irf8 or promote osteoclastogenesis. The methyl donor for Dnmt3a is S-adenosylmethionine (SAM), and hence the authors analyzed its concentrations7. They found that SAM levels were significantly increased after RANKL stimulation, and that increased SAM concentrations augmented Dnmt3a activity and methylation at the Irf8 locus7. SAM is generated from methionine and ATP, and Nishikawa et al.7 attributed the increase in SAM to increased cellular ATP levels. They then linked this to RANKL-induced increased flux through the tricarboxylic acid cycle and oxidative phosphorylation, which is in accord with increased mitochondrial biogenesis previously observed in osteoclastogenesis7,9. This change in cell respiration resulted in elevated concentrations of ATP, leading to increased SAM production7. A role for cellular metabolism in regulating the production of substrates for epigenetic regulators has been established4, and the current study supports a RANKL-induced metabolic-epigenetic coupling in which mitochondrial biogenesis and efficient ATP generation are linked to de novo DNA methylation by Dnmt3a.
This study also has important translational and therapeutic implications, as treatment of mice with the theaflavin derivative TF-3, which selectively inhibits Dnmt3 methyltransferase activity, abrogated bone loss in the OVX model of osteoporosis7. Together with two recent studies showing that inhibition of BET bromodomain-containing epigenetic regulators by the small-molecule compounds JQ-1 and IBET-151 suppresses pathological bone remodeling in OVX, inflammatory arthritis, and tumor models10,11, these results support a role for epigenetic therapy in bone diseases. Although these inhibitors did not exhibit apparent toxicity, given the broad expression and importance of their targets in fundamental cell processes, a thorough examination of side effects and off-target effects is warranted.
The work by Nishikawa et al.7 establishes Dnmt3a-mediated DNA methylation and silencing of Irf8 to be an important epigenetic mechanism that promotes osteoclastogenesis. Epigenetic mechanisms such as DNA methylation can thus potentially stabilize components of myeloid lineage differentiation to make them difficult to reverse by environmental cues. Such ‘irreversible’ phenotypes could be important for committed functions such as bone resorption. This work also shows that epigenetic regulation of osteoclast differentiation is linked to cell metabolism and is susceptible to therapeutic targeting. It will be necessary to identify additional epigenetic mechanisms that regulate osteoclastogenesis, such as tissue-specific remodeling of the enhancer landscape, which has an important role in myeloid cell differentiation12,13. Future work in this area to identify key epigenetic regulators and osteoclast-specific enhancers, as well as the signaling pathways, metabolic programs and transcription factors that regulate them, will dramatically enhance a new therapeutic space for drug development to treat pathological bone resorption.
Footnotes
COMPETING FINANCIAL INTERESTS
The author declares no competing financial interests.
References
- 1.Nakashima T, Hayashi M, Takayanagi H. Trends Endocrinol Metab. 2012;23:582–590. doi: 10.1016/j.tem.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Zhao B, et al. Nat Med. 2009;15:1066–1071. doi: 10.1038/nm.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao B, Ivashkiv LB. Arthritis Res Ther. 2011;13:234. doi: 10.1186/ar3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaelin WG, Jr, McKnight SL. Cell. 2013;153:56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Asagiri M, et al. J Exp Med. 2005;202:1261–1269. doi: 10.1084/jem.20051150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yasui T, et al. J Bone Miner Res. 2011;26:2665–2671. doi: 10.1002/jbmr.464. [DOI] [PubMed] [Google Scholar]
- 7.Nishikawa K, et al. Nat Med. 2015;21:281–287. doi: 10.1038/nm.3774. [DOI] [PubMed] [Google Scholar]
- 8.Zhao B, Grimes SN, Li S, Hu X, Ivashkiv LB. J Exp Med. 2012;209:319–334. doi: 10.1084/jem.20111566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishii KA, et al. Nat Med. 2009;15:259–266. doi: 10.1038/nm.1910. [DOI] [PubMed] [Google Scholar]
- 10.Lamoureux F, et al. Nat Commun. 2014;5:3511. doi: 10.1038/ncomms4511. [DOI] [PubMed] [Google Scholar]
- 11.Park-Min KH, et al. Nat Commun. 2014;5:5418. doi: 10.1038/ncomms6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gosselin D, et al. Cell. 2014;159:1327–1340. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lavin Y, et al. Cell. 2014;159:1312–1326. doi: 10.1016/j.cell.2014.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]