

Abstract
The development of new forms of treatment of advanced renal cell carcinoma over the past two decades has been primarily focused on targeting the VHL/HIF pathway. The recent identification of mutations of chromatin remodeling genes in clear cell renal carcinoma (RCC), of genomic heterogeneity and of a Warburg-like metabolic phenotype in advanced disease has had a profound effect on our understanding of the evolution of clear cell RCC and on potential approaches to personalized therapy. Early approaches to therapy for patients with advanced type 1 papillary renal cell carcinoma that have centered around the MET/HGF pathway will expand as more genomic information becomes available. Sporadic and familial Type 2 papillary renal cell carcinoma are characterized by enhanced aerobic glycolysis and share an antioxidant response phenotype. In fumarate hydratase-deficient RCC, fumarate-induced succination of KEAP1 activates Nrf2 signaling. CUL3 and Nrf2 mutations as well as an Nrf2 activation phenotype are found in sporadic type 2 papillary RCC. Therapeutic approaches designed to target the Nrf2 pathway as well as to impair blood flow and glucose delivery in these cancers that are highly dependent on a robust tumor vasculature and on ready availability of glucose for energy production and glycolysis are in development.
Background
Renal cell carcinoma affects over 270,000 individuals annually worldwide and there are nearly 120,000 deaths each year due to this disease (1). Patients with relatively small, localized (T1) disease treated surgically have excellent five and ten year survival rates (ninety five percent). However, in patients with advanced disease (T4 or M1), the two year survival was less than twenty percent before the advent of targeted agents directed against the HIF/VEGF pathway. Our ability to better manage patients with advanced disease has been largely driven by a better understanding of the genetic and biochemical alterations underlying these tumors. Over the past two decades, it has become clear that renal cell carcinoma is not a single disease but is made up of a number of different types of cancer that occur in this organ, each with a different histology, a different clinical course, responding differently to therapy and caused by different genes. The morphologic classification of RCC is on its way to becoming obsolete as we discover the genomic and molecular drivers of these diseases. Much of what has been learned about the genetic basis of renal cell carcinoma (RCC) has come from study of the inherited forms of renal cell carcinoma, including von Hippel-Lindau (clear cell RCC), hereditary papillary renal carcinoma (type 1 papillary RCC), and hereditary leiomyomatosis renal cell carcinoma (type 2 papillary RCC) (2, 3). Over the past decade, significant progress has been made in the development of targeted therapeutic approaches for patients with advanced renal cell carcinoma (4). However, despite the advent of these new agents, most patients with advanced renal cell carcinoma progress on therapy and many will eventually die of this disease.
Clear cell renal cell carcinoma is the most common type of renal cell carcinoma and accounts for 75% of cases. The VHL tumor suppressor gene, located on the short arm of chromosome 3, is the gene for the inherited form of clear cell renal cell carcinoma associated with von Hippel-Lindau (5) and has been found to be mutated or methylated in a high percentage of tumors (~90%) from patients with clear cell renal cell carcinoma (6, 7). The VHL protein forms a complex with elongin C, elongin B and CUL2 to target the hypoxia inducible factors, HIF1α and HIF2α for ubiquitin-mediated degradation (8–10). This is an oxygen sensing process; in normoxia HIF prolyl hydroxylase (PHD) transfers hydroxyl groups to two prolines in HIF which enables the VHL complex to target and degrade HIF. In hypoxia, PHD does not hydroxlylate HIF, the VHL complex cannot target and degrade HIF, and HIF accumulates. HIF1α and HIF2α are transcription factors that regulate the transcription of a number of hypoxia responsive genes, such as vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF), and glucose transporters GLUT1 and GLUT4. Understanding the VHL/HIF pathway has provided the foundation for the development of a number of targeted therapeutic approaches for the treatment of patients with advanced clear cell renal cell carcinoma, leading to the development and FDA approval of several agents which target this pathway. Temsorilimus and everolimus, which target the mTOR pathway, have also been approved for advanced RCC. While these agents have had remarkable clinical effect, up to a 45% response rate and increased progression free survival rates, virtually all eventually develop progressive disease (11).
Papillary renal cell carcinoma, which accounts for 15% of renal cell carcinoma, is made up of a heterogeneous group of cancer that encompasses multiple histologic subtypes, including type 1 papillary RCC and type 2 papillary RCC. Although inhibition of the VEGF or mTOR signaling pathways has led to some clinical benefits, the effect is marginal and the prognosis remains poor for patients with advanced disease (12).
On the Horizon
Clear cell renal cell carcinoma
Targeting the VHL/HIF pathway
Why is targeting the VHL/HIF pathway in clear cell RCC only partially successful clinically? Is it because we need to develop more effective ways to target this pathway or because we need to identify and target other genes associated with the development of this disease. The use of more target-specific, potent VEGFR inhibitors hasn’t been shown to improve efficacy but may be associated with a better toxicity profile. Concurrent inhibition of VEGFR and MET, Tie2, FGFR and mTOR pathways are being evaluated; however, complete remissions and long-term responses are rare with these strategies (11). Functional studies have long suggested that HIF2α (versus HIF1α) is the critical downstream target of VHL (13, 14) and that inhibition of HIF2α can suppress the ability of a VHL −/− tumor to form tumors in xenograft models (15, 16) and HIF2α single nucleotide polymorphisms have been linked to an increased risk of developing renal cell carcinoma (17, 18). Genetic and functional studies have implicated HIF1α as a chromosome 14q kidney cancer suppressor gene (19) and chromosome 14q loss has been found to be associated with poor prognosis (20). A number of strategies have been developed to target HIF2α. The Molecular Targets Laboratory at the National Cancer Institute has isolated a number of agents which have been shown to inhibit HIF2α transcriptional activity which will need to be further studied in in-vitro and in-vivo models to determine specificity and effectiveness (21–24). Zimmer et al. have reported small molecule inhibitors of HIF2α translation based on enhancing the binding activity of IRP1 to the iron responsive element (IRE) in the 5′-untranslated region of the HIF2α message (25, 26). Utilizing a similar approach, tempol, a stable nitroxide that activates IRP1 towards IRE binding, has been shown to decrease HIF2α and its downstream targets in VHL −/− RCC cell line models (27). The role of this agent alone or in combination with other anti-tumor agents will need to be evaluated in in-vitro and in-vivo models. Additional approaches involving autophagic degradation of HIF2α (28), targeting proteosomal degradation of VHL missense mutations (29), and targeting the hypoxia associated factor (HAF)-mediated regulation of HIF2α-dependent transcription during hypoxia are being evaluated (30, 31).
Chromatin remodeling gene mutations
Dalgliesh et al. opened a new chapter in our understanding of the genetic basis of clear cell RCC and the role of systematic screens to determine genomic architecture when they reported the identification of inactivating mutations of genes involved in histone modification, such as the histone H3 lysine 36 methyltransferase SETD2, and the histone H3 lysine 4 demethylase KDM5C (32). This was followed shortly by the identification of truncating mutations of the SWI/SNF chromatin remodeling complex gene, PBRM1, and BAP1, a deubiquitase that is the catalytic subunit of the Polycomb repressive deubiquitinase complex, in clear cell RCC, highlighting the fundamental role of aberrant chromatin biology in this disease (33, 34). The Cancer Genome Atlas (TCGA) performed a comprehensive molecular characterization of more than 400 clear cell renal tumors and identified 19 significantly mutated genes, including VHL and other chromosome 3 genes such as PBRM1, SETD2 and BAP1, as well as KDM5C, PTEN, MTOR, TP53 and PIK3CA. Mutations in the chromatin regulators, SETD2, BAP1 and PBRM1 were each differentially associated with altered expression patterns with a distinct set of down stream effects, reflecting different roles for chromatin remodeling in the transcriptome (35). A number of findings have highlighted the importance of driver mutations of chromatin remodeling genes in ccRCC. In large studies somatic mutations of BAP1 have been correlated with decreased overall survival and a straightforward immunohistochemical assay has been developed to assess BAP1 levels (7, 35–38). In addition, germline BAP1 mutations have been found to be the driver mutations in families characterized by an increased incidence of early onset, bilateral, multifocal clear cell renal carcinoma (39, 40). Intensive efforts are currently underway to determine the functional consequences of inactivation of chromatin remodeling genes in clear cell renal cell carcinoma to provide the foundation for the development of effective forms of therapy targeting these gene pathways. Simon et al. have recently identified alterations in chromatin organization and transcript profiles in ccRCC associated with SETD2 mutations in a large cohort of primary human kidney tumors (41) as a first step in developing actionable targets in SETD2-deficient clear cell RCC. The findings that SETD2 has been shown to be required for DNA double-strand break repair and activation of the p53-mediated checkpoint (42), that SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability, (43) and that cells lacking SETD2 activity display microsatellite instability and an elevated spontaneous mutation frequency, characteristic of DNA mismatch repair-deficient cells (44) opens the door to therapeutic approaches targeting genomic instability in SETD2-deficient renal cell carcinomas.
Genomic heterogeneity in clear cell RCC
In studies that have profound implications for both understanding clear cell RCC tumor evolution as well as for the development of effective strategies for therapy of this disease, Gerlinger et al. performed multi-region exome sequencing (M-seq) in large tumors from ten patients (all except two of whom presented with metastatic disease) and identified pronounced intratumoral heterogeneity (45, 46). VHL gene mutation, which was found in each sample, was considered to be a “truncal” mutation as it was uniformly present in all segments. PBRM1 was found to be mutated in a portion of the tumors and was also considered to be truncal in those subsets. Other driver mutations, found in the chromatin remodeling genes BAP1, KDM5C and SETD2, genes in the phosphoinositide 3-kinase (PI3K)-mTOR pathway (mTOR, PIK3CA, TSC2, PTEN) and TP53 were found only in “branch” segments of the tumors and each portion of the tumors were found to have a unique spectrum of branch mutations. As more portions of the tumors were samples, more tumor heterogeneity was identified; suggesting that even these studies underestimate the true extent of heterogeneity of driver mutations in clear cell RCC (45–47).
These studies raise a number of very fundamental questions. First, what is the best way to detect driver mutations in clear cell RCC? One approach could involve performing M-seq on the primary kidney tumor to identify druggable driver mutations for therapy. Another approach could involve performing M-seq on tumor material from metastatic tumors, considering that the tumor that has already metastasized might be most likely to contain the critical driver mutations. The second and more critical question is; how should we develop a therapy for a tumor with such genomic heterogeneity. One approach would be to continue to focus on developing more effective approaches to targeting the primary truncal mutation (the VHL pathway). When more information about the effect of mutations in chromatin remodeling genes such as SETD2 and BAP1 becomes available, approaches targeting these pathways will be evaluated.
Targeting the metabolic basis of clear cell RCC
Integrative analysis in the TCGA study revealed that high grade, high stage clear cell RCC from patient with poor survival demonstrated evidence of a metabolic shift, involving downregulation of genes involved in the TCA cycle, decreased PTEN and AMPK protein levels, upregulation of the glutamine transporter and pentose phosphate pathway genes and increased acetyl-CoA carboxylase protein (35). The finding of a metabolic shift involving dependence on the pentose phosphate shunt, decreased AMPK, increased glutamine transport and fatty acid production is consistent the findings of VHL-deficient clear cell renal cell carcinoma lines preferentially use glutamine dependent reductive carboxylation for lipid biosynthesis (48) which is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation (49). The balance between reductive carboxylation and glucose catabolism in VHL-deficient cells is coordinated by nicotinamide transhydrogenase (NNT) (50). The finding of a Warburg shift in clear cell RCC, consistent with that seen in fumarate hydratase-deficient renal cell carcinoma (51), provides a number of opportunities for targeted therapies (Fig. 1). A number of potential targets are available, including glutaminase, AMPK, FAS, and LDHA. Agents targeting AMPK, such as metformin and AICAR, have been shown to have anti-tumor activity in in-vitro RCC cell line models (52). Glutaminase inhibitors have shown efficacy in VHL-deficient RCC cell line models (49) and are currently being evaluated in clinical trials. Intense efforts are underway to identify an effective LDHA inhibitor appropriate for clinical use.
Figure 1. Potential therapeutic targets: clear cell renal cell carcinoma.
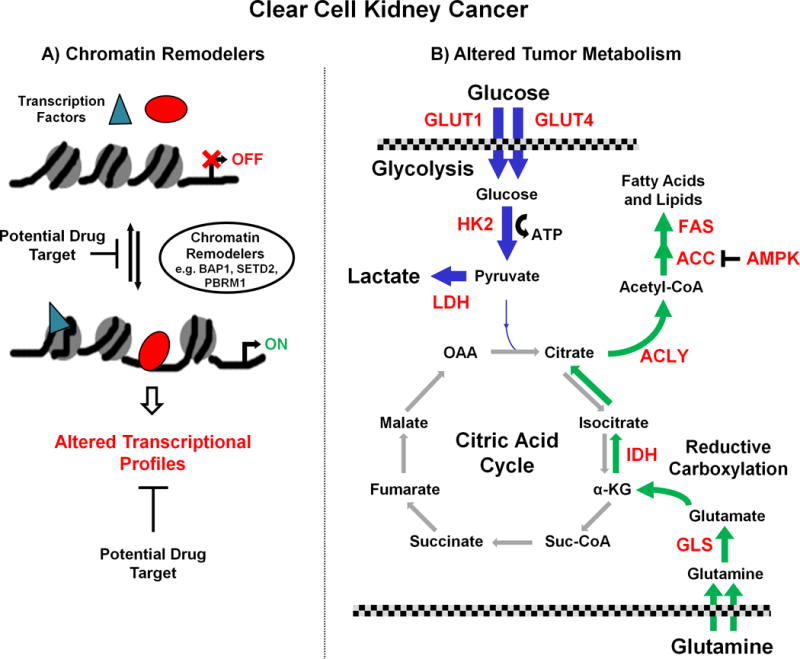
Potentially drug targetable pathways associated with clear cell RCC include chromatin remodeling and altered tumor metabolism. A, mutations in several chromatin remodeling genes such as PBRM1, SETD2 and BAP1 are common in clear cell RCC and result in altered transcription profiles due to the specific effects of loss of each of these genes. As knowledge of these pathways increases, potential therapies could target downstream transcriptional gain of function or loss of function events. B, higher grade, high stage low survival clear cell tumors are associated with a metabolic shift consistent with a suppression of oxidative phosphorylation and a subsequent dependence upon glycolysis for energy. This would result in a dependence on glucose for ATP and for the conversion of pyruvate to lactate for excretion while suppressing AMP-activated protein kinase (AMPK) to aid fatty acid production and growth. The enzymes and transporters involved in these processes, such as Hexokinase 2 (HK2), lactate dehydrogenase A (LDHA) and glucose transporters 1/4 (GLUT1/4), are potential targets (highlighted in red). Additionally, the increased need for fatty acids for growth requires an alternative carbon metabolite to enter the citric acid cycle to produce the necessary citrate. These tumors would increase their glutamine uptake and conversion to glutamate for entry into the Krebs cycle and both the glutaminase (GLS) and fatty acid synthesis enzymes, such as ATP citrate lyase (ACLY), acetyl-CoA carboxylase (ACC) and Fatty acid synthase (FAS), are potential therapeutic targets. Due to the suppression of the normal flow in the citric acid cycle the alternative reductive carboxylation pathway in used to convert α-ketoglutarate (α-KG) to citrate via the isocitrate dehydrogenase (IDH) enzymes that provide further potential targets.
Papillary renal cell carcinoma
Type 1 papillary renal cell carcinoma
Type 1 papillary renal cell carcinoma occurs in a sporadic, non-inherited as well as a hereditary form. Hereditary papillary kidney (HPRC) cancer is an autosomal dominant hereditary cancer syndrome in which affected individuals are at risk for the development bilateral, multifocal type 1 papillary renal cell carcinoma (53). The gene for hereditary papillary renal cell carcinoma, localized to the long arm of chromosome 7, was found to be the proto-oncogene MET (54). Activating mutations in the tyrosine kinase domain of MET are found in the germline of patients affected with HPRC (54, 55). Kidney tumors from patients affected with both HPRC and sporadic papillary renal cell carcinoma are characterized by polysomy of chromosome 7 and non-random duplication of the mutant MET allele have been found in tumors from patients affected with HPRC (56). Hereditary and sporadic type 1 papillary renal carcinoma share a distinct morphologic phenotype (57) and MET mutations have been found in 13% of sporadic type 1 papillary tumors.
The identification of oncogenic MET mutations in patients affected with hereditary papillary renal carcinoma and in a subset of patients with sporadic papillary RCC led the conduct of a multicenter phase II study of foretinib, an inhibitor of Met, VEGFR2, RON and AXL tyrosine kinases, in patients with sporadic as well as HPRC-associated papillary renal cell carcinoma. The overall response rate was 13.5%, with a median progression free survival of 9.3 months, which was considerably higher than that seen in historical controls treated with agents targeting mTOR or VEGFR pathways (58). In a subgroup analysis, 5/10 (50%) of HPRC patients with germline MET mutations were found to have a partial response to therapy, versus 5/57 (9%) of non-HPRC patients. Sufficient pathology review was not available to determine if type 1 papillary responded better than type 2 papillary RCC (58). A clinical trial evaluating the effect of INC280, an agent that selectively targets Met, in patients with papillary renal cell carcinoma. Trials are currently being planned to evaluate the effect of a number of agents which target Met in patients with both papillary and non-papillary renal cell carcinoma. The TCGA papillary renal cell carcinoma project is currently underway to provide more insights into the genetic characteristics of both type 1 as well as type 2 papillary renal cell carcinoma and will hopefully identify driver mutations that will guide the conduct of future trials in this disease.
Type 2 papillary renal cell carcinoma
Type 2 papillary kidney cancer is an aggressive and often lethal form of renal cell carcinoma (2, 3). Like type 1 papillary RCC, there is both a sporadic as well as familial form of type 2 papillary RCC. The hereditary form of type 2 papillary RCC occurs in Hereditary Leiomyomatosis and Renal Cell Carcinoma (HLRCC). HLRCC is an autosomal dominant hereditary cancer syndrome in which patients are at risk for the development of cutaneous and uterine leiomyomas and a very aggressive, lethal form of type 2 papillary renal cell carcinoma that has a propensity to spread when the primary tumor is very small (59–61). HLRCC is characterized by germline mutation of the gene for the Krebs cycle enzyme, fumarate hydratase (62).
Warburg model of cancer
In fumarate hydratase (FH)-deficient renal cell carcinoma oxidative phosphorylation is impaired and the cells undergo a shift to aerobic glycolysis, consistent with the Warburg effect (63, 64). FH-deficient renal cell carcinoma cells are unusually dependent on glucose for proliferation and glycolysis to generate the increased ATP needed for rapid proliferation (65). In FH-deficient RCC, fumarate accumulates and inhibits HIF prolyl hydroxylase (PHD), resulting in accumulation of HIF1 and increased transcription of its targets such as VEGF and GLUT1, which lead to the increased vascularity and glucose transport needed to provide increased nutrients for rapid growth and proliferation and for ATP production needed in cells characterized by a decrease in oxidative phosphorylation (Fig. 2) (66). Diminished AMPK levels have been found in FH-deficient RCC cells, resulting in reduced phosphoryhlation of acetyl CoA carboxylase (pACC), which would be expected to increase fatty acid synthesis. In addition, phospho-S6 ribosomal protein, a downstream mTOR effector, was also found to be activated in FH-deficient RCC cells (51, 67). Finally, FH-deficient renal cell carcinoma is characterized by a glutamine-dependent reductive carboxylation in which glutamine is the primary carbon source for lipid biosynthesis (Fig. 2) (68).
Figure 2. Potential therapeutic targets: papillary type 2 renal cell carcinoma.
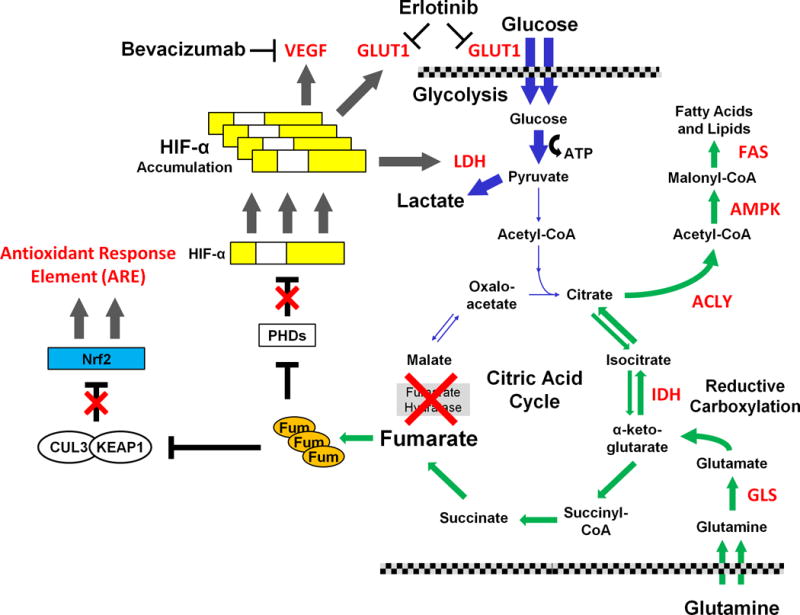
Loss of fumarate hydratase (FH) is associated with the familial form of type 2 papillary RCC and results in the activation of several pathways. The loss of FH activity suppresses flow through the citric acid cycle in the normal direction and impairs the cell’s ability to use oxidative phosphorylation, forcing the cells to be dependent upon glucose and glycolysis for energy production. The loss of canonical tricarboxylic acid cycle function results in usage of the reductive carboxylation pathway and increased glutamine uptake for fatty acid synthesis and, in a similar manner to clear cell RCC, provides a series of potential targets for therapy (highlighted in red) involved in the conversion of glutamine to fatty acids.
The highly increased levels of fumarate (Fum) result in inhibition of α-ketoglutarate depenedent enzymes such as the prolyl hydroxylases (PHDs). In normoxia, the PHDs hydroxylate the HIF-α transcription factors to allow for VHL-dependent degradation. In fumarate hydratase deficient RCC the HIF1α accumulates and activates downstream targets. This results in increased levels of vascular epithelial growth factor (VEGF), lactate dehydrogenase A (LDHA) and the glucose transporter, GLUT1, that support the high levels glycolysis required by this aggressive form of renal cell carcinoma. A therapeutic approach utilizing the combination of bevacizumab and erlotinib is currently being evaluated. In addition, the elevated levels of fumarate result in succination of multiple proteins including the KEAP1 protein, which, as part of an E3 ubiquitin ligase complex with CUL3, targets the NRF2 transcription factor for degradation. Succination of KEAP1 inactivates KEAP1 and inhibits NRF2 degradation, resulting in activation of the NRF2 antioxidant response element (ARE) transcription pathway. While mutation of fumarate hydratase is rarely seen in sporadic type 2 papillary RCC, recent studies have reported activation of the NRF2 pathway in sporadic type 2 PRCC due to inactivating mutations of CUL3 or activating mutations of NRF2. An intense effort is underway to develop therapeutic agents that target the NRF2 ARE pathway.
KEAP1/Nrf2 antioxidant response pathway
As FH-deficient renal cell carcinoma is characterized by increased oxidative stress and elevated levels of reactive oxygen, and effective antioxidant response is critical for continued growth (69). In FH-deficient renal cell carcinoma increased levels of fumarate induce succination of the Kelch-like ECH-associated protein 1 (KEAP1), resulting in stabilization of nuclear factor E2-related factor 2 (Nrf2), increasing expression of antioxidant response element (ARE) genes (Fig. 2) (70, 71). Ooi et al. found a similar antioxidant response phenotype shared between hereditary and sporadic type 2 papillary RCC (71) and that CUL2 and Nrf2 mutations confer an Nrf2 activation phenotype in sporadic type 2 papillary RCC (72).
Therapeutic approaches for type 2 papillary renal cell carcinoma
A number of potential therapeutic approaches are currently being evaluated in pre-clinical as well as clinical trials. Screening studies are underway to identify agents which target Nrf2. The effect of agents such as metformin, which activate AMPK and its downstream target, SIRT1, on Nrf2 activity is also being evaluated. LDHA inhibition has been shown to have anti-tumor activity against FH-deficient RCC cells (73) and an effort is underway to identify clinically evaluable LDHA inhibitors. As mentioned above, FH-deficient renal cell carcinoma is characterized by increased levels of HIF1α, which results in increased expression of genes such as vascular endothelial growth factor (VEGF) and the glucose transporter GLUT1, which are critical for the increased vasculature and glucose transport needed to support aerobic glycolysis (Fig. 2). In order to evaluate the effectiveness of agents such as bevacizumab and erlotinib, which could target the vasculature as well as impair glucose transport and glycolysis (74), a Phase 2 trial of bevacizumab and erlotinib in patients with advanced HLRCC-associated type 2 papillary renal cell carcinoma as well as sporadic papillary renal cell carcinoma is currently underway (NIH study trial registration number NCT01130519; clintrials.gov).
Summary
Although enormous progress has been made over the past twenty years in our understanding of the genetic basis and development of targeted therapeutics for clear cell renal cell carcinoma, we still have a long way to go. For clear cell RCC we need better approaches for targeting both the VHL/HIF2α as well as chromatin remodeling gene pathways. Given the profound genomic heterogeneity in clear cell RCC, it is most likely that successful strategies will need to target both truncal as well as branch driver mutations. Ongoing genomic studies in papillary RCC will hopefully identify novel driver mutations and pathways for this disease. For type 1 papillary RCC the MET pathway seems particularly promising. For type 2 papillary RCC targeting the tumor vascularity and glucose transport/glycolysis and targeting the KEAP1/NRF2 pathway also appear to be promising approaches. Ongoing metabolomic and functional studies will hopefully identify novel metabolic pathways to target in both clear cell as well as papillary renal cell carcinoma.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 2.Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol. 2010;7:277–85. doi: 10.1038/nrurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linehan WM. Genetic basis of kidney cancer: role of genomics for the development of disease-based therapeutics. Genome Res. 2012;22:2089–100. doi: 10.1101/gr.131110.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jonasch E, Motzer RJ. Ten years of progress in renal cell carcinoma. J Natl Compr Canc Netw. 2012;10:690–3. doi: 10.6004/jnccn.2012.0071. [DOI] [PubMed] [Google Scholar]
- 5.Latif F, Tory K, Gnarra JR, Yao M, Duh F-M, Orcutt ML, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–20. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 6.Nickerson ML, Jaeger E, Shi Y, Durocher JA, Mahurkar S, Zaridze D, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res. 2008;14:4726–34. doi: 10.1158/1078-0432.CCR-07-4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013;45:860–7. doi: 10.1038/ng.2699. [DOI] [PubMed] [Google Scholar]
- 8.Duan DR, Pause A, Burgess WH, Aso T, Chen DY, Garrett KP, et al. Inhibition of transcription elongation by the VHL tumor suppressor protein. Science. 1995;269:1402–6. doi: 10.1126/science.7660122. [DOI] [PubMed] [Google Scholar]
- 9.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–8. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 10.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–72. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 11.Voss MH, Hsieh JJ, Motzer RJ. Novel approaches targeting the vascular endothelial growth factor axis in renal cell carcinoma. Cancer J. 2013;19:299–306. doi: 10.1097/PPO.0b013e31829d5cff. [DOI] [PubMed] [Google Scholar]
- 12.Linehan WM, Srinivasan R, Garcia JA. Non-clear cell renal cancer: disease-based management and opportunities for targeted therapeutic approaches. Semin Oncol. 2013;40:511–20. doi: 10.1053/j.seminoncol.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD. The contribution of VHL substrate binding and HIF1-alpha to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell. 2002;1:247–55. doi: 10.1016/s1535-6108(02)00044-2. [DOI] [PubMed] [Google Scholar]
- 14.Kondo K, Klco J, Nakamura E, Lechpammer M, Kaelin WG., Jr Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell. 2002;1:237–46. doi: 10.1016/s1535-6108(02)00043-0. [DOI] [PubMed] [Google Scholar]
- 15.Kondo K, Kim WY, Lechpammer M, Kaelin WG., Jr Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 2003;1:E83. doi: 10.1371/journal.pbio.0000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zimmer M, Doucette D, Siddiqui N, Iliopoulos O. Inhibition of hypoxia-inducible factor is sufficient for growth suppression of VHL−/− tumors. Mol Cancer Res. 2004;2:89–95. [PubMed] [Google Scholar]
- 17.Purdue MP, Johansson M, Zelenika D, Toro JR, Scelo G, Moore LE, et al. Genome-wide association study of renal cell carcinoma identifies two susceptibility loci on 2p21 and 11q13.3. Nat Genet. 2011;43:60–5. doi: 10.1038/ng.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen C, Kaelin WG., Jr The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol. 2013;23:18–25. doi: 10.1016/j.semcancer.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen C, Beroukhim R, Schumacher SE, Zhou J, Chang M, Signoretti S, et al. Genetic and functional studies implicate HIF1a as a 14q kidney cancer suppressor gene. Cancer Discov. 2011;1:223–35. doi: 10.1158/2159-8290.CD-11-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Monzon FA, Alvarez K, Peterson L, Truong L, Amato RJ, Hernandez-McClain J, et al. Chromosome 14q loss defines a molecular subtype of clear-cell renal cell carcinoma associated with poor prognosis. Mod Pathol. 2011;24:1470–9. doi: 10.1038/modpathol.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woldemichael GM, Vasselli JR, Gardella RS, McKee TC, Linehan WM, McMahon JB. Development of a cell-based reporter assay for screening of inhibitors of hypoxia-inducible factor 2-induced gene expression. J Biomol Screen. 2006;11:678–87. doi: 10.1177/1087057106289234. [DOI] [PubMed] [Google Scholar]
- 22.Bokesch HR, Gardella RS, Rabe DC, Bottaro DP, Linehan WM, McMahon JB, et al. A new hypoxia inducible factor-2 inhibitory pyrrolinone alkaloid from roots and stems of Piper sarmentosum. Chem Pharm Bull (Tokyo) 2011;59:1178–9. doi: 10.1248/cpb.59.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grkovic T, Whitson EL, Rabe DC, Gardella RS, Bottaro DP, Linehan WM, et al. Identification and evaluation of soft coral diterpenes as inhibitors of HIF-2alpha induced gene expression. Bioorg Med Chem Lett. 2011;21:2113–5. doi: 10.1016/j.bmcl.2011.01.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKee TC, Rabe D, Bokesch HR, Grkovic T, Whitson EL, Diyabalanage T, et al. Inhibition of hypoxia inducible factor-2 transcription: isolation of active modulators from marine sponges. J Nat Prod. 2012;75:1632–6. doi: 10.1021/np300211x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zimmer M, Ebert BL, Neil C, Brenner K, Papaioannou I, Melas A, et al. Small-molecule inhibitors of HIF-2a translation link its 5′UTR iron-responsive element to oxygen sensing. Mol Cell. 2008;32:838–48. doi: 10.1016/j.molcel.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zimmer M, Lamb J, Ebert BL, Lynch M, Neil C, Schmidt E, et al. The connectivity map links iron regulatory protein-1-mediated inhibition of hypoxia-inducible factor-2a translation to the anti-inflammatory 15-deoxy-delta12,14-prostaglandin J2. Cancer Res. 2010;70:3071–9. doi: 10.1158/0008-5472.CAN-09-2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sourbier C, Srivastava G, Ghosh MC, Ghosh S, Yang Y, Gupta G, et al. Targeting HIF2α translation with Tempol in VHL-deficient clear cell renal cell carcinoma. Oncotarget. 2012;3:1472–82. doi: 10.18632/oncotarget.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu XD, Yao J, Tripathi DN, Ding Z, Xu Y, Sun M, et al. Autophagy mediates HIF2alpha degradation and suppresses renal tumorigenesis. Oncogene. 2014 Jul 7; doi: 10.1038/onc.2014.199. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ding Z, German P, Bai S, Reddy AS, Liu XD, Sun M, et al. Genetic and pharmacological strategies to refunctionalize the von Hippel Lindau R167Q mutant protein. Cancer Res. 2014;74:3127–36. doi: 10.1158/0008-5472.CAN-13-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koh MY, Darnay BG, Powis G. Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and ubiquitinates hypoxia-inducible factor 1alpha, leading to its oxygen-independent degradation. Mol Cell Biol. 2008;28:7081–95. doi: 10.1128/MCB.00773-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koh MY, Lemos R, Jr, Liu X, Powis G. The hypoxia-associated factor switches cells from HIF-1alpha- to HIF-2alpha-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011;71:4015–27. doi: 10.1158/0008-5472.CAN-10-4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–3. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varela I, Tarpey P, Raine K, Huang D, Ong CK, Stephens P, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–42. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo G, Gui Y, Gao S, Tang A, Hu X, Huang Y, et al. Frequent mutations of genes encoding ubiquitin-mediated proteolysis pathway components in clear cell renal cell carcinoma. Nat Genet. 2011;12:1–3. doi: 10.1038/ng.1014. [DOI] [PubMed] [Google Scholar]
- 35.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013;499:43–9. doi: 10.1038/nature12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hakimi AA, Ostrovnaya I, Reva B, Schultz N, Chen YB, Gonen M, et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: a report by MSKCC and the KIRC TCGA research network. Clin Cancer Res. 2013;19:3259–67. doi: 10.1158/1078-0432.CCR-12-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pena-Llopis S, Vega-Rubin-de-Celis S, Liao A, Leng N, Pavia-Jimenez A, Wang S, et al. BAP1 loss defines a new class of renal cell carcinoma. Nat Genet. 2012;44:751–9. doi: 10.1038/ng.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joseph RW, Kapur P, Serie DJ, Eckel-Passow JE, Parasramka M, Ho T, et al. Loss of BAP1 protein expression is an independent marker of poor prognosis in patients with low-risk clear cell renal cell carcinoma. Cancer. 2014;120:1059–67. doi: 10.1002/cncr.28521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farley MN, Schmidt LS, Mester JL, Pena-Llopis S, Pavia-Jimenez A, Christie A, et al. A novel germline mutation in BAP1 predisposes to familial clear-cell renal cell carcinoma. Mol Cancer Res. 2013;11:1061–71. doi: 10.1158/1541-7786.MCR-13-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Popova T, Hebert L, Jacquemin V, Gad S, Caux-Moncoutier V, Dubois-d’Enghien C, et al. Germline BAP1 Mutations Predispose to Renal Cell Carcinomas. Am J Hum Genet. 2013;92:974–80. doi: 10.1016/j.ajhg.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simon JM, Hacker KE, Singh D, Brannon AR, Parker JS, Weiser M, et al. Variation in chromatin accessibility in human kidney cancer links H3K36 methyltransferase loss with widespread RNA processing defects. Genome Res. 2014;24:241–50. doi: 10.1101/gr.158253.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carvalho S, Vitor AC, Sridhara SC, Martins FB, Raposo AC, Desterro JM, et al. SETD2 is required for DNA double-strand break repair and activation of the p53-mediated checkpoint. Elife. 2014;3:e02482. doi: 10.7554/eLife.02482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pfister SX, Ahrabi S, Zalmas LP, Sarkar S, Aymard F, Bachrati CZ, et al. SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Rep. 2014;7:2006–18. doi: 10.1016/j.celrep.2014.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li F, Mao G, Tong D, Huang J, Gu L, Yang W, et al. The histone mark H3K36me3 regulates human DNA mismatch repair through its interaction with MutSalpha. Cell. 2013;153:590–600. doi: 10.1016/j.cell.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46:225–33. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ricketts CJ, Linehan WM. Intratumoral heterogeneity in kidney cancer. Nat Genet. 2014;46:214–5. doi: 10.1038/ng.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2011;481:380–4. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gameiro PA, Yang J, Metelo AM, Perez-Carro R, Baker R, Wang Z, et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013;17:372–85. doi: 10.1016/j.cmet.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gameiro PA, Laviolette LA, Kelleher JK, Iliopoulos O, Stephanopoulos G. Cofactor balance by nicotinamide nucleotide transhydrogenase (NNT) coordinates reductive carboxylation and glucose catabolism in the tricarboxylic acid (TCA) cycle. J Biol Chem. 2013;288:12967–77. doi: 10.1074/jbc.M112.396796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tong WH, Sourbier C, Kovtunovych G, Jeong SY, Vira M, Ghosh M, et al. The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell. 2011;20:315–27. doi: 10.1016/j.ccr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Woodard J, Joshi S, Viollet B, Hay N, Platanias LC. AMPK as a therapeutic target in renal cell carcinoma. Cancer Biol Ther. 2010;10 doi: 10.4161/cbt.10.11.13629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zbar B, Tory K, Merino MJ, Schmidt LS, Glenn GM, Choyke P, et al. Hereditary papillary renal cell carcinoma. J Urol. 1994;151:561–6. doi: 10.1016/s0022-5347(17)35015-2. [DOI] [PubMed] [Google Scholar]
- 54.Schmidt LS, Duh FM, Chen F, Kishida T, Glenn GM, Choyke P, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997;16:68–73. doi: 10.1038/ng0597-68. [DOI] [PubMed] [Google Scholar]
- 55.Schmidt LS, Junker K, Weirich G, Glenn G, Choyke P, Lubensky I, et al. Two North American families with hereditary papillary renal carcinoma and identical novel mutations in the MET proto-oncogene. Cancer Res. 1998;58:1719–22. [PubMed] [Google Scholar]
- 56.Zhuang Z, Park WS, Pack S, Schmidt LS, Pak E, Pham T, et al. Trisomy 7 - harboring non-random duplication of the mutant MET allele in hereditary papillary renal carcinomas. Nat Genet. 1998;20:66–9. doi: 10.1038/1727. [DOI] [PubMed] [Google Scholar]
- 57.Lubensky IA, Schmidt LS, Zhuang Z, Weirich G, Pack S, Zambrano N, et al. Hereditary and sporadic papillary renal carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol. 1999;155:517–26. doi: 10.1016/S0002-9440(10)65147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Choueiri TK, Vaishampayan U, Rosenberg JE, Logan TF, Harzstark AL, Bukowski RM, et al. Phase II and biomarker study of the dual MET/VEGFR2 inhibitor foretinib in patients with papillary renal cell carcinoma. J Clin Oncol. 2013;31:181–6. doi: 10.1200/JCO.2012.43.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, Pukkala E, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A. 2001;98:3387–2. doi: 10.1073/pnas.051633798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Merino MJ, Torres-Cabala CA, Zbar B, Chian-Garcia CA, Linehan WM. Hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCC): clinical, histopathological and molecular features of the first American families described. Mod Pathol. 2003;16:739. [Google Scholar]
- 61.Grubb RL, III, Franks ME, Toro J, Middelton L, Choyke L, Fowler S, et al. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol. 2007;177:2074–80. doi: 10.1016/j.juro.2007.01.155. [DOI] [PubMed] [Google Scholar]
- 62.Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–10. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 63.Warburg O, Posener K, Negelein E. On the metabolism of cancer cells. Biochem Z. 1924;152:319–44. [Google Scholar]
- 64.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 65.Yang Y, Valera VA, Padilla-Nash HM, Sourbier C, Vocke CD, Vira MA, et al. UOK 262 cell line, fumarate hydratase deficient (FH−/FH−) hereditary leiomyomatosis renal cell carcinoma: in vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer Genet Cytogenet. 2010;196:45–55. doi: 10.1016/j.cancergencyto.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell. 2005;8:143–53. doi: 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 67.Linehan WM, Rouault TA. Molecular pathways: fumarate hydratase-deficient kidney cancer–targeting the Warburg effect in cancer. Clin Cancer Res. 2013;19:3345–52. doi: 10.1158/1078-0432.CCR-13-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481:385–8. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sudarshan S, Sourbier C, Kong HS, Block K, Romero VV, Yang Y, et al. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and HIF-1 alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol. 2009;15:4080–90. doi: 10.1128/MCB.00483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell. 2011;20:524–37. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell. 2011;20:511–23. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 72.Ooi A, Dykema K, Ansari A, Petillo D, Snider J, Kahnoski R, et al. CUL3 and NRF2 mutations confer an NRF2 activation phenotype in a sporadic form of papillary renal cell carcinoma. Cancer Res. 2013;73:2044–51. doi: 10.1158/0008-5472.CAN-12-3227. [DOI] [PubMed] [Google Scholar]
- 73.Xie H, Valera VA, Merino MJ, Amato AM, Signoretti S, Linehan WM, et al. LDH-A inhibition, a therapeutic strategy for treatment of hereditary leiomyomatosis and renal cell cancer. Mol Cancer Ther. 2009;8:626–35. doi: 10.1158/1535-7163.MCT-08-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Engelman JA, Cantley LC. A sweet new role for EGFR in cancer. Cancer Cell. 2008;13:375–6. doi: 10.1016/j.ccr.2008.04.008. [DOI] [PubMed] [Google Scholar]