

Abstract
Motivation: Automated screening approaches are able to rapidly identify a set of small molecules inducing a desired phenotype from large small-molecule libraries. However, the resulting set of candidate molecules is usually very diverse pharmacologically, thus little insight on the shared mechanism of action (MoA) underlying their efficacy can be gained.
Results: We introduce a computational method (Drug-Set Enrichment Analysis—DSEA) based on drug-induced gene expression profiles, which is able to identify the molecular pathways that are targeted by most of the drugs in the set. By diluting drug-specific effects unrelated to the phenotype of interest, DSEA is able to highlight phenotype-specific pathways, thus helping to formulate hypotheses on the MoA shared by the drugs in the set. We validated the method by analysing five different drug-sets related to well-known pharmacological classes. We then applied DSEA to identify the MoA shared by drugs known to be partially effective in rescuing mutant cystic fibrosis transmembrane conductance regulator (CFTR) gene function in Cystic Fibrosis.
Availability and implementation: The method is implemented as an online web tool publicly available at http://dsea.tigem.it.
Contact: dibernardo@tigem.it
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
Large collections of small-molecules can be automatically screened against a desired phenotypic effect. Automated experimental screening approaches include high-content screening (HCS) (Bickle, 2010) and high-throughput screening (HTS) (Bajorath, 2002), with different advantages and limitations, and a screening capacity that ranges from thousands to millions of compounds per assay.
Screening assays can be performed either to identify lead compounds binding a specific molecular target, or inducing a specific phenotype of interest. A common drawback of automated molecular screening is the opacity of the hit compound selection mechanism. Indeed, the set of positive hits following an HTS or HCS typically includes small-molecules with unknown mode-of-action (MoA) or whose MoA are so different from each other, that no hint on the shared molecular mechanisms underlying their efficacy can be gained (Sams-Dodd, 2005).
The difficulty in characterizing a set of screening hits resides in the complexity of their interactions within the cell. Molecules binding the same molecular target can induce different phenotypes caused by unknown off-targets. On the contrary, molecules binding different targets can induce the same phenotype, when they act in the same pathway (Sams-Dodd, 2005). Nonetheless, among the heterogeneous effects induced by the hit compounds in the cell, there could exist a common mechanism responsible for their efficacy in the screening selection.
Here, we introduce a new method, named drug-set Enrichment Analysis (DSEA), that aims at identifying the mechanism(s) of action shared by a set of compounds in terms of the molecular pathways targeted by all, or most of them.
We define a pathway as a set of genes. We collected a large database of pathways by merging nine different publicly available collections, including generic gene sets (co-localized genes, co-regulated genes, protein complex subunits, etc.) and disease-related gene sets.
DSEA looks for shared pathways in a set of drugs by analysing transcriptional responses induced by each of the compounds of interest in one or more cell lines. To this end, we exploited the Connectivity Map (cMap, Lamb et al., 2006) dataset consisting of about 7000 microarrays following treatment of four different cell lines with 1309 drugs.
The main hypothesis underlying DSEA is that if pharmacologically different drugs induce the same phenotype of interest, then some of the molecular pathways they target must be shared by most of them. Although this is not necessarily true in general, it is a reasonable assumption. DSEA is designed to search transcriptional responses of different, but phenotypically-related, drugs for shared pathways whose genes are upregulated (or downregulated) by most of the drugs in the set. In this way, pathways relevant for the phenotype of interest should emerge, while drug-specific pathways, which are unrelated to the phenotype of interest, should cancel out.
To validate the method, we thoroughly tested the ability of DSEA in identifying the shared pathways for five different drug-sets whose MoA has already been well characterized: Histone Deacetylase Inhibitors (HDIs), Cyclin Dependent Kinase Inhibitors (CDKIs), Heat Shock Protein 90 Inhibitors (HSP90Is), Topoisomerase Inhibitors (TIs), Cardiac Glycosides (CGs). Finally, we applied DSEA to a set of eleven drugs, belonging to different pharmacological classes, that have been shown to act as (weak) correctors of the mutant CFTR protein defect (ΔF508) causative of cystic fibrosis.
2 Methods
2.1 Data preparation
We downloaded raw data files from the cMap (Lamb et al., 2006), a compendium of 7056 Affymetrix microarrays (.CEL files) obtained with three different chipsets (HG-U133A, HT_HG-U133A, HT_HG-U133A_EA). Expression values for all the samples were computed using the R package affy v.1.40.0 (Gautier et al., 2004) with MAS5 normalization. The probes of each chipset were re-annotated to 12 012 genes using the Brainarray CDF packages v.16.0.0 (Dai et al., 2005). The combined matrix (12 012 × 7056), after removing non-common (control) probes, was quantile-normalized (see Fig. 1a). Fold change (FC) values were obtained as log-ratios between the values of the treatment samples and the corresponding control samples (averaged over replicates) thus reducing the data matrix to size 12 012 × 6100. After converting FC values to ranks, we built Prototype Ranked Lists (PRLs) by merging all the samples corresponding to the same drug, as described in Iorio et al. (2010), thus obtaining a 12 012 genes × 1309 drugs matrix of PRLs (see Fig. 1b).
Fig. 1.

Data preparation pipeline. (a) Raw genome wide expression profiles are collected from the cMap and preprocessed. (b) Control-treatment fold change values are computed and converted to ranks. Profiles referring to the same small molecule in different experimental conditions are merged together. (c) Gene expression ranks are converted to pathway Enrichment Scores. (d) The ESs are converted to row-wise ranks
2.2 A Pathway-based connectivity map
We collected set of genes (pathways) from nine publicly available databases (see Table 1): Biological Processes (GO-BP), Molecular Function (GO-MF) and Cellular Component (GO-CC) from BioMart (Durinck et al., 2009), excluding pathways with less than 5 or more than 500 genes, KEGG, Reactome, Biocarta, Canonical Pathways, Genetic and Chemical Perturbation (as collected in MSigDB, Subramanian et al., 2005) and MIPS Corum (Ruepp et al., 2010). For each gene set, we removed the genes not included in the set of 12012 Affymetrix probe-mapped genes. In addition, we defined a gene-based collection (Single-Gene Sets, SGS) by building 12 012 fictitious gene sets containing only one gene. We provided this addition database just as a resource for the user who wishes to perform DSEA analysis in a gene-wise fashion, although we discourage its use being DSEA designed to work with gene-sets rather than single genes. To convert the 12 012 genes × 1309 drugs PRL matrix to a pathway-oriented matrix, we proceeded as follows: given a pathway database of interest, for each pathway i in the database, and each PRL j, we computed a signed Enrichment Score ESij and a p-value using the Kolmogorov–Smirnov (KS) test (Subramanian et al., 2005). The two-sample KS statistic is defined as the maximum distance between two empirical distribution functions. Along the lines of the Gene Set Enrichment Analysis method (GSEA, Subramanian et al., 2005), we apply a signed version of the KS statistic to compare gene ranks. The ES associated with the KS statistic is thus defined as follows:
| (1) |
where F1 and F2 are the two empirical distribution functions corresponding to the ranks of the genes included in a set of interest (F1) against those that are not included (F2), and s is a function returning –1 or + 1 according to the sign of at the point where their absolute difference is maximal. Note that a P-value for the KS test can be computed analytically without resorting to random permutations. In particular, we used our signed variant (available online at https://github.com/franapoli/signed-ks-test) of the R function ks.test to compute the signed KS statistic (and the corresponding p-values, used in the next Subsection). We thus obtained, for each database, one Enrichment Score matrix ES whose rows correspond to pathways and whose columns correspond to drugs (see Fig. 1c).
Table 1.
Gene set databases currently supported by DSEA
| Source | Name | Description | # |
|---|---|---|---|
| BioMart | GO BP | Gene Ontology—Biological Processes | 3262 |
| BioMart | GO MF | Gene Ontology—Molecular Function | 939 |
| BioMart | GO CC | Gene Ontology—Cellular Component | 556 |
| MSigDB | CP | Expert-defined Canonical Pathways | 243 |
| MSigDB | KEGG | Kyoto Encyclopedia of Genes and Genomes | 186 |
| MSigDB | Biocarta | Community-fed molecular relationships | 217 |
| MSigDB | Reactome | Open-source, open access, manually curated and peer-reviewed pathway database | 674 |
| MSigDB | CGP | Genetic and Chemical Perturbations | 2427 |
| Mips | CORUM | Comprehensive Resource of Mammalian protein complexes | 1343 |
| – | SGS | Sets containing single genes mapped from Affymetrix chip U133A | 12012 |
We also added a collection of fictitious sets each containing a single gene from the 12012 obtained after probe reannotation of the cMap raw data. We collected existing gene sets from a number of publicly available databases.
In the rest of the paper, we will make two other different uses of the KS test: to compute the DSEA itself in Section 2.3, and to validate it in Section 3.2.
2.3 Drug-set enrichment analysis
DSEA quantifies the extent at which a set of drugs consistently upregulates (or downregulates) one or more pathways. Starting from the ES matrix, we first sorted each row i according to the Enrichment Scores ESij of the pathway i across the drugs (see Fig. 1d), obtaining a rank-based matrix R. Each element Rij in R represents the rank of drug j when sorting drugs according to their effect on pathway i. The significance of a drug-set for each pathway is assessed by applying the same procedure showed previously to compute the ESij scores, but comparing drug ranks (distributed across the row corresponding to a given pathway) as opposed to gene ranks (distributed across the column corresponding to a given drug). In this case, the sign of the ES indicates whether a pathway is activated or inhibited by the drugs in the set.
DSEA can be thought of as the dual of GSEA: if in GSEA a drug-induced expression profiles can be represented as a ranked list of differentially expressed genes, in DSEA we modeled a pathway as a ranked list of drugs.
From a methodological point of view, DSEA is able to highlight pathways that are significantly modulated by most of the drugs in the input drug-set relative to the other drugs in the database. This means that if drugs in a drug-set tend to modulate the same pathway more than the other drugs in the database, this pathway will be found by DSEA, even if the modulation exerted by the single drugs on the pathway is weak. This mechanism is key to identify a shared mode of action, even when it is not apparent when considering the individual drugs in the set.
3 Results
3.1 Drug-set enrichment analysis (DSEA)
We developed the Drug-Set Enrichment Analysis (DSEA) algorithm in order to identify the shared molecular pathways modulated by all, or most of, the compounds in a given set.
DSEA exploits a compendium (the cMap, Lamb et al., 2006) of Gene Expression Profiles (GEPs) following treatment with 1309 small molecules (mostly FDA approved drugs). DSEA works by applying the following steps: (i) Figure 1a,b—GEPs for each small molecule are merged into ranked lists of differentially expressed genes (following treatment of multiple cell-lines and at different dosages) into a unique Prototype Ranked List (PRL, Iorio et al., 2010); (ii) Figure 1c—each gene-wise PRL is converted to a pathway-wise PRL, by computing the Enrichment Score (ES) of each pathway through a GSEA approach (see Section 2), using the list of genes in the pathway as the gene-set, and the gene-wise PRL as the ranked list of genes. (Subramanian et al., 2005). Each pathway thus has a specific ES for each small-molecule; (iii) Figure 1d—the resulting pathway-by-small-molecule matrix is then sorted row-wise, so that each pathway is now associated to a ranked list of small-molecules: from the one most activating the pathway to the one most inhibiting it.
Given a query-set of small-molecules, DSEA checks for each pathway whether small-molecules tend to be significantly ranked at the top (or the bottom) of the list, by applying a Kolmogorov-Smirnov (KS) test, as shown in Figure 2. An Enrichment Score for the drug-set and a p-value can thus be computed for each pathway exactly, without the need of random permutations. The final output of DSEA is a list of pathways ranked by the KS P-value, which are significantly modulated by the majority of the small-molecules in the drug-set.
Fig. 2.

The DSEA method. Pathways are defined as ranked lists of drugs. DSEA performs a statistical test to assess whether the drugs in a set are significantly ranked at top (or bottom) of the row corresponding to a given pathway. Each row is ranked according to how much the drug in the column upregualtes (or downregulates) the genes in the pathway. The toy example shows how a set of three drugs is found to consistently downregulate one pathway (top arrow), while upregulating another one (bottom arrow)
3.2 Validation
To validate the method, we applied DSEA to five drug-sets consisting of compounds belonging to five distinct pharmacological classes, as summarized in Table 2. Prior knowledge about each drug-set allowed us to assess whether the shared pathways found by DSEA within each class were correct.
Table 2.
Drug-sets chosen to validate the DSEA method
| Drug-set | Affected activity | Phrmacological class | Validation target |
|---|---|---|---|
| Histone deacetylase inhibitors (HDI) | Transcription | Scriptaid, trichostatin A, valproic acid, vorinostat, HC toxin, bufexamac | HAT1 |
| Cyclin dependant kKinase inhibitors (CDKI) | Cell cycle | Alsterpaullone, GW-8510, H-7, staurosporine | CDK1 |
| Heat shock protein 90 inhibitors (HSP90I) | Protein folding | Geldanamycin, monorden, tanespimycin, alvespimycin | HSP90AA1 |
| Topoisomerase inhibitors (TI) | Cell cycle | Doxorubicin, etoposide, camptothecin, irinotecan, genistein, ofloxacin, Mitoxantrone, flumequine, luteolin | TOP2A/B |
| Cardiac glycosides (CG) | Na+-K+ pump | Digitoxigenin, digoxigenin, digoxin, ouabain | ATP1A1 |
Column 1: Pharmacological class; column 2: molecular processes known to be targeted by the drugs; column 3: Drugs in the set; column 4: targets chosen for the validation process (see main text). A golden-standard was designed for each pharmacological class by collecting all the pathways containing the corresponding gene shown in the last column.
To this end, we defined a golden standard for each drug-set as follows: we selected the known target gene for each drug-set (see Table 2). For drugs with more than one known target, we chose the first member (alphabetical order) of the target protein family. In the case of Topoisomerase Inhibitor (TI) drug-set, we chose TOP2 because six out of nine drugs in the set were TOP2 inhibitors. For the Histone Deacetylase Inhibitor (HDI) drug-set, we chose HAT1 in addition to HDAC1.
For each drug-set, we then added to the golden-standard all the Gene Ontology (GO) pathways containing the chosen drug target. A summary of the golden standard for each drug-set is reported in Suppl. Table S1.
To evaluate the performance of DSEA, we checked whether the golden-standard pathways were significantly enriched at the top of the ranked list of pathways given as output by DSEA by applying again a KS test.
As shown in Table 3, for each drug-set, DSEA ranked the golden-standard pathways significantly at least in one GO database. Suppl. Table S3 shows the same type of validation for each of the 10 pathway databases.
Table 3.
Validation results
| GO-BP | GO-MF | GO-CC | |
|---|---|---|---|
| CDKI | 1.34E− | 0.2834 | 0.1407 |
| HDI | 0.3483 | 0.0007 | 0.5182 |
| HSP90I | 0.0065 | 0.64 | 0.1582 |
| TI | 0.03868 | 0.1556 | 0.6597 |
| CG | 0.002 | 0.3764 | 0.2673 |
The P-values assess if the golden-standard pathways within the GO-BP, GO-MF and GO-CC databases are ranked significantly at the top by DSEA. P-values < 0.005 are highlighted in bold.
Interestingly for the HDI drug-set, when using HDAC1 as the target gene for the golden standard pathways, we did not find any significant enrichment, however when using the dual enzyme, HAT1, we did find the golden standard pathways to be significantly shared by the drugs in the drug-set (Table 3). These pathways are related to the process complementary to deacetylation, that is acetylation. The acetylation-deacetylation balance is known as acetylation homeostasis and the existence of a HAT-HDAC coupling through a common signal has been suggested (Dokmanovic et al., 2007).
In order to exclude that the results obtained in Table 3 were due to a hidden bias in the golden standard pathways, we generated for each of the 5 drug-sets, 1000 random drug-sets with the same size as the corresponding original drug-set. We then ran DSEA on each random drug-set and checked whether the golden-standard pathways of the original drug-set were significantly enriched at the top of the resulting ranked list of pathways. Since these random drug-sets consist of unrelated drugs, the golden standard pathways should not be significantly enriched and the corresponding KS P-values should be uninformative. The results are summarized in Figure 3. Observe that in this significance analysis, we treated p-values as random variables, thus their expected value should be close to 0.5 (Sackrowitz and Samuel-Cahn, 1999, and Suppl. Fig. S14). It is clear from Figure 3 that using random drug-sets, DSEA ranks the golden-standard pathways at random positions.
Fig. 3.
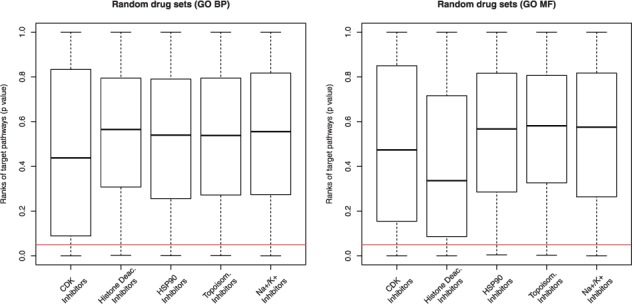
Golden-standard pathways significance for random drug-sets. The box-plots show DSEA validation against the golden-standard when using 1000 random drug-sets containing the same number of drugs as in the original drug-sets. The horizontal line indicates the 0.05 significance threshold. The P-value obtained by chance is close to 0.5, as expected (black segment in each box shows the median P-value)
3.2.1 Robustness and convergence
Hit compounds selected from automated drug screening techniques may contain false-positive hits, exhibiting a mode of action inconsistent with the other compounds in the selected set. For this reason, we investigated the effects of adding noise to the drug-sets used for validation before running DSEA.
In order to assess the robustness of the method with respect to varying degrees of false-positive hits included in the drug-set, we added 1–10 random drugs to each of the 5 drug-sets and run DSEA on the resulting augmented drug-sets. More precisely, we repeated this process 1000 times thus producing a total of 5 x 10 x 1000 = 50 000 perturbed drug-sets and ran DSEA on each of them.
As before, we used the golden standard to evaluate the p-values for the enrichment of golden standard pathways in the ranked list given as output by DSEA.
As shown in Figure 4 adding up to 3 random drugs to the HSP90I drug-set, i.e. 75% of the initial set size, the golden standard pathways are still significant showing the robustness of DSEA to false positives. Similar results were obtained for the other drug-sets: HSP90I and CG show robustness up to 75% false positives, CDKI and HDI, up to 250% and 167%, respectively. Only in the case of TIs adding a single false positive will cause the result to become not significant (refer Supplementary Figs S4–S8). We hypothesize that the sub-set of drugs in the TI drug-set not targeting TOP2 (3 out of 9 in the set) may partly contribute to decrease the robustness of DSEA in this case.
Fig. 4.
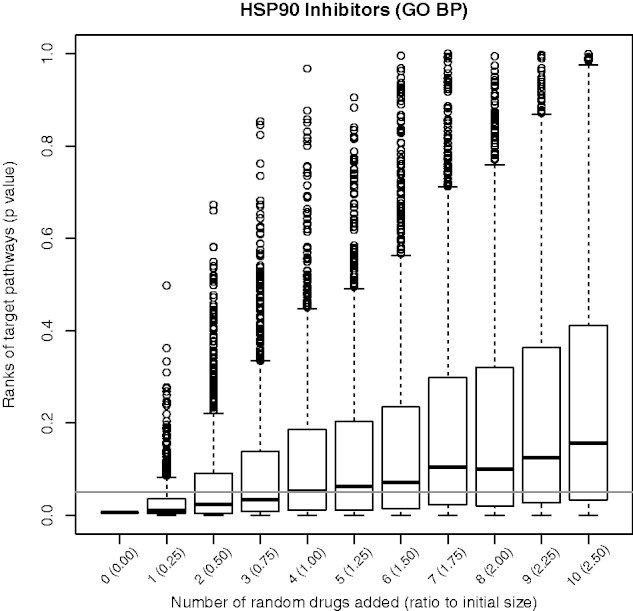
DSEA Robustness. Golden-standard pathways’ significance (P-values on the y-axis) for the HSP90I drug-set with an increasing number of random drugs (reported on the x axis) added to the drug-set. The horizontal line indicates the 0.05 significance threshold. DSEA correctly identifies the golden-standard pathways even when up to three random drugs are added to the drug-set (75% of the drug-set)
To test the convergence properties of DSEA, we ran the analysis by varying the number of drugs in the drug-sets, in order to understand how many drugs were needed in order for the golden standard pathways to become significant. Specifically, for each of the five drug-sets, we generated all the possible combinations of subsets of one drug, two drugs and so on, up the total number of drugs in the drug-set, and then ran DSEA on each subset. Figure 5 shows the results for the HDI drug-set. It can be observed how the P-value of the golden-standard pathways exponentially decreases when more drugs are included in the drug-set, thus demonstrating the power of DSEA in finding pathways shared in common by multiple drugs. We also demonstrated that the same convergence property holds across the other four drug-sets (Supplementary Figs S9–S13).
Fig. 5.
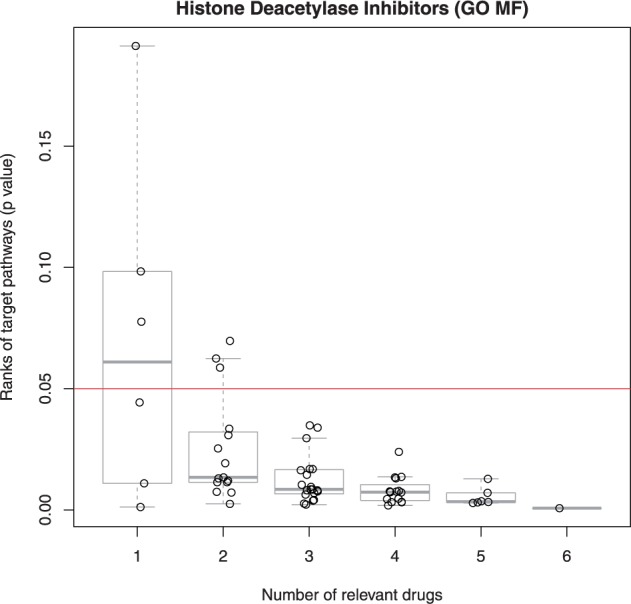
DSEA Convergence. Golden-standard pathways’ significance (P-values on the y axis) for subsets of the HDI drug-set (subset size on the x axis). The horizontal line indicates the 0.05 significance threshold. For HDI subsets greater than 3, DSEA ranks the golden-standard pathways significantly (P < 0.05)
3.3 Example of application to antineoplastic agents
The cMap data were generated from experiments on cancer cell lines. The multi-factorial nature of cancer is reflected by the heterogeneity of the pharmacological approaches to its treatment. The World Health Organization (WHO) defined 5 main categories of antineoplastic agents: Alkylating agents, damaging DNA to impair replication; Antimetabolites, interfering with cancer cell metabolism; Alkaloids, causing metaphase arrest; Cytotoxic antibiotics and related substances, mainly affecting the function or synthesis of nucleic acids; other, with different known or unknown mode of action. We tested the ability of the DSEA in finding a common effect across all the drugs in the cMap that have been annotated as antineoplastic agents (code L01) by the WHO. The drug-set includes 23 drugs unevenly distributed across the five different subclasses. DSEA ranked in the GO-BP database as the most significant pathway (out of 3262): DNA damage response, signal transduction by p53 class mediator resulting in cell cycle arrest. This result is in line with the common mode of action of antineoplastic drugs, particularly alkylating agents, which are the most enriched class of drugs in this drug-set. The result of the analysis for all of the pathways is available as Supplementary Data.
3.4 Application to cystic fibrosis: correctors of DF508-CFTR trafficking defects
CF is one of the most common genetic diseases among people of Caucasian origin (O’Sullivan and Freedman, 2009). It mostly affects lungs causing inflammation, tissue scarring and severe breathing difficulties, thus substantially impacting the patient life-span and quality of life. CF is caused by mutations in the gene coding for the CFTR (CF transmembrane conductance regulator) protein. The most frequent mutation is the deletion of phenylalanine 508 (DF508). Wild-type CFTR translocates to the plasma membrane where it acts as a chloride channel. Mutant DF508CFTR is unable to fold correctly and, although partially functional, it is tagged for degradation (O’Sullivan and Freedman, 2009). No therapeutic treatment is currently available for this disorder. Nevertheless, thanks to world-wide efforts of the academic and industrial research community, some drugs with a mild ‘corrector’ activity for DF508CFTR have been found, largely by HTS studies (Hanrahan et al., 2013). However, each of these compounds has a different known MoA, completely unrelated to CFTR function. Hence, the mechanism by which these drugs are able to correct DF508CFTR function are unknown.
To test the usefulness of DSEA in a ‘real-life’ scenario, we applied DSEA to a drug-set consisting of drugs reported to act as DF508-CFTR correctors in Cystic Fibrosis (CF).
We included 11 drugs in the drug-set (Table 4) according to the following criteria: (i) a DF508CFTR corrector activity is reported in literature, and (ii) GEPs were available in the cMap dataset. As expected these drugs are very heterogenous and no obvious relation to the correction of the DF508CFTR trafficking defects exists.
Table 4.
Drugs with a DF508CFTR corrector activity according to the literature
| Drug | Class | Use / MoA |
|---|---|---|
| Chloramphenicol (Carlile et al., 2007) | Antibiotics | Inhibits bacterial protein synthesis by preventing peptidyl transferase activity. |
| Chlorzoxazone (Carlile et al., 2007) | Muscle Relaxants | Inhibits degranulation of mast cells and prevents the release of histamine and slow-reacting substance of anaphylaxis. |
| Dexamethasone (Caohuy et al., 2009) | Glucocorticoid Agonists | Its anti-inflammatory properties are thought to involve phospholipase A2 inhibitory proteins, lipocortins. |
| Doxorubicin (Maitra et al., 2001) | Topoisomerase Inhibitors | DNA intercalator stabilizing the DNA-topoisomerase II complex. |
| Glafenine (Robert et al., 2010) | NSAID | Non-Steroidal Anti-Inflammatory. An anthranilic acid derivative with analgesic properties. |
| Liothyronine (Carlile et al., 2007) | Synthetic hormones | Increases the basal metabolic rate, affect protein synthesis and increase the body’s sensitivity to catecholamines. |
| Entinostat (Hutt et al., 2010) | HDAC Inhibitors | Inhibits preferentially HDAC 1, also HDAC 3. |
| Scriptaid (Hutt et al., 2010) | HDAC Inhibitors | Inhibits HDAC1, HDAC3 and HDAC8. |
| Strophanthidin (Carlile et al., 2007) | Cardiac Glycosides | Inhibits Na+/K+ ATPase. Also known to inhibit the interaction of MDM2 and MDMX. |
| Thapsigargin (Egan et al., 2002) | Calcium Channel Blockers | Inhibits non-competitively the sarco/endoplasmic Ca2+ ATPase. |
| Trichostatin-A (Hutt et al., 2010) | HDAC Inhibitors | Inhibits HDAC1, HDAC3, HDAC8 and HDAC7. |
DSEA results for the GO-BP, GO-MF, GO-CC databases are reported in Table 5 and for all the other pathway databases in online Supplementary Data.
Table 5.
Top 10 enriched pathways for DF508CFTR-correctors
| GO-BP | GO-MF | GO-CC | |||||||
|---|---|---|---|---|---|---|---|---|---|
| # | Term | ES | P | Term | ES | P | Term | ES | P |
| 1 | Natural killer cell activation | 0.68 | Metallocarboxypeptidase activity | 0.62 | Chloride channel complex | 0.62 | |||
| 2 | Potassium ion export | 0.68 | Hormone activity | 0.60 | Dendrite membrane | 0.59 | |||
| 3 | Smooth muscle contraction | 0.67 | 4 Iron, 4 sulfur cluster binding | −0.59 | mRNA cleavage factor complex | −0.58 | |||
| 4 | Positive regulation of IL-8 biosynthetic process | 0.65 | Heparin binding | 0.57 | Signal recognition particle | 0.57 | |||
| 5 | Positive regulation of cAMP-mediated signaling | 0.62 | Insulin receptor substrate binding | −0.57 | Axonemal dynein complex | 0.56 | |||
| 6 | Keratinocyte differentiation | 0.62 | Exonuclease activity | −0.57 | Nuclear membrane | −0.56 | |||
| 7 | Potassium ion transport | 0.61 | DNA N-glycosylase activity | −0.55 | Transcription factor TFIIIC complex | −0.56 | |||
| 8 | Regulation of pH | 0.61 | Mitogen-activated protein kinase kinase binding | −0.55 | Cell surface | 0.55 | |||
| 9 | Interferon-gamma production | 0.61 | Cytokine activity | 0.55 | Voltage-gated potassium channel complex | 0.54 | |||
| 10 | GABA signaling pathway | 0.60 | Spindle | −0.55 | Integrin complex | 0.54 |
The top-ranking GO-CC gene set, chloride channel complex, clearly identifies the main common feature of the chosen drug-set. The top 10 enriched pathways according to DSEA for each GO category resulting from the analysis of the 11 small molecules reported as DF508CFTR-correctors.
Strikingly the most significant pathway ranked in the GO-CC database according to DSEA is the chloride channel complex. This gene-set comprises 38 genes, including CFTR itself. Hence, DSEA predicts that one mode of action shared in common by the 11 drugs is the upregulation of chloride channel genes’ expression (since the ES score associated to chloride channel complex for the drug-set is positive as reported in Table 5). Note that this effect would have never been detected by analysing GEPs of each individual drug separately, as the median rank of the chloride channel complex across the 11 drugs is 196. The 11 drugs belong to very different pharmacological classes, therefore the effect on the chloride channel gene expression is detected by DSEA only because it is a common ‘side-effect’ shared by most of them.
To assess whether the DSEA-predicted shared MoA, i.e. up regulation of chloride channel genes is reasonable, we searched the literature of each of these drugs for evidence of chloride channel gene upregulation. Known effects on CFTR expression have been reported for cardiac glycosides (Srivastava et al., 2004, Strophanthidin in Table 5), HDAC inhibitors (Hutt et al., 2010, Entinostat, Scriptaid, Tricostatin-A) and Doxorubicin, a topoisomerase inhibitor (Maitra et al., 2001). These observations support the results of the DSEA analysis.
Additional signalling pathways, which are known regulators of ion channels activity, are also notable in the DSEA results, such as hormone activity, insulin receptor substrate binding, cytokine activity, which are ranked 2, 5, 9, in GO-CC; and signal recognition particle, ranked 4 in GO-CC.
We also investigated expected pathways which were not found by DSEA. Of interest is the absence of references to protein folding and ER quality control pathways, which seems to exclude a direct role of these 11 drugs as chemical chaperons.
Experimental validations would certainly help in confirming the validity of our hypothesis. However, a more in-depth experimental analysis of these drugs falls out of the scope of our work.
Overall these results confirm the usefulness of this new approach when investigating the shared MoA among a set of unrelated drugs resulting from automated screening efforts.
4 Conclusions
We introduced DSEA, a computational approach to help elucidating the mechanism of action of a set of drugs resulting from automated screening techniques, also available online at http://dsea.tigem.it.
Hit compounds are selected from automated screening if they are able to induce a phenotype of interest. Usually, these selected drugs belong to different pharmacological classes, therefore the molecular mechanisms mediating their effectiveness on the screened phenotype is not immediately obvious. DSEA aims at identifying these mechanisms by looking for recurrent pathways modulated by most of drugs in the set. This is achieved by analysing the transcriptional response elicited by drugs, as available in the cMap database.
With DSEA, we present a new perspective in which drugs represent features of pathways (or genes). The most relevant pathway is thus the one that is most dysregulated by the drugs in the set, as compared with the other drugs in the database. The DSEA analysis is thus able to highlight pathways that are targeted by most of the drugs in the set. Applying DSEA after an automated screening study can thus support the formulation of hypotheses explaining the efficacy of the positive hits.
Beyond High Content Screening, a broad range of drug set generating applications that could take advantage of the DSEA can be imagined. Similarity based methods, like Transcriptional Drug Networks (Carrella et al., 2014) or Virtual Screening (Bajorath, 2002) could exploit DSEA to provide additional biological insights about a drug neighbourhood. Prior knowledge about drugs can be another method to define drug sets. In fact, it is the method we used to define the drug sets for the cancer and cystic fibrosis examples.
In particular, for the cancer application, we defined a set by simply using ATC codes. Although a very heterogeneous class of drugs was analyzed, the DSEA highlighted, as top in GO-BP, a pathway that is very related to mechanisms of action commonly used by antineoplastic agents. In the case of the cystic fibrosis application, instead, we derived the set of DF508CFTR correctors by searching relevant literature. DSEA provided a possible explanation of their corrective effect as mediated by overexpression of CFTR and other chloride channel genes. It is worth observing that expression profiles in cMap were mostly obtained in MCF7 cells, which are very different from bronchial epithelial cells where CFTR is active. Nevertheless, also in this case DSEA was able to detect a strong signal related to the chloride channel complex, which was ranked as the first most significant pathway in the GO-CC category.
The DSEA method was developed and has been validated in a pharmacological context. However, it can be used to analyse any biological condition inducing a measurable transcriptional phenotype, including different cell types, diseases and genetic perturbations. Moreover, the method can be easily applied to other experimental techniques, such as RNA-seq. The DSEA web site provides all the raw and processed data used by the tool, together with pointers to external gene set databases. Moreover, relevant code is maintained on GitHub (https://github.com/franapoli/signed-ks-test). These resources are meant to facilitate the expansion of the DSEA database of pathways and profiles, to encourage further development of the methods and to ensure replicability of our results.
Supplementary Material
Acknowledgement
The authors would like to thank Ramanath Hegde for providing the referenced list of CFTR correctors.
Funding
This study has been funded by: the Italian Ministry of Health (GR-2009-1596824), Fondazione Telethon Grant (TGM11SB1) and the EU FP7 Grant NANOSOLUTIONS (FP7/2007-2013 309329).
Conflict of Interest: none declared.
References
- Bajorath J. (2002) Integration of virtual and high-throughput screening. Nat. Rev. Drug Dis., 1, 882–894. [DOI] [PubMed] [Google Scholar]
- Bickle M. (2010) The beautiful cell: high-content screening in drug discovery. Anal. Bioanal. Chem., 398, 219–226. [DOI] [PubMed] [Google Scholar]
- Caohuy H., et al. (2009) Rescue of DeltaF508-CFTR by the SGK1/Nedd4‐2 signaling pathway. J. Biol. Chem., 284, 25241–25253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlile G.W., et al. (2007) Correctors of protein trafficking defects identified by a novel high-throughput screening assay. Chembiochem Eur. J. Chem. Biol., 8, 1012–1020. [DOI] [PubMed] [Google Scholar]
- Carrella D., et al. (2014) Mantra 2.0: an online collaborative resource for drug mode of action and repurposing by network analysis. Bioinformatics, 30, 1787–1788. [DOI] [PubMed] [Google Scholar]
- Dai M., et al. (2005) Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res., 33, e175–e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokmanovic M., et al. (2007) Histone deacetylase inhibitors: overview and perspectives. Mol. Cancer Res., 5, 981–989. [DOI] [PubMed] [Google Scholar]
- Durinck S., et al. (2009) Mapping identifiers for the integration of genomic datasets with the r/bioconductor package biomaRt. Nat. Protocols, 4, 1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan M.E., et al. (2002) Calcium-pump inhibitors induce functional surface expression of F508-CFTR protein in cystic fibrosis epithelial cells. Nat. Med., 8, 485–492. [DOI] [PubMed] [Google Scholar]
- Gautier L., et al. (2004) affy–analysis of affymetrix GeneChip data at the probe level. Bioinformatics (Oxford, England), 20, 307–315. [DOI] [PubMed] [Google Scholar]
- Hanrahan J.W., et al. (2013) Novel pharmacological strategies to treat cystic fibrosis. Trends Pharmacol. Sci., 34, 119–125. [DOI] [PubMed] [Google Scholar]
- Hutt D.M., et al. (2010) Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat. Chem. Biol., 6, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F., et al. (2010) Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci., 107, 14621–14626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J., et al. (2006) The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science, 313, 1929–1935. [DOI] [PubMed] [Google Scholar]
- Maitra R., et al. (2001) Increased functional cell surface expression of CFTR and DeltaF508-CFTR by the anthracycline doxorubicin. Am. J. Physiol. Cell Physiol., 280, C1031–C1037. [DOI] [PubMed] [Google Scholar]
- O’Sullivan B.P., Freedman S.D. (2009) Cystic fibrosis. Lancet, 373, 1891–1904. [DOI] [PubMed] [Google Scholar]
- Robert R., et al. (2010) Correction of the Delta phe508 cystic fibrosis transmembrane conductance regulator trafficking defect by the bioavailable compound glafenine. Mol. Pharmacol., 77, 922–930. [DOI] [PubMed] [Google Scholar]
- Ruepp A., et al. (2010) CORUM: the comprehensive resource of mammalian protein complexes–2009. Nucleic Acids Res., 38, D497–D501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sackrowitz H., Samuel-Cahn E. (1999) P values as random variables expected P values. Am. Stat., 53, 326–331. [Google Scholar]
- Sams-Dodd F. (2005) Target-based drug discovery: is something wrong? Drug Dis . Today, 10, 139–147. [DOI] [PubMed] [Google Scholar]
- Srivastava M., et al. (2004) Digitoxin mimics gene therapy with CFTR and suppresses hypersecretion of IL-8 from cystic fibrosis lung epithelial cells. Proc. Natl. Acad. Sci. USA, 101, 7693–7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA, 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.