

Abstract
Motivation: Recent technological advances revealed that an unexpected large number of proteins interact with transcripts even if the RNA-binding domains are not annotated. We introduce catRAPID signature to identify ribonucleoproteins based on physico-chemical features instead of sequence similarity searches. The algorithm, trained on human proteins and tested on model organisms, calculates the overall RNA-binding propensity followed by the prediction of RNA-binding regions. catRAPID signature outperforms other algorithms in the identification of RNA-binding proteins and detection of non-classical RNA-binding regions. Results are visualized on a webpage and can be downloaded or forwarded to catRAPID omics for predictions of RNA targets.
Availability and implementation: catRAPID signature can be accessed at http://s.tartaglialab.com/new_submission/signature.
Contact: gian.tartaglia@crg.es or gian@tartaglialab.com
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
RNA-binding proteins (RBPs) use RNA-binding domains (RDs) to recognize target RNAs and to regulate co-/post-transcriptional processes. Examples of classical RDs include RNA-recognition motif (RRM), double-stranded RNA-binding domain (dsRRM), K-homology (KH), RGG box and the Pumilio/FBF (PUM) domain (Lunde et al., 2007). In addition to classical RDs, recent experimental studies on HeLa (Castello et al., 2012), HEK298 (Baltz et al., 2012) and mESC (Kwon et al., 2013) cells, indicate that a number of RNA-interacting proteins contain non-classical RDs (ncRDs) for which annotation is not yet available. Discovery of new RDs is a challenging task: domain-detection tools, such as HMMER (Finn et al., 2011) and BLAST (Camacho et al., 2009) rely on sequence similarity searches to identify annotated RDs and fail to recognize newly discovered RBPs. Similarly, other methods such as RNApred (Kumar et al., 2011) predict RNA-binding ability using features of annotated RDs that might be different in ncRDs. Alternatives to identify RNA-binding regions include BindN+ (Wang et al., 2010), PPRInt (Kumar et al., 2008) and RNAbindR+ (Walia et al., 2014), but the algorithms have been trained to identify single amino acids and not contiguous regions. catRAPID signature overcomes these limitations by (i) predicting the propensity of a protein to interact with RNA and (ii) identifying RNA-binding regions through physico-chemical properties instead of sequence patterns. The algorithm is an extension of the catRAPID approach (Bellucci et al., 2011) to predict protein-RNA interactions and the cleverSuite algorithm (Klus et al., 2014) to classify protein groups using physico-chemical features.
2 Algorithm and performances
To build catRAPID signature we exploited a number of physico-chemical properties reported in our previous publication (Klus et al., 2014):
We used each physico-chemical property [e.g. structural disorder (Castello et al., 2012)] to build a signature, or profile, containing position-specific information arranged in a sequential order from the N- to the C-terminus;
We computed Pearson correlation coefficient between signatures of annotated human RDs and same-length regions taken from RNA-binding proteins as well as negative controls (Supplementary Table S1 and online Documentation);
We identified a number of discriminating physico-chemical properties, their associated RDs and correlation cutoffs (Supplementary Table S2 and online Documentation).
For each protein, we calculated the fraction of residues with correlation coefficients above the cutoffs that are associated with physico-chemical properties and RDs (Table S2; online Documentation), which we then used to train catRAPID signature. Using a Support Vector Machine with RBF-kernel (online Documentation), we built a method for the (i) identification of ribonucleoproteins and (ii) prediction of RNA-binding regions:
catRAPID signature shows an AUC = 0.76 for discrimination of 950 RBPs from 950 negative cases (10-fold cross-validation; Supplementary Fig. S1, Supplementary Data). On an independent test set (Table S3) comprising 47 mouse proteins harboring ncRDs and same number of negatives (Kwon et al., 2013), we obtained accuracy = 0.71, sensitivity = 0.70, specificity = 0.72 and precision = 0.70. By contrast, conventional pattern recognition methods such as HMMER and BLAST show poor sensitivity (Table S3). Our algorithm outperforms RNApred in both specificity and precision (0.25 and 0.52, respectively; Table S3). Moreover, catRAPID signature reliably detects ribonucleoproteins across different kingdoms, including M. pulmonis, E. coli, C. albicans, S. cerevisiae, A. thaliana and A. oryza (Supplementary Fig. S2; online Documentation).
The training for the identification of RNA-binding regions has been done on 1115 annotated RNA-binding regions. As negative counterpart we randomly selected 1115 non-binding regions of the same length from each RBP (AUC = 0.80 in 10-fold cross-validation; Supplementary Fig. S1). On 102 ncRDs versus 102 negative mouse proteins, catRAPID signature outperforms other algorithms: accuracy = 0.67, sensitivity = 0.76, specificity = 0.60 and precision = 0.65 (Supplementary Table S4). By contrast, RNABindR + shows accuracy = 0.48, sensitivity = 0.53, specificity = 0.42 and precision = 0.48. Similar performances were obtained for BindN + and PPRInt (Supplementary Table S4). In addition, we observed high performances on a protein dataset whose RNA-binding sites have been determined through X-ray and NMR (Supplementary Fig. S3 and online Documentation).
3 Server description and example
The input of the server is a FASTA sequence. To illustrate the output with an example, we studied the RNA-binding ability of Fragile X Mental Retardation Protein FMRP. catRAPID signature predicts that FMRP binds to RNA (overall interaction score = 0.85; Fig. 1A; Fig. S4) and correctly identifies two peaks corresponding to the KH domains and one peak in the RGG box (Ascano et al., 2012) [Fig. 1A,B and C; ‘classical’ score = 0.73]. In addition, catRAPID signature indicates that the N-terminus (amino acids 1-215; Fig. 1B) has RNA-binding ability (‘putative’ score = 0.74), which is in agreement with very recent evidence revealing the presence of a novel KH domain (Myrick et al., 2015). Comparing experimental targets [number of PAR-CLIP binding sites ≥ 1] (Ascano et al., 2012) with transcriptome-wide predictions of FMRP N-terminus [amino acids 1–215; Fig. 1D] (Agostini et al., 2013) we observed a significant enrichment in predicted interaction propensities (P-value < 1 −9 calculated with Kolmogorov–Smirnov test on 105 × 103 transcripts of which 7 × 103 positives), which suggests that the N-terminus contributes to the RNA-binding ability of the full-length FMRP.
Fig. 1.
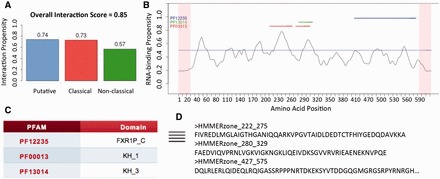
RNA-binding ability of Fragile X Mental Retardation Protein FMRP. (A) The server reports the propensity of FMRP for the putative (0.74), classical (0.73) and non-classical (0.57) RBP classes, as well as an overall prediction score (0.85); (B) The profile shows protein regions and their propensity to interact with RNA. catRAPID signature correctly identifies two peaks corresponding to the central KH domains, a region in the RGG box [amino acids 527-552] at the C-terminus (Ascano et al., 2012) and a recently discovered RD at the N-terminus (Myrick et al., 2015). (C) Annotated RDs are shown in a table and linked to PFAM webpages; (D) Annotated and predicted RNA-binding sequences can be downloaded and/or forwarded to catRAPID omics (Agostini et al., 2013) for further analysis
4 Conclusions
As newly discovered RDs are not annotated, traditional domain-detection tools fail their identification. catRAPID signature addresses this limitation by detecting binding regions through physico-chemical features. Our algorithm will be helpful to investigate components of ribonucleoprotein complexes and to identify RNA-binding regions.
Supplementary Material
Acknowledgements
The authors would like to thank Prof R. Guigó and Dr C. Notredame for stimulating discussions.
Funding
The research leading to these results has received funding from the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013) / ERC grant agreement n°RIBOMYLOME_309545. We acknowledge support of the Spanish Ministry of Economy and Competitiveness, ‘Centro de Excelencia Severo Ochoa 2013-2017', SEV-2012-0208 and FEDER funds (European Regional Development Fund) under the project number BFU2014-55054-P. R. Delli Ponti is supported by the MINECO's pre-doctoral grant Severo Ochoa 2013–2017 (SVP-2014-068402).
Conflict of Interest: none declared.
References
- Agostini F., et al. (2013) catRAPID omics: a web server for large-scale prediction of protein-RNA interactions. Bioinformatics, 29, 2928–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascano M., et al. (2012) FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature, 492, 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltz A.G., et al. (2012) The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell, 46, 674–690. [DOI] [PubMed] [Google Scholar]
- Bellucci M., et al. (2011) Predicting protein associations with long noncoding RNAs. Nat. Methods, 8, 444–445. [DOI] [PubMed] [Google Scholar]
- Camacho C., et al. (2009) BLAST+: architecture and applications. BMC Bioinformatics, 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A., et al. (2012) Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell, 149, 1393–1406. [DOI] [PubMed] [Google Scholar]
- Finn R.D., et al. (2011) HMMER web server: interactive sequence similarity searching. Nucleic Acids Res., 39, W29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klus P., et al. (2014) The cleverSuite approach for protein characterization: predictions of structural properties, solubility, chaperone requirements and RNA-binding abilities. Bioinformatics, 30, 1601–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M., et al. (2008) Prediction of RNA binding sites in a protein using SVM and PSSM profile. Proteins, 71, 189–194. [DOI] [PubMed] [Google Scholar]
- Kumar M., et al. (2011) SVM based prediction of RNA-binding proteins using binding residues and evolutionary information. J. Mol. Recognit., 24, 303–313. [DOI] [PubMed] [Google Scholar]
- Kwon S.C., et al. (2013) The RNA-binding protein repertoire of embryonic stem cells. Nat. Struct. Mol. Biol., 20, 1122–1130. [DOI] [PubMed] [Google Scholar]
- Lunde B.M., et al. (2007) RNA-binding proteins: modular design for efficient function. Nat. Rev. Mol. Cell Biol., 8, 479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myrick L.K., et al. (2015) Human FMRP contains an integral tandem Agenet (Tudor) and KH motif in the amino terminal domain. Hum. Mol. Genet., 24, 1733–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walia R.R., et al. (2014) RNABindRPlus: a predictor that combines machine learning and sequence homology-based methods to improve the reliability of predicted RNA-binding residues in proteins. PLoS ONE, 9, e97725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., et al. (2010) BindN+ for accurate prediction of DNA and RNA-binding residues from protein sequence features. BMC Syst Biol, 4Suppl 1, S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.