

Abstract
The secreted disulfide catalyst Quiescin sulfhydryl oxidase-1 (QSOX1) affects extracellular matrix organization and is overexpressed in various adenocarcinomas and associated stroma. Inhibition of extracellular human QSOX1 by a monoclonal antibody decreased tumor cell migration in a cell co-culture model and hence may have therapeutic potential. However, the species specificity of the QSOX1 monoclonal antibody has been a setback in assessing its utility as an anti-metastatic agent in vivo, a common problem in the antibody therapy industry. We therefore used structurally guided engineering to expand the antibody species specificity, improving its affinity toward mouse QSOX1 by at least four orders of magnitude. A crystal structure of the re-engineered variant, complexed with its mouse antigen, revealed that the antibody accomplishes dual-species targeting through altered contacts between its heavy and light chains, plus replacement of bulky aromatics by flexible side chains and versatile water-bridged polar interactions. In parallel, we produced a surrogate antibody targeting mouse QSOX1 that exhibits a new QSOX1 inhibition mode. This set of three QSOX1 inhibitory antibodies is compatible with various mouse models for pre-clinical trials and biotechnological applications. In this study we provide insights into structural blocks to cross-reactivity and set up guideposts for successful antibody design and re-engineering.
Keywords: anti-metastatic agents, dual-specificity, laminin, monoclonal antibodies, structure-guided engineering
Introduction
Monoclonal antibody therapy has become an integral part of cancer diagnostics and treatment (Scott et al., 2012). The success of antibody-based therapy stems from the high specificity and affinity that antibodies offer compared with other antitumor agents. In addition, tumors express on their cell surfaces many potential targets for antibody therapeutics. Antibodies supplied extracellularly can both neutralize the function of their cell-surface antigen and recruit the immune system for a more extensive antitumor response (Hudis, 2007; Weiner, 2010). The realization that tumor stroma has a major role in supporting tumor development and metastasis inspired antibody-based cancer therapies targeting extracellular matrix (ECM) components in addition to targeting tumor cells directly. Examples for such agents are antibodies that affect the extracellular glycoprotein tenascin (Rizzieri et al., 2004) or fibroblast activation protein, found in stromal fibroblasts of most human carcinomas (Scott et al., 2003).
ECM proteins are good candidates for antibody therapy because they are both accessible and abundant in most tissues, making the same ECM components a target in various cancers. A major component of the ECM that is overexpressed, reorganized and cross-linked in tumorigenesis is collagen. Collagen cross-linking by the enzyme lysyl oxidase (LOX) is increased in several cancers and contributes to matrix stiffening, thereby promoting cell adhesion and migration (Levental et al., 2009). Inhibitors of LOX activity, including monoclonal antibodies, significantly inhibited tumor growth and metastasis in gastric carcinoma (Peng et al., 2009; Rodriguez et al., 2010). Laminin is another abundant scaffolding ECM protein that interacts with integrins to mediate cell adhesion and migration, a requirement for metastasis. Indeed, laminin is overexpressed in various cancers, and its chain isotypes serve as tumor biomarkers (Nagato et al., 2005; Huang et al., 2008; Beroukhim et al., 2009; Kosanam et al., 2013). Like collagen cross-linking and integrin blocking (Zhaofei et al., 2008), laminin incorporation into the matrix may serve as a complementary target for antibody-based cancer therapeutics.
We have recently shown that laminin incorporation into the ECM is affected by the disulfide catalyst Quiescin sulfhydryl oxidase-1 (QSOX1) (Ilani et al., 2013). The enzyme QSOX1 is a fusion of two thioredoxin (Trx) domains and an Erv-fold sulfhydryl oxidase module (Fig. 1a) (Heckler et al., 2008). QSOX1 contains two CXXC motifs as redox-active sites that cooperate to relay electrons from reduced thiols of substrate proteins to molecular oxygen. Mechanistically, after oxidizing the substrate, the Trx active site transfers two electrons to the Erv CXXC motif through an inter-domain disulfide intermediate (Supplementary Fig. S1). The electrons proceed to the adjacent flavin adenine dinucleotide cofactor, which in turn reduces oxygen to hydrogen peroxide, leaving QSOX1 oxidized and ready for another catalytic cycle. Unlike other disulfide catalysts, QSOX1 is localized downstream of the endoplasmic reticulum (ER). It is found in the Golgi apparatus and secreted from quiescent fibroblasts into the ECM (Coppock et al., 2000), where it affects ECM composition and especially laminin incorporation (Ilani et al., 2013). Specifically, QSOX1 affects the incorporation of laminin isoforms that contain an α4 chain (Ilani et al., 2013), a known marker for tumor progression (Nagato et al., 2005; Huang et al., 2008; Beroukhim et al., 2009). Together with the overproduction of QSOX1 in various adenocarcinomas (Antwi et al., 2009; Soloviev et al., 2013) and associated stroma (Finak et al., 2008), these findings point to a possible role of QSOX1 in stimulating tumor cell migration via laminin incorporation.
Fig. 1.
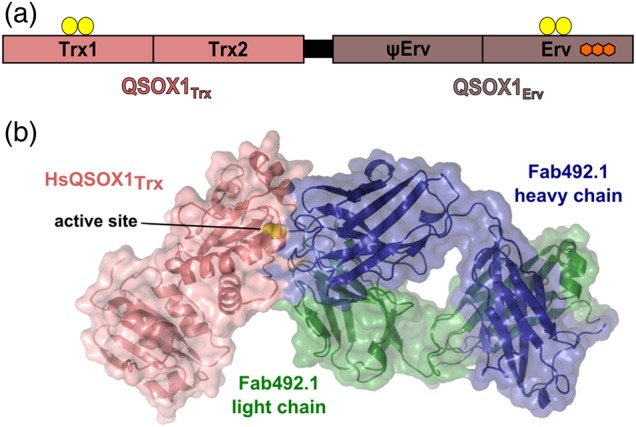
QSOX1 domain organization and inhibition by MAb492.1. (a) The two modules of mammalian QSOX1 enzymes are linked by a flexible linker (black). The amino-terminal module, QSOX1Trx, is composed of two Trx-fold domains, the first of which has a redox-active CXXC motif (yellow balls). A second redox-active CXXC motif is located in the carboxy-terminal Erv domain. This domain binds an FAD cofactor (fused hexagons). The domain labeled ψErv is a degenerate Erv domain lacking active-site cysteines and an FAD cofactor. (b) Surface presentation of a complex of a Fab fragment from MAb492.1 (Fab492.1) and HsQSOX1Trx (PDB ID: 4IJ3) showing that Fab492.1 inhibits HsQSOX1 by burying the Trx CXXC active site (yellow spheres).
The multistep catalytic cycle of QSOX1 implies that obscuring any one of the several sites on the protein by interaction with antibody may accomplish inhibition. We developed an inhibitory monoclonal antibody, MAb492.1, which blocks substrate access to the Trx CXXC redox-active site of human QSOX1 (HsQSOX1) (Fig. 1b) (Grossman et al., 2013). MAb492.1 efficiently inhibited HsQSOX1 activity, and consequently inhibited adhesion and migration of cancer cells to and through fibroblasts from corresponding tissues (Ilani et al., 2013). We proposed that MAb492.1 may serve as an anti-metastatic drug in antibody-based cancer therapy (Grossman et al., 2013; Ilani et al., 2013).
In certain cases, an antibody targeting a human protein is sufficient for pre-clinical trials in animal models due to the availability of xenograft and tumorgraft models (Warburton et al., 2004). However, a major disadvantage of these models is the artificial interaction of the tumor with surrounding tissues. Moreover, eventual recruitment of mouse fibroblasts will create an inhomogeneous tumor microenvironment composed of both mouse and human constituents. Hence, when targeting a secreted protein such as QSOX1, mouse models with a natural organization of murine cancer cells and stroma, such as genetically modified mice that develop spontaneous tumors (Francia et al., 2011), may be preferable for studying metastasis development.
In this study we report the structure-guided modification of the MAb492.1 antibody to introduce the desired reactivity against the mouse QSOX1 ortholog. The re-engineered antibody was constructed and characterized in parallel with an inhibitory monoclonal antibody raised against mouse QSOX1 in a QSOX1-null animal. These two reagents were developed using different strategies, leading to inhibitors with different binding modes and distinct species cross-reactivity, but both are compatible with mouse models for in vivo QSOX1 inhibition experiments and biotechnological applications. Our structural analysis of the binding of these antibodies to QSOX1 orthologs provides insights into the challenges in modifying antibody specificity and ways to overcome them.
Materials and methods
Plasmid construction
HsQSOX1 and MmQSOX1 mutants were made by restriction-free cloning based on the published HsQSOX1 (Grossman et al., 2013) and MmQSOX1 (Alon et al., 2012) expression plasmids. MmQSOX1Trx and MmQSOX1Erv span residues 36–275 and 289–550, respectively, of Mus musculus QSOX1. The RnQSOX1 construct was previously described (Gat et al., 2014). The CpQSOX1 protein spans residues 34–547 of Cavia porcellus QSOX1. A synthetic gene (Genescript) codon-optimized for CpQSOX1 production in E. coli was cloned between the NdeI and BamHI sites of the pET-15b vector (Novagen). For production of biotinylated MmQSOX1Trx, an AviTag (Kay et al., 2009) was added at the carboxy terminus.
QSOX1 expression and purification
Recombinant HsQSOX1 (Grossman et al., 2013) and RnQSOX1 (Gat et al., 2014) were expressed and purified as described. MmQSOX1, MmQSOX1Erv and CpQSOX1 were prepared as for HsQSOX1. MmQSOX1Trx used for crystallization was produced in the BL21 (DE3) E. coli strain. Cells were grown in LB media to OD600 nm 0.5 at 37°C. Isopropyl-1-thio-β-d-galactopyranoside (IPTG) was added to a final concentration of 0.5 mM, and the cultures were grown for a further 40 h at 15°C. Cells were lysed in 20 mM Tris buffer, pH 8.5, 500 mM NaCl, 20 mM imidazole, sonicated, and centrifuged for 1 h at 40 000 × g. The supernatant was applied to a Ni-NTA column (GE healthcare), and protein was eluted in 20 mM Tris buffer, pH 8.5, 500 mM NaCl, and a gradient of imidazole (20–500 mM). Eluted protein was further purified by size-exclusion chromatography on a Superdex 75 16/60 column in 10 mM Tris buffer, pH 8, 100 mM NaCl. Purified MmQSOX1Trx was concentrated to 12 mg/ml, and immediately before crystallization was mixed with thrombin (3 units/mg MmQSOX1Trx). Biotinylated MmQSOX1Trx for yeast-surface display screening was co-expressed with an expression plasmid for biotin ligase. Upon induction of protein expression with 500 μM IPTG, biotin was added to the growth medium at a concentration of 50 μM, and the cultures were grown for a further 24 h at 20°C. Purification of biotinylated MmQSOX1Trx was as for MmQSOX1Trx. Biotinylation was verified by enzyme-linked immunosorbent assay (ELISA).
Oxygen consumption assays for testing QSOX1 enzymes activity and inhibition
Recombinant mammalian QSOX1 enzymes (100 nM) were assayed with 200 μM dithiothreitol (DTT) and various MAb492.1 concentrations in a Clarke-type oxygen electrode (Hansatech Instruments) as reported (Grossman et al., 2013). Reactions were initiated by DTT injection, and oxygen consumption rates were obtained from initial slopes. For testing inhibition of MmQSOX1 and HsQSOX1 mutants by MAb492.1 and MAb316.1, respectively, 50 nM enzyme was assayed with 200 μM DTT, with and without 250 nM antibody. Measurements were performed three times, and resulting rates were averaged. For each mutant, the rate in the presence of inhibitory antibody was divided by the rate in the absence of antibody to get percent activity.
Generation and selection of MAb316.1
Hybridomas were generated and supernatants of ∼1000 clones were screened for MmQSOX1 binding as described (Ilani et al., 2013). Twenty top binders were tested for MmQSOX1 inhibition in the in vitro oxygen consumption assay, in which hybridoma supernatants were mixed with 100 nM MmQSOX1 and 200 μM DTT. Two inhibitory clones in addition to three strong binders were chosen for sub-cloning. Each of the sub-clones was tested for binding by ELISA. Approximately 40 sub-clones were chosen for inhibition assays. Supernatants of sub-clones 316.1 inhibited MmQSOX1 activity repeatedly, and so were chosen for further studies. MAb316.1 used for inhibition assays was produced in a miniPERM bioreactor (Sarstedt) in serum-free medium (DCCM) and was purified as described previously (Ilani et al., 2013). Variable region sequencing of both monoclonal antibodies was performed as described previously (Grossman et al., 2013). Briefly, the variable region was reverse transcribed from hybridomal mRNA and amplified by PCR using sets of degenerate primers (Zhou et al., 1994; Benhar and Reiter, 2002). Amplified fragments of the heavy chain and light chain were then cloned into the pGEM-T Easy vector, sequenced and analyzed in the ImMunoGeneTics database (Lefranc et al., 2005). A productively rearranged sequence was confirmed for each fragment and verified on the protein level by liquid chromatography–tandem mass spectrometry of purified MAb316.1 (Supplementary Table SII).
Screening of scFv492.1 mutants with yeast-surface display
The scFv492.1 construct, without the His-tag and thrombin cleavage site (Grossman et al., 2013), was cloned between the NdeI and BamHI sites in the yeast display plasmid pETCON (Fleishman et al., 2011). Mutants of scFv492.1 were prepared on the basis of this plasmid by in vivo recombination in EBY100 yeast using the LiAc method. Yeast growth and induction of scFv expression were done as described previously (Chao et al., 2006), except that for induction 10 g raffinose per liter were added to the media and the cultures were grown for 16 h at 20°C. Yeast were labeled with anti-cMyc (Santa Cruz Biotechnology) at a dilution 1:50 to monitor scFv expression, and with biotinylated MmQSOX1Trx (for concentrations, see Table II) for 45 min. After washing, secondary labeling was performed with streptavidin–allophycocyanin (streptavidin–APC) from Jackson Immunoresearch Laboratories, Inc. at a 1:50 dilution and with goat anti-mouse IgG1 secondary antibody Alexa fluor 488 conjugate (Life Technologies) at a 1:100 dilution, for the anti-cMyc labeling. Alternatively, in the first rounds of screening (Table I), yeast were labeled with anti-cMyc, washed, and then labeled with goat anti-mouse IgG1 secondary antibody Alexa fluor 488 conjugate and with biotinylated MmQSOX1Trx pre-loaded with streptavidin-APC in a 1:4 molar ratio (Chao et al., 2006). Display of scFv clones and MmQSOX1Trx binding was monitored by Alexa 488 and APC fluorescence, respectively, using an Accuri C6 flow cytometer. Anti-fluorescein scFv (PDB ID: 1X9Q) was used as a positive control for scFv display. Yeast displaying scFv492.1 labeled with biotinylated HsQSOX1Trx were used as a positive control for binding.
Table II.
Inhibition constants for antibodies targeting QSOX1
| Antibody/enzyme | HsQSOX1 | MmQSOX1 |
|---|---|---|
| MAb492.1 | 0.9 ± 0.1 nM | No inhibition at 1 μM |
| MAb492gen | 1.6 ± 0.6 nM | 2.2 ± 0.5 nM |
| MAb316.1 | No inhibition at 1 μM | 16 ± 2 nM |
Table I.
MAb492.1 residues mutated to generate MAb492gen
| Template name | Template for mutations | Residuea | Mutation | Strategy | MmQSOX1Trx concentration used for binding tests | Positive mutationsb |
|---|---|---|---|---|---|---|
| wt | scFv492.1 | Y92 (light chain) | N/D | 25 of the possible combinatorial mutants involving these three residues were tested | 500 nM pre-loaded with streptavidin-APC | S93A |
| S93 (light chain) | G/A | |||||
| Y100 (heavy chain) | D/N/G/S | |||||
| a | scFv492.1 S93A | Y99-S102 (heavy chain) | All possible amino acids | Library in potential size of 204 clones subjected to three rounds of selection | 250 nM pre-loaded with streptavidin- APC | Y99S Y100K G101D S102P |
| b | scFv492.1 S93A Y99S Y100K G101D S102P |
Y92-P95 (light chain) | All possible amino acids | Library in potential size of 204 clones subjected to three rounds of selection | 250 nM pre-loaded with streptavidin- APC | – |
| T30-Y32 (heavy chain) | All possible amino acids | Library in potential size of 203 clones subjected to three rounds of selection | 250 nM pre-loaded with streptavidin- APC | – | ||
| W52-D54, R56 (heavy chain) | All possible amino acids | Library in potential size of 204 clones subjected to three rounds of selection | 250 nM pre-loaded with streptavidin- APC | – | ||
| D58 (heavy chain) | E/Q | Tested point mutations for MmQSOX1Trx binding | 250 nM pre-loaded with streptavidin- APC | D58E | ||
| c | scFv492.1 S93A Y99S Y100K G101D S102P D58E |
S30-T31 (light chain) | All possible amino acids | Library in potential size of 202 clones subjected to three rounds of selection | 250 nM pre-loaded with streptavidin- APC | T31G |
| H49-S50, Y52 (light chain) |
All possible amino acids | Library in potential size of 203 clones subjected to three rounds of selection | 250 nM pre-loaded with streptavidin- APC | H49S S50M Y52Q |
||
| d | scFv492.1 S93A Y99S Y100K G101D S102P D58E T31G H49S S50M Y52Q |
All | All | Library was created by epPCR and its size was evaluated as 5 × 107 clones. Library was subjected to three rounds of selection | 250 nM (not pre-loaded) |
G33S N35I S100M (heavy chain) |
aNumbering is according to structure of HsQSOX1Trx–Fab492.1 complex (PDB ID: 4IJ3).
bMutations that improved MmQSOX1Trx binding.
Small scFv libraries were constructed by fully randomizing specific residue positions (Table I) using the NNS codon. Oligonucleotides with NNS codons in desired positions and wild-type flanking regions were ordered from Sigma. NdeI and BamHI sites were deleted from the N- and C-termini of the scFv construct in the pETCON plasmid and inserted flanking the desired region for randomization. The plasmid was restricted, and the oligonucleotide pool was inserted through in vivo recombination using the LiAc method. In all libraries constructed, the number of colonies obtained was at least an order of magnitude larger than the potential size of the library (20number of randomized positions). Libraries were induced and labeled as described above. Cells were sorted using a FACSAria III Cell Sorter in three iterative rounds of enrichment. In the first sorting round, the top 5% cells found within the green and red fluorescence area were collected into growth media. In the following sorting rounds, the top 1–4% cells were collected. Plasmids from the last cycle of FACS enrichment were sequenced. Eighteen colonies enriched from the library constructed in CDR H3 were sequenced, yielding two distinct sequences. Each of the two sequences was tested separately for MmQSOX1Trx binding, and the better binder (Table I) was chosen for further study. Enrichment of the library constructed in L1 yielded a single clone, verified by sequencing 12 colonies. Enrichment of the library constructed in L2 yielded eight clones, which were tested for MmQSOX1Trx binding separately. We incorporated the enriched L1 sequence to the top binder. After verifying that the combined clone showed increased MmQSOX1Trx binding compared with the clones enriched directly from the L1 and L2 libraries, this clone was subjected to epPCR on the entire scFv sequence using the Agilent GeneMorph II Random Mutagenesis kit. Recombination in vivo was performed by electroporation, yielding a library of size 5 × 107. The library was subjected to three rounds of selection as described above, recovering scFv492gen.
Recombinant MAb492gen production
The light-chain and heavy-chain variable regions of scFv492gen were amplified and cloned separately into mammalian expression vectors for human IgG1 antibodies (Tiller et al., 2008). MAb492gen was expressed by transient co-transfection of the two plasmids in human embryonic kidney (HEK) 293T cells using polyethylenimine. Cell supernatants were collected and replaced with fresh medium every 4 days. Expression and secretion to the medium was verified by western blot. MAb492gen was purified from the media using protein G (Ilani et al., 2013). Purified MAb492gen with human constant regions was used for inhibition assays.
Inhibitory constant determination
Colorimetric assays of RNase A oxidation were performed as described previously to obtain IC50 values (Grossman et al., 2013). Ki values were obtained in oxygen consumption assays as described previously (Grossman et al., 2013).
Laminin staining and cell adhesion assay
The assays were conducted essentially as described previously (Ilani et al., 2013). Instead of human fibroblasts, mouse fibroblasts (provided by Dr. Y. Hanna) were grown for 3 days in the presence of various antibodies. HEK 293T cells were used as adhering cells.
Analytical size-exclusion chromatography
MsQSOX1, its fragments, or MAb316.1 was loaded onto a Superdex 200 column (GE HealthCare) equilibrated with phosphate-buffered saline at a flow rate of 0.8 ml/min. MsQSOX1–antibody complexes were injected after a 30 min co-incubation at 4°C. Elution of proteins was monitored by absorbance at 280 nm.
Fab–MmQSOX1Trx complex formation
MAb492.1 light chain and heavy chain were amplified from hybridomal cDNA (Grossman et al., 2013) and cloned into the MAb492gen expression vectors, separately. Variable regions of scFv492gen were amplified and cloned into these vectors instead of the 492.1 variable regions. A stop codon was inserted after the CH1 segment of the heavy chain, to obtain a coding sequence for a Fab fragment (Fab492gen). Fab492gen was expressed and purified as was MAb492gen. The heavy and light variable regions of MAb316.1 were amplified from hybridomal cDNA and cloned separately into the above-mentioned MAb492gen expression vectors. MAb316.1 was expressed and purified as for MAb492gen. A Fab fragment from MAb316.1 (Fab316.1) was prepared by papain digestion as described previously (Grossman et al., 2013). Purified Fab492gen or Fab316.1 was mixed with purified MmQSOX1Trx at a 1 : 2 ratio for 30 min at 4°C to form complexes, which were purified from excess MmQSOX1Trx by size-exclusion chromatography. Prior to crystallization the complexes were concentrated to 10 mg/ml.
Protein crystallization
Crystals were grown by hanging-drop vapor diffusion at 293 K. MmQSOX1Trx crystals used for seeding were grown over a well solution containing 0.2 M ammonium sulfate, 0.1 M sodium acetate, pH 4.6, 21% w/v polyethylene glycol (PEG) 4 kDa. These crystals were crushed and seeded into drops of MmQSOX1Trx grown over a well solution containing 5% w/v dimethyl sulfoxide, 0.1 M sodium acetate, pH 4.6, 7% w/v PEG monomethyl ether 2 kD. Crystals were transferred to a solution containing 0.1 M sodium acetate, pH 4.6, 15% w/v PEG monomethyl ether 2 kDa, 25% glycerol and flash frozen in a nitrogen stream at 100 K. Crystals of the MmQSOX1Trx–Fab316.1 complex were grown over a well solution containing 50 mM ammonium sulfate, 0.1 M bis–tris methane buffer, pH 5.5, 22% w/v PEG 3.35 kD. Crystals were transferred to a solution with the same content and 25% w/v glycerol for freezing. Crystals of the MmQSOX1Trx–Fab492gen complex were grown over a well solution containing 50 mM CaCl2, 0.1 M 2-(N-morpholino)ethanesulfonic acid buffer, pH 6, 22.5% w/v PEG 6 kDa, and were transferred to the same solution containing 25% w/v glycerol prior to freezing.
Data collection
Diffraction data for MmQSOX1Trx, MmQSOX1Trx–Fab316.1 complex and MmQSOX1Trx–Fab492gen complex were collected at 100 K on a RU-H3R generator (Rigaku) equipped with a RaxisIV++ image plate system and Osmic mirrors. For MmQSOX1Trx, data were collected to 2.05 Å resolution from a crystal of space group P21. MmQSOX1Trx–Fab316.1 complex data were collected to 2.2 Å resolution from a crystal of space group P212121. Diffraction data for MmQSOX1Trx–Fab492gen were collected to 2.3 Å resolution from a crystal of space group P21. All data sets were processed and scaled using DENZO and SCALEPACK (Broennimann et al., 2006).
Structure solution
All three structures were determined by molecular replacement using Phaser (McCoy et al., 2007). The HsQSOX1Trx structure (PDB ID: 3Q6O) was used as a search model for MmQSOX1Trx, and model rebuilding and mutagenesis was done in Coot (Emsley and Cowtan, 2004). The resulting MmQSOX1Trx structure (PDB ID: 5D8I) was used as a search model for both MmQSOX1Trx–Fab complexes. After translation and rotation functions were found for MmQSOX1Trx, a search for the constant region of Fab316.1 was performed using an identical constant region (PDB ID: 3D85, chain A spanning residues 108–213 and chain B spanning residues 116–217). The variable region of Fab316.1 heavy chain was searched with a model having 87% identity (PDB ID: 4Q0X, chain H spanning residues 1–118 without CDR H3), and the light-chain variable region was searched with a 94% identical model (PDB ID: 3AB0, chain C spanning residues 1–106). For Fab492gen, three search models from Fab492.1 (PDB ID: 4IJ3, chains B and C) were used: the constant region, the light-chain variable region without the mutated residues in Fab492gen, and the heavy-chain variable region without CDRs H1 and H3. For both complexes, addition of CDR loops and model rebuilding were done using Coot (Emsley and Cowtan, 2004). Refinement was performed using Phenix (Afonine et al., 2005). Validation was done using MolProbity (Lovell et al., 2003), according to which there are no Ramachandran outliers in the structures reported herein.
Accession numbers
Protein Data Bank: Coordinates and structure factors have been deposited with the accession codes 5D8I for MmQSOX1Trx, 5D93 for the Fab316.1–MmQSOX1Trx complex and 5D96 for the Fab492gen–MmQSOX1Trx complex.
Results
The QSOX1 inhibitor MAb492.1 is species-specific
MAb492.1, which inhibits HsQSOX1 activity in vitro and in cell culture at a near-stoichiometric concentration (Grossman et al., 2013), was tested on other mammalian QSOX1 enzymes to find a suitable model for assessing QSOX1 inhibition in vivo. Three mammalian QSOX1 enzymes, from common experimental organisms and having 78–79% sequence identity with HsQSOX1, were chosen: Mus musculus QSOX1 (MmQSOX1), Rattus norvegicus QSOX1 (RnQSOX1) and Cavia porcellus (CpQSOX1). Enzyme activity was assessed in an oxygen consumption assay on the model substrate DTT. According to the assay, MAb492.1 had no effect on MmQSOX1, RnQSOX1 or CpQSOX1, even at 1 μM, a 10:1 molar ratio of antibody to enzyme (Fig. 2a).
Fig. 2.

MAb492.1 is species specific. (a) Turnover numbers of various mammalian QSOX1 enzymes in the absence and presence of 250 nM or 1 μM MAb492.1. Activity was evaluated using an oxygen consumption assay. (b) Sequence alignment of HsQSOX1 and other mammalian QSOX1 orthologs in the region bound by MAb492.1. Residues involved in interactions with MAb492.1 are in bold. Residues from MmQSOX1, RnQSOX1 and CpQSOX1 that differ from the corresponding HsQSOX1 residues are colored in orange. (c) Superposition of the Fab492.1–HsQSOX1Trx complex structure (PDB ID: 4IJ3) and the structure of MmQSOX1Trx (PDB ID: 5D8I). The two chains of MmQSOX1Trx from the crystal asymmetric unit are light (chain B) and dark (chain A) gray, and HsQSOX1Trx is pink. Right, close-up of the expected clash between CDR H3 and residues TLPG(138–141) from MmQSOX1. Left, close-up of the expected clash between CDR L3 and Asn117 from MmQSOX1. Only chain B of MmQSOX1Trx is shown for simplicity. (d) Percent activity for different MmQSOX1 mutants in the presence of MAb492.1. Oxygen consumption measurements were conducted with 100 nM enzyme, 250 nM MAb492.1 and 200 μM DTT. Percent activity was calculated according to the results of the same measurements in the absence of antibody. Error bars represent standard deviations from an average of three measurements.
To understand the molecular basis for MAb492.1 species restriction, we compared the epitope on HsQSOX1 with the corresponding regions of RnQSOX1, CpQSOX1 and particularly MmQSOX1 by crystallizing and solving the structure of the Trx module of MmQSOX1 (MmQSOX1Trx) to 2.05 Å resolution (Supplementary Table SI). Two MmQSOX1Trx molecules were present in the crystal asymmetric unit. The atomic coordinates of these two molecules were overlaid on the previously solved structure of a complex between HsQSOX1Trx and Fab492.1 (Grossman et al., 2013). Although the regions near the CXXC redox-active site are identical in sequence among QSOX1 orthologs, a few other positions of contact between Fab492.1 and HsQSOX1 differ (Fig. 2b). In particular, HsQSOX1 P116, which fits well into a cleft between hydrophobic complementary determining regions (CDRs) L3, H2 and H3 of Fab492.1, is replaced with alanine in other mammalian QSOX1 enzymes. The MmQSOX1Trx structure (Fig. 2c) showed that this alanine residue cannot fill the hydrophobic cleft in a hypothetical complex between MmQSOX1 and Fab492.1. Moreover, the replacement of proline by alanine affects the position of the backbone nearby, such that a clash would form between MmQSOX1 N117 and CDR L3 (Fig. 2c, left). Another region that is not conserved among the QSOX1 enzymes examined is VFPV(135–138) from HsQSOX1. The corresponding TLPG(138–141) loop in one of the MmQSOX1 molecules is displaced from the antibody CDRs L1 and L2 and is found closer to CDR H3 than the VFPV(135–138) loop of HsQSOX1 (Fig. 2c, right). As a result, MAb492.1 CDR H3 would clash sterically with this loop, eliminating potential interactions of MmQSOX1 with CDRs L1 and L2.
To confirm that the structural differences noted between HsQSOX1 and MmQSOX1 are the cause of MAb492.1 species specificity, three MmQSOX1 mutants that mimic HsQSOX1 in distinct positions were constructed. MAb492.1 inhibition was tested on these mutants to identify the residues that interfere with MAb492.1–MmQSOX1 complex formation. The first mutant, A116P, was inhibited by about 60% (Fig. 2d), and the second mutant, TLPG(138–141)VFPV, by about 50%. The third mutant, which comprises both the above mutations, was inhibited by MAb492.1 to the same extent as HsQSOX1 (100%), confirming the identity of residues that determine the specificity of MAb492.1 toward HsQSOX1.
Generation and characterization of a murine antibody inhibitor targeting MmQSOX1
The murine immune system used to elicit MAb492.1 (Ilani et al., 2013) is likely to be biased toward antibodies that recognize nonself epitopes on HsQSOX1. We therefore exploited QSOX1 knock-out (QSOX1-KO) mice produced in our laboratory to generate surrogate antibodies against MmQSOX1, which is a foreign antigen for these animals. Hybridoma supernatants were screened for binding of MmQSOX1 using a standard ELISA. Top binders were tested for MmQSOX1 inhibition, and five were chosen for sub-cloning (see Materials and Methods). Antibody sub-clones derived from one particular clone inhibited MmQSOX1, and one sub-clone, designated as MAb316.1 and classified to be of the IgG1 isotype, was selected for further study. The sequences of the MAb316.1 variable regions (see Materials and Methods) are presented in Supplementary Table SII.
Generation of a MAb492.1 variant targeting MmQSOX1
In parallel to obtaining an antibody targeting MmQSOX1Trx from hybridoma clones, we developed a variant of MAb492.1 that inhibits MmQSOX1. According to the observation that mutating four residues of MmQSOX1 was sufficient to achieve inhibition by MAb492.1 (Fig. 2d), we reasoned that inhibiting MmQSOX1 would be possible by making a set of mutations in the MAb492.1 CDRs or surrounding regions. Candidate residues for mutation to improve MmQSOX1Trx binding were identified based on the structures of MmQSOX1Trx and the Fab492.1–HsQSOX1Trx complex (Table I). We tested specific point mutations and constructed small libraries, varying up to four CDR residues in specific locations, from which binding mutants were enriched.
Screening was performed using yeast-surface display of single-chain variable fragments (scFv) (Chao et al., 2006) (see Materials and Methods). An scFv variant of MAb492.1 (scFv492.1) had been constructed, expressed in E. coli, and shown to inhibit HsQSOX1 successfully (Grossman et al., 2013), laying the groundwork for using yeast-surface display to select for MmQSOX1 inhibitors. An scFv variant carrying 10 mutations in the CDRs achieved by specific rational mutations and consecutive rounds of library sorting and enrichment (Table I) bound MmQSOX1Trx on the surface of yeast (Fig. 3a). Since mutations in other CDR residues did not improve MmQSOX1Trx binding further, we performed one round of affinity maturation on the scFv492.1 mutant for fine-tuning. To this end we constructed a gene library (total size of 5 × 107 clones) by error-prone polymerase chain reaction (epPCR), with an average of three mutations per gene, and subjected the library to three rounds of flow-cytometry selection for MmQSOX1Trx binding. A mutant with a total of 12 mutations on scFv492.1, designated as scFv492gen (Supplementary Fig. S2), was isolated and characterized, yielding an apparent Kd of 65 nM on the surface of yeast (Supplementary Fig. S3). Based on the scFv492gen sequence, a recombinant full-length antibody, MAb492gen, was produced in mammalian cells.
Fig. 3.

Evaluation of binding and inhibition constants of antibodies targeting MmQSOX1. (a) Histograms of fluorescence (APC-conjugated streptavidin) reporting MmQSOX1Trx binding to the surface of yeast displaying scFv492.1 or a scFv492.1 mutant (mutant d, Table I). Each histogram represents 50 000 yeast cells. The scFv492.1 mutant shows an increase in APC fluorescence compared with wild-type scFv492.1. (b) Dose–response curve of MAb492gen to 150 or 25 nM MmQSOX1, based on results from a colorimetric assay quantifying RNase A oxidation. The inhibitory activity is expressed as absorbance at 412 nm, representing free thiols that reacted with 5,5′-dithiobis-(2-nitrobenzoic acid). Error bars represent standard deviations from an average of three measurements. The IC50 values were determined by nonlinear regression analysis and yielded values close to the MmQSOX1 concentration. (c) Inhibition curve of MAb492gen to 50 nM MmQSOX1, based on results from oxygen electrode assays at a range of MAb492gen concentrations. Inhibitory activity is expressed as the ratio of the inhibited rate to the uninhibited rate (vi/v0). Error bars represent standard deviations from an average of three measurements. The Ki value was determined by nonlinear regression analysis. (d) Same as in (c), only for MAb316.1.
Comparison of inhibition constants
We next quantified MmQSOX1 inhibition by MAb492gen and compared it with inhibition by the natural antibody MAb316.1. In one assay, MmQSOX1 activity was measured by the number of free thiols remaining at the end of the oxidation reaction of denatured and reduced RNase A. Various MAb492gen concentrations were scanned against two MmQSOX1 concentrations. In both cases, the IC50 values were similar to the MmQSOX1 concentration used in the assay (Fig. 3b), indicating tight-binding inhibition. In a second assay we measured MmQSOX1 catalytic activity on DTT in the oxygen consumption assay, providing data that were fitted to a model of tight-binding inhibition (Bieth, 1995) to yield an inhibitory constant (Ki) of 2.2 ± 0.5 nM (Fig. 3c and Table II). Like MAb492gen, MAb316.1 exhibited tight-binding inhibition of MmQSOX1, but with a slightly larger Ki value of 16 ± 2 nM (Fig. 3d and Table II).
Antibodies inhibit MmQSOX1 activity in cell culture
We examined MmQSOX1 inhibition by MAb316.1 and MAb492gen in cell culture using two readouts of QSOX1 activity (Ilani et al., 2013). QSOX1 and laminin are secreted from quiescent fibroblasts into the ECM, where QSOX1 promotes laminin incorporation, and consequently the adherence of epithelial cells to the fibroblast monolayer. Thus, intact laminin networks in the ECM and cell adherence indicate functional QSOX1 activity in the ECM (Ilani et al., 2013). We first verified that MmQSOX1 is secreted from confluent mouse embryonic fibroblasts (MEFs) (Supplementary Fig. S4). Next, we stained extracellular laminin secreted by MEFs grown for 3 days in the presence of MAb492.1, MAb316.1 or MAb492gen. As in human fibroblast cultures (Ilani et al., 2013), an extensive laminin network was observed only in ECM produced with active QSOX1 (Fig. 4a and Supplementary Fig. S4). We then performed a quantitative assay of epithelial cell adhesion to MEF monolayers. Fluorescently labeled epithelial cells were added to MEFs grown in the absence or presence of the antibodies, and adherent cells were quantified. As expected, more epithelial cells adhered to MEFs grown in the absence of inhibitory antibody compared with MEFs grown in the presence of either 250 nM or 1 μM antibody targeting MmQSOX1 (MAb316.1 and MAb492gen). MAb492.1, which inhibits HsQSOX1 but not MmQSOX1, did not affect the laminin network or epithelial cell adherence in the mouse fibroblast cultures (Fig. 4a and b and Supplementary Fig. S4).
Fig. 4.

MmQSOX1 inhibition in cell culture. (a) Representative images of laminin immunostaining in cultures treated with no antibody, MAb492.1, MAb316.1 or MAb492gen. Scale bar is 40 µm. Additional images are presented in Supplementary Fig. S4. (b) Quantification of adhesion of fluorescently labeled epithelial cells to a mouse fibroblast monolayer grown in the absence or presence of MAb492.1, MAb316.1 or MAb492gen. Error bars are standard error of the mean. Sample images are shown in Supplementary Fig. S4.
MAb492gen binds MmQSOX1 due to alterations at the interface of the heavy and light chains
After attaining an MAb492.1 mutant that inhibits MmQSOX1, we used X-ray crystallography to examine the structural effects of mutations that enabled antigen recognition. A complex between Fab492gen and MmQSOX1Trx was formed, and its structure was solved to 2.3 Å resolution (Supplementary Table SI). Two complexes were found in the asymmetric unit, deviating from one another mainly in the angle between the constant and the variable regions of the Fab fragment. As expected, the MAb492gen epitope on MmQSOX1 corresponds to the HsQSOX1 region bound by MAb492.1 (Fig. 5a). MmQSOX1Trx bound by MAb492gen takes the conformation of chain B from the unbound MmQSOX1Trx, in which the TLPG(138–141) loop approaches the CXXC motif. MAb492gen differs from MAb492.1 primarily around this QSOX1Trx region (Fig. 5b). The complex structure revealed how a combination of relieving steric clashes and introducing new favorable interactions enabled recognition of MmQSOX1.
Fig. 5.

Structure of the Fab492gen–MmQSOX1Trx complex. (a) Surface presentation of the Fab492gen–MmQSOX1Trx complex (PDB ID: 5D96) showing that Fab492gen binds MmQSOX1 in the same mode as Fab492.1 with respect to HsQSOX1Trx, by burying the Trx CXXC active site (yellow spheres). (b) Top view of MAb492.1 and MAb492gen CDRs. Coloring of Fab492gen chains is as in (a). The Fab492gen–MmQSOX1Trx complex was overlaid on the Fab492.1–HsQSOX1Trx complex by aligning MmQSOX1Trx and HsQSOX1Trx. (c) Cartoon presentation of CDRs L2 and H3 in Fab492gen, showing their close interaction. Residues in stick presentation were mutated in the development of Fab492gen from Fab492.1. The dashed black line represents a hydrogen bond. (d) and (e) Comparison of the CDR L1–QSOX1Trx interaction in Fab492gen–MmQSOX1Trx and Fab492.1–HsQSOX1Trx complexes. Coloring is as in (a) and (b). One hydrogen bond is possible between Fab492.1 and HsQSOX1Trx, and none between Fab492.1 and MmQSOX1Trx (d). Two hydrogen bonds provide interactions in the Fab492gen–MmQSOX1Trx complex (e).
The YYGS-to-SMDP mutation of CDR H3 in Fab492gen both eliminated a clashing tyrosine and caused a rearrangement of the backbone conformation (Fig. 5b and Supplementary Fig. S5), resulting in the first significant improvement in binding. Changing the structure of the CDR H3 loop also allowed closer approach of CDR L2, together with the rest of the light chain, toward the heavy chain (Fig. 5b). This movement appears to have helped relieve the expected clash between CDR L3 and N117 in MmQSOX1 (Fig. 2c).
A second effect of the slight change in the relative orientations of the heavy and light chain variable regions observed in the Fab492gen–MmQSOX1Trx structure was the introduction of new interactions. Some interactions were generated between the heavy and light chains, and some with the target. Mutating H49 in CDR L2 to serine enabled an interaction with the new CDR H3 SMDP loop (Fig. 5c), potentially contributing to pre-organization of the paratope. Mutating T31 in CDR L1 to glycine enabled formation of hydrogen bonds between CDR L1 and the MmQSOX1 TLPG(138–141) loop that could not form between MAb492.1 and MmQSOX1 (Fig. 5d and e).
MAb316.1 exhibits a new inhibition mode of QSOX1
MAb492.1 and MAb492gen bind the N-terminal QSOX1 Trx domain. Since MAb316.1 was generated in mice injected with full-length MmQSOX1, we sought to localize its epitope. We used size-exclusion chromatography to compare the elution profiles of MmQSOX1, or each of its constituent modules, to their elution profiles when mixed with MAb316.1. In size-exclusion chromatography, this antibody shifted the elution of both full-length MmQSOX1 and the Trx module to higher apparent molecular weights (Fig. 6a). In contrast, migration of the MmQSOX1 Erv module, responsible for the sulfhydryl oxidase activity, was not affected, pointing at the Trx module as the MAb316.1 binding region in MmQSOX1.
Fig. 6.

MAb316.1 binds the Trx domain of MmQSOX1. (a) Elution profiles of MAb316.1 and MmQSOX1 or its fragments from analytical size-exclusion chromatography. MAb316.1 binds the Trx module. (b) Surface presentation of the Fab316.1–HsQSOX1Trx complex (PDB ID: 5D93) showing that Fab316.1 does not bury the CXXC motif, accessible at the complex surface (yellow). (c) Superposition of the Fab316.1–HsQSOX1Trx complex (PDB ID: 5D93) and MmQSOX1 C76A/C455S (PDB ID: 3T58). Only the variable region of the light chain is shown (gray) from Fab316.1. Yellow spheres represent the interdomain disulfide between the Trx and Erv active-site cysteines. MmQSOX1Erv clashes with the Fab316.1 light chain (black circle), suggesting that Fab316.1 inhibits MmQSOX1 by interfering with formation of the inter-domain electron-transfer intermediate.
To localize the MAb316.1 epitope to specific residues in MmQSOX1Trx, we co-crystallized Fab316.1 with MmQSOX1Trx and solved the structure to 2.2 Å resolution (Supplementary Table SI). Two MmQSOX1Trx–Fab316.1 complexes were present in the crystal asymmetric unit. The complex structure revealed a distinct QSOX1 inhibition mode from the one displayed by MAb492.1 and MAb492gen. Fab316.1 envelopes the helix containing the Trx redox-active site at its amino terminus but does not block access to the CXXC motif itself (Fig. 6b). It seems that a small substrate like DTT might be able to reduce the Trx active site even in the presence of MAb316.1. Nevertheless, MAb316.1 would physically prevent formation of the inter-domain electron-transfer intermediate of MmQSOX1 (Alon et al., 2012) (Fig. 6c), thus interrupting a different step in the catalytic cycle than MAb492.1 and MAb492gen (transition from state 2 to 3, rather than 1 to 2 in Supplementary Fig. S1).
MAb492gen is the only QSOX1 inhibitory antibody cross-reactive with mouse and human orthologs
A QSOX1-targeting antibody with dual species specificity, i.e. which inhibits HsQSOX1 as well as MmQSOX1, would be a valuable tool that could be used in animal models, and in turn, with minimal engineering, in clinical trials. We therefore tested for MAb492gen and MAb316.1 inhibition of HsQSOX1. MAb316.1 up to a concentration of 1 μM did not inhibit HsQSOX1 (Fig. 7a). In contrast, though no selection for binding of HsQSOX1 was performed during its development, MAb492gen retained tight-binding activity against HsQSOX1, having a Ki value of 1.6 ± 0.6 nM for HsQSOX1 (Fig. 7b and Table II) compared with 0.9 ± 0.1 nM previously measured for MAb492.1 (Grossman et al., 2013).
Fig. 7.

HsQSOX1 inhibition by MAb316.1 and MAb492gen. (a) Turnover numbers of HsQSOX1 in the absence and presence of 250 nM or 1 μM MAb316.1. Activity was evaluated using an oxygen consumption assay. Error bars represent standard deviations from an average of three measurements. (b) Inhibition curve of MAb492gen calculated from oxygen electrode assays of 50 nM HsQSOX1 in the presence of various MAb492gen concentrations. Inhibitory activity is expressed as the ratio of the inhibited rate to the uninhibited rate (vi/v0). Error bars represent standard deviations from an average of three measurements. The Ki value was determined by nonlinear regression analysis. (c) Superposition of HsQSOX1Trx (PDB ID: 4IJ3, chain a) and Fab316.1–MmQSOX1Trx complex (PDB ID: 5D93). Residues that differ in sequence between the two QSOX1Trx orthologs, namely the VFPV(135–138) loop from HsQSOX1 and the corresponding TLPG(138–141) loop from MmQSOX1, are in stick presentation. (d) The percent activity of MmQSOX1, HsQSOX1 and its mutant VFPV(135–138)TLPG, designated TLPG, in the presence of MAb316.1. Measurements were conducted in an oxygen consumption assay with 100 nM enzyme, 250 nM MAb492.1, and 200 μM DTT. Percent activity was calculated according to the results of the same measurements in the absence of antibody. Error bars represent standard deviations from an average of three measurements.
A comparison of the three available QSOX1–antibody complex structures suggests features that prevent or contribute to target species specificity. Lack of binding of HsQSOX1Trx by MAb316.1 can be explained once again by the VFPV(135–138) loop, which was a major specificity determinant for MAb492.1. In the Fab316.1–MmQSOX1Trx complex, however, the corresponding TLPG(138–141) loop is not directly in the interface, but rather affects the position of the conserved downstream residues AGA(139–142). These downstream residues in HsQSOX1 are expected to clash sterically with CDR H2 of MAb316.1 (Fig. 7c). The constellation of aromatic residues (light chain: Y31, Y90, Y93; heavy chain: Y33, W50, Y52) that mediates interaction between Fab316.1 and MmQSOX1 presumably does not accommodate minor differences in backbone position between the mouse and human QSOX1 orthologs. Mutating HsQSOX1 to mimic MmQSOX1 by converting TLPG(138–141) to VFPV(138–141) made MAb316.1 inhibition of HsQSOX1 possible, confirming this region as the block to cross-reactivity (Fig. 7d).
As opposed to MAb316.1 and MAb492.1, MAb492gen accommodates both the TLPG(138–141) loop from MmQSOX1 and presumably the corresponding VFPV(135–138) from HsQSOX1 between its H3, L1 and L2 CDRs, resulting in dual specificity (Supplementary Fig. S5). One key difference between MAb492gen and the species-specific antibodies (MAb492.1 and MAb316.1) is its lower content of tyrosines and other aromatic residues in the CDRs. Tyrosines are highly prevalent and effective in antibody CDRs (Koide and Sidhu, 2009), where they can make van der Waals, hydrogen bonding and cation–π interactions. In MAb492gen, however, three tyrosines were replaced by more flexible residues. Another difference is the introduction of a charged residue, D101, into CDR H3, which becomes buried at the QSOX1–antibody interface. D101 forms water-mediated interactions with the MmQSOX1 TLPG(138–141) loop, while also hydrogen bonding to the light chain (Fig. 5c). The implications of these differences will be discussed further below. CDRs that were maintained in MAb492gen (CDRs H1 and H2) are those that interact with the CXXC motif region, the universal catalytic element in QSOX1 Trx domains.
Discussion
When monoclonal antibodies raised as potential drugs target both human and animal orthologs of the same antigen, a single agent can be used in the drug-approval process, from animal studies to clinical trials. Very often, however, species-specific antibodies are obtained, regardless of the method used to produce them. Although differences in antigen structure might be small among orthologs, they are exploited by the immune system to bind the foreign antigen and avoid self-reactivity. In the large antigen surface area buried by antibodies, minor differences between orthologs can cause steric clashes that cannot be remedied by a simple corresponding change in the antibody. Nevertheless, in some cases, a point mutation in the antibody can be sufficient to tune antibody species specificity (Farady et al., 2009). In other cases, substantial antibody re-engineering or design is required (Werther et al., 1996; Liang et al., 2006; Garcia-Rodriguez et al., 2007). Re-engineering methods involving random mutagenesis such as saturation mutagenesis (Chowdhury and Pastan, 1999) or epPCR (Martineau, 2002) do not require a structure of the antigen–antibody complex, but they risk shifting the antibody binding site to a different epitope. For some antibodies, modulating their species specificity is too labor-intensive and time-consuming, or considered to be too great a challenge, so surrogate antibodies are developed instead for animal studies (Wakefield et al., 2011).
In the case study presented herein, we used two different methods to obtain an inhibitory antibody targeting MmQSOX1: natural antibody production independent of MAb492.1 and rational modification of MAb492.1 using structural data. In the structure-based method, the key step was to recognize the specific residues that prevent cross-reactivity. We identified the amino acids by sequence alignment and structure comparison, and validated the effect of these residues by mutagenesis (Fig. 2d). Mutating four residues of MmQSOX1 was sufficient to achieve inhibition by MAb492.1, but the opposite direction, in which mutations were introduced into MAb492.1 to produce an inhibitor of wild-type MmQSOX1, required stronger manipulation. In contrast to modifying MmQSOX1 to mimic HsQSOX1, modification of the antibody had to be made without a guiding structure. Furthermore, the four QSOX1 residues controlling reactivity with MAb492.1 affected the position of the QSOX1 polypeptide backbone, hinting that substantial compensating mutations affecting CDR loop position and/or structure would be needed to convert MAb492.1 into a cross-reactive reagent. In an attempt to affect CDR backbone conformation, we randomized several residues from the same CDR together, but in specific locations, and only up to four residues each time. This careful approach was taken to increase the likelihood of gaining functionality against MmQSOX1 without losing binding to the targeted epitope.
Based on the Fab492gen–MmQSOXTrx structure, we suspect that the first mutations introduced, in CDR L3 and H3, prevented collisions between MmQSOX1 and the antibody. CDR H3 is the most variable CDR in sequence and structure among antibodies and is usually responsible for most of the paratope (Xu and Davis, 2000). It was therefore not surprising that replacing four residues in this CDR (YYGS-to-SMDP) (Supplementary Fig. S2) resulted in significant improvement in binding. Particularly noteworthy is the MAb492gen H3 residue Asp101. Asp101 forms water-mediated interactions with the MmQSOX1 TLPG(138–141) loop and with a structurally conserved neighboring strand. The bulky aromatic residues within the corresponding YYGS sequence of MAb492.1 H3 were apparently unable to accommodate the deviation of MmQSOX1 TLPG(138–141) from HsQSOX1 VFPV(135–138). Though the structural variability of this QSOX1 loop likely positions backbone hydrogen bond donor and acceptor groups differently in space, the water network through which Asp101 interacts with its target in the Fab492gen-MmQSOXTrx complex suggests that MAb492gen can accommodate these differences through a re-organized water structure.
The next steps in the construction of MAb492gen produced little improvement in binding MmQSOX1, but a final stage proved highly beneficial. The minimal improvement involved libraries constructed to randomize residues in CDR H1 and H2. Though both these CDRs are in tight contact with MmQSOX1, they bind structurally conserved regions in HsQSOX1 and MmQSOX1, explaining the lack of improvement. Having exhausted other options, we then changed our strategy from eliminating clashes to forming new interactions. Mutations in CDR L1 and L2 resulted in inter-chain interactions that were not present in MAb492.1, and consequently in new and improved antibody–MmQSOX1 interactions. Because L2 in MAb492.1 does not form tight contacts with HsQSOX1, these mutations did not jeopardize HsQSOX1 binding, resulting in a dual-species specific antibody.
The parallel strategy, generating MAb316.1 in QSOX1 KO mice, was conducted independently of the re-engineering of MAb492.1. In principle, eliciting an antibody in an organism completely lacking the target molecule could produce cross-reactive antibodies since there is no bias against conserved epitopes. However, neither is there bias toward them. MAb316.1 does not directly bind the specificity determinants that prevent MAb492.1 from targeting MmQSOX1. Nevertheless, one of these determinants propagates backbone conformational differences into the MAb316.1 epitope, such that conserved residues occupy slightly different positions in MmQSOX1 and HsQSOX1. The network of aromatic residues in MAb316.1 provides excellent shape complementarity to MmQSOX1 but is unforgiving of the minor backbone differences between QSOX1 orthologs. In principle, it should have been possible using QSOX1-KO mice to obtain an antibody that recognizes only structurally invariant regions of QSOX1 and thus exhibits cross-species reactivity. In practice, one can screen for desirable properties in a pool of natural antibodies, but ultimately the antibodies obtained will be limited by what was produced by natural variation and what entered the screening pipeline.
In antibody engineering, desired properties can be introduced and combined at will, but it is often a challenge to realize the engineering goals. We demonstrate here the importance of using structural information and identifying the specificity determinants in the common challenge of changing or broadening antibody specificity. We provide a case study of overcoming an antibody species-specificity barrier with simple tools while highlighting our key insights. We found that relieving steric clashes was necessary but not sufficient to extend binding to a distinct ortholog of the target protein and that additional, favorable interactions had to be introduced. These interactions were accomplished by altering the association between the heavy and light antibody chains, which recruited CDRs L1 and L2 into the antibody–MmQSOX1 interface. Dual specificity was enabled by substituting the constrained rings of aromatic residues with more flexible side chains, including those that can participate in water-bridged polar interactions. CDRs that recognize structurally conserved elements were left in their natural state, preserving specificity to QSOX1 antigens.
Supplementary data
Funding
This work was funded by European Research Council under the European Union's Seventh Framework Programme, grant number 310649 and by the Center of Research Excellence (I-CORE) in Structural Cell Biology. Funding to pay the Open Access publication charges for this article was provided by the European Research Council.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge Dr. O. Leitner and H. Hamawi from the Weizmann Institute Antibody Unit for performing all technical aspects of natural antibody elicitation and production. T. Mehlman and D. Merhav from the Weizmann Institute Mass Spectrometry Unit verified antibody sequences using LC-MS/MS. Dr. Y. Friedman gave valuable advice on yeast surface display handling and library construction. We thank Dr. A. Alon and S. Warszawski for useful discussions. Dr. A. Sharp, E. Ariel, and Dr. Z. Porat from the Weizmann Institute FACS Unit assisted in library sorting. Dr. R. Diskin provided useful advice on recombinant antibody expression and provided the vectors.
References
- Afonine P.V., Grosse-Kunstleve R.W., Adams P.D. (2005) CCP4 Newsletter, 42, contribution 8.
- Alon A., Grossman I., Gat Y., Kodali V.K., DiMaio F., Mehlman T., Haran G., Baker D., Thorpe C., Fass D. (2012) Nature, 488, 414–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antwi K., Hostetter G., Demeure M.J., Katchman B.A., Decker G.A., Ruiz Y., Sielaff T.D., Koep L.J., Lake D.F. (2009) J. Proteome Res., 8, 4722–4731. [DOI] [PubMed] [Google Scholar]
- Benhar I., Reiter Y. (2002) Curr. Protoc. Immunol. Chapter 10:Unit 10.19B. [DOI] [PubMed] [Google Scholar]
- Beroukhim R., Brunet J.P., Di Napoli A. et al. (2009) Cancer Res., 69, 4674–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieth J.G. (1995) Methods Enzymol., 248, 59–84. [DOI] [PubMed] [Google Scholar]
- Broennimann Ch., Eikenberry E.F., Henrich B. et al. (2006) J. Synchrotron Radiat., 13, 120–130. [DOI] [PubMed] [Google Scholar]
- Chao G., Lau W.L., Hackel B.J., Sazinsky S.L., Lippow S.M., Wittrup K.D. (2006) Nat. Protoc., 1, 755–768. [DOI] [PubMed] [Google Scholar]
- Chowdhury P.S., Pastan I. (1999) Nat. Biotechnol., 17, 568–572. [DOI] [PubMed] [Google Scholar]
- Coppock D., Kopman C., Gudas J., Cina-Poppe D.A. (2000) Biochem. Biophys. Res. Commun., 269, 604–610. [DOI] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. (2004) Acta Crystallogr. D. Biol. Crystallogr., 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Farady C.J., Sellers B.D., Jacobson M.P., Craik C.S. (2009) Bioorg. Med. Chem. Lett., 19, 3744–3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G., Bertos N., Pepin F. et al. (2008) Nat. Med., 14, 518–527. [DOI] [PubMed] [Google Scholar]
- Fleishman S.J., Whitehead T.A., Ekiert D.C., Dreyfus C., Corn J.E., Strauch E.M., Wilson I.A., Baker D. (2011) Science, 332, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia G., Cruz-Munoz W., Man S., Xu P., Kerbel R.S. (2011) Nat. Rev. Cancer, 11, 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rodriguez C., Levy R., Arndt J.W., Forsyth C.M., Razai A., Lou J., Geren I., Stevens R.C., Marks J.D. (2007) Nat. Biotechnol., 25, 107–116. [DOI] [PubMed] [Google Scholar]
- Gat Y., Vardi-Kilshtain A., Grossman I., Major D.T., Fass D. (2014) Protein Sci., 23, 1102–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman I., Alon A., Ilani T., Fass D. (2013) J. Mol. Biol., 425, 4366–4378. [DOI] [PubMed] [Google Scholar]
- Heckler E.J., Alon A., Fass D., Thorpe C. (2008) Biochem., 47, 4955–4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., Ji G., Wu Y., Wan B., Yu L. (2008) J. Cancer Res. Clin. Oncol., 134, 705–714. [DOI] [PubMed] [Google Scholar]
- Hudis C.A. (2007) N. Engl. J. Med., 357, 39–51. [DOI] [PubMed] [Google Scholar]
- Ilani T., Alon A., Grossman I., Horowitz B., Kartvelishvily E., Cohen S.R., Fass D. (2013) Science, 341, 74–76. [DOI] [PubMed] [Google Scholar]
- Kay B.K., Thai S., Volgina V.V. (2009) Methods Mol. Biol., 498, 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide S., Sidhu S.S. (2009) ACS Chem. Biol., 4, 325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosanam H., Prassas I., Chrystoja C.C. et al. (2013) Mol. Cell. Proteomics, 12, 2820–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc M.P., Giudicelli V., Kaas Q., Duprat E., Jabado-Michaloud J., Scaviner D., Ginestoux C., Clément O., Chaume D., Lefranc G. (2005) Nucleic Acids Res., 33, 593–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental K.R., Yu H., Kass L. et al. (2009) Cell, 138, 891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang W.C., Wu X., Peale F.V. et al. (2006) J. Biol. Chem., 281, 951–961. [DOI] [PubMed] [Google Scholar]
- Lovell S.C., Davis I.W., Arendall W.B. III, de Bakker P.I., Word J.M., Prisant M.G., Richardson J.S., Richardson D.C. (2003) Proteins, 50, 437–450. [DOI] [PubMed] [Google Scholar]
- Martineau P. (2002) Methods Mol. Biol., 178, 287–294. [DOI] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. (2007) J. Appl. Crystallogr., 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagato S., Nakagawa K., Harada H., Kohno S., Fujiwara H., Sekiguchi K., Ohue S., Iwata S., Ohnishi T. (2005) Int. J. Cancer, 117, 41–50. [DOI] [PubMed] [Google Scholar]
- Peng L., Ran Y.L., Hu H. et al. (2009) Carcinogenesis, 30, 1660–1669. [DOI] [PubMed] [Google Scholar]
- Rizzieri D.A., Akabani G., Zalutsky M.R. et al. (2004) Blood, 104, 642–648. [DOI] [PubMed] [Google Scholar]
- Rodriguez H.M., Vaysberg M., Mikels A., McCauley S., Velayo A.C., Garcia C., Smith V. (2010) J. Biol. Chem., 285, 20964–20974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott A.M., Wiseman G., Welt S. et al. (2003) Clin. Cancer. Res., 9, 1639–1647. [PubMed] [Google Scholar]
- Scott A.M., Wolchok J.D., Old L.J. (2012) Nat. Rev. Cancer, 12, 278–287. [DOI] [PubMed] [Google Scholar]
- Soloviev M., Esteves M.P., Amiri F., Crompton M.R., Rider C.C. (2013) PLoS One, 8, e57327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiller T., Meffre E., Yurasov S., Tsuiji M., Nussenzweig M.C., Wardemann H. (2008) J. Immunol. Methods, 329, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield I., Stephens S., Foulkes R., Nesbitt A., Bourne T. (2011) Toxicol. Sci., 122, 170–176. [DOI] [PubMed] [Google Scholar]
- Warburton C., Dragowska W.H., Gelmon K., Chia S., Yan H., Masin D., Denyssevych T., Wallis A.E., Bally M.B. (2004) Clin. Cancer Res., 10, 2512–2524. [DOI] [PubMed] [Google Scholar]
- Weiner G.J. (2010) Semin. Hematol., 47, 115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werther W.A., Gonzalez T.N., O'Connor S.J. et al. (1996) J. Immunol., 157, 4986–4995. [PubMed] [Google Scholar]
- Xu J.L., Davis M.M. (2000) Immunity, 13, 37–45. [DOI] [PubMed] [Google Scholar]
- Zhaofei L., Fan W., Xiaoyuan C. (2008) Drug Dev. Res., 69, 329–339.20628538 [Google Scholar]
- Zhou H., Fisher R.J., Papas T.S. (1994) Nucleic Acids Res., 22, 888–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.