

Abstract
The merits of exercise on muscle health and well-being are numerous and well documented. However, the mechanisms underlying the robust adaptations induced by exercise, particularly on mitochondria, are less clear and much sought after. Recently, an evolutionary conserved cellular recycling mechanism known as autophagy has been implicated in the adaptations to acute and chronic exercise. A basal level of autophagy is constantly ongoing in cells and tissues, ensuring cellular clearance and energy homeostasis. This pathway can be further induced, as a survival mechanism, by cellular perturbations, such as energetic imbalance and oxidative stress. During exercise, a biphasic autophagy response is mobilized, leading to both an acute induction and a long-term potentiation of the process. Posttranslational modifications arising from upstream signaling cascades induce an acute autophagic response during a single bout of exercise by mobilizing core autophagy machinery. A transcriptional program involving the regulators Forkhead box O, transcription factor EB, p53, and peroxisome proliferator coactivator-1α is also induced to fuel sustained increases in autophagic capacity. Autophagy has also been documented to mediate chronic exercise-induced metabolic benefits, and animal models in which autophagy is perturbed do not adapt to exercise to the same extent. In this review, we discuss recent developments in the field of autophagy and exercise. We specifically highlight the molecular mechanisms activated during acute exercise that lead to a prolonged adaptive response.
Keywords: mitophagy, physical activity, lysosome, PGC-1, mitochondria
chronic exercise training leads to pleiotropic benefits that are unmatched by any other intervention, be they pharmacological or nutritional. For decades scientists have been searching for the molecular forces driving these robust adaptations, with the hopes of unearthing pharmacological and lifestyle interventions for the treatment and prevention of various pathologies. In a society that is aging and is becoming simultaneously more sedentary, the ability to harness the beneficial effects of exercise is becoming increasingly critical.
The merits of exercise on whole body metabolism and general health are numerous. Regular exercise improves glucose homeostasis, cardiovascular health, muscle mass maintenance, and even mood and cognition (1, 6, 88, 92). On a cellular level, exercise is beneficial for bioenergetics, nutrient delivery, and uptake, resulting in tissues of superior fitness, ready to endure the brunt of prolonged exercise and thus better cope with the stresses of day-to-day life (28, 33). Energetic distress during exercise induces cellular remodeling in an attempt to satisfy the newly elevated energy requirements. Inefficient, nascent, and dysfunctional mitochondria, as well as oxidized and damaged proteins must be recycled to allow for the synthesis of new and improved machinery. Much emphasis has been placed on searching for the mechanisms involved in the synthesis of new organelles and proteins following exercise. However, a growing body of evidence suggests that organelle and protein degradation are just as important as synthesis for cellular renovation (26, 52, 54, 111). Indications of the involvement of cellular recycling programs in exercise-mediated adaptations have recently emerged. Indeed, both of the major cellular proteolytic pathways, the ubiquitin (Ub) proteasome and autophagy-lysosome systems, are activated with an acute bout of endurance exercise in various organs and tissues in humans and rodents (19, 26, 35, 36, 54, 95).
Autophagy is a cellular recycling mechanism responsible for the disposal of toxic protein aggregates as well as nascent organelles. Autophagy has been conserved throughout evolution, and this process is vital for cellular turnover. Three different types of autophagy have been described, and they differ mainly in their mode of substrate delivery to the lysosome. Microautophagy refers to the degradation of substrates in the immediate vicinity of the lysosome through lysosomal membrane invaginations. This process is responsible for the degradation of specific proteins and cytosolic materials that are in close proximity to this organelle, or on the lysosomal membrane. Little is known about the role and regulation of this process, but it may be involved in mediating lysosomal signaling (51). Chaperone-mediated autophagy, is a targeted degradation mechanism through which dedicated chaperones, such as, heat shock cognate protein-70 recognize a common KFERQ motif on selected substrates and directly deliver them to lysosomes for degradation (41). Macroautophagy (hereafter termed autophagy) is the best understood of the three and is the focus of this review. During this process, substrates, along with any cytosolic materials surrounding them, are engulfed within double membrane vesicles known as autophagosomes, which are subsequently delivered to the lysosome for degradation. The autophagosomes themselves, along with the proteins responsible for substrate recognition and delivery, are also recycled. Posttranslational modifications lead to an acute activation of this process, which is then followed by the potentiation of a longer-term cellular remodeling transcriptional program. Once activated, autophagy is thought to proceed in five distinct steps. These include 1) induction, 2) nucleation and expansion, 3) cargo selection, 4) fusion, and 5) degradation and efflux (Fig. 1). These steps are characterized by the involvement of specific molecular complexes.
Fig. 1.

Exercise-induced autophagy signaling. During muscle contraction, levels of calcium, NAD+, AMP, and ROS increase. These molecular messengers activate their respective downstream effectors, CaMK/calcineurin, SIRT1, AMPK, and p38. These, in turn, initiate both acute autophagy and potentiate a longer term transcriptional program. AMPK can lead to the activation of the autophagy induction complex by ULK1 phosphorylation, as well as inhibition of mTOR, which normally acts to block autophagy. Autophagy induction is followed by nucleation and expansion of the autophagosome mediated by the beclin-1 complex and the lipidation of LC3 to LC3-II by a series of conjugation reactions by a series of autophagy-related proteins we termed the LC3 lipidation complex. The beclin-1 complex can also be antagonized by Bcl-2. During the cargo selection process, dysfunctional organelles (such as mitochondria) or protein aggregates are tagged for degradation by ubiquitin (Ub) and are subsequently recognized by the adaptor protein p62, which binds to Ub and coalesces the materials to be degraded into the growing autophagosome by interacting with LC3. Mitochondria can be tagged for degradation by ubiquitination of substrates on the outer mitochondrial membrane by the E3 Ub ligase parkin, or by mitochondria-specific receptors BNIP3/L, independent of Ub. The precise mechanism of cargo selection following exercise is yet to be elucidated. Once the cargo is engulfed, the mature autophagosome is sealed and delivered to the lysosome. The two then fuse together, and the contents are degraded by various hydrolases within the lysosomal lumen. Basic building blocks, such as animo acids (AAs) are then released and can negatively feed back on the pathway by reactivating mTOR. Long-term potentiation of an autophagy transcriptional pathway also occurs with exercise. Calcineurin can dephosphorylate the master regulator of the autophagy-lysosome system, TFEB, and allow its localization into the nucleus. p38 and ROS can also induce a p53-mediated transcriptional program, although that is yet to be confirmed. AMPK, p38, and SIRT1 can also activate FoxO-dependent gene transcription. Moreover, CaMK and calcineurin induce PGC-1α expression, while SIRT1, AMPK, and p38 upregulate PGC-1α activity. Once active in the nucleus, PGC-1α co-activates transcription factors, resulting in the increased expression of autophagy and lysosomal genes. See text for definition of acronyms.
AUTOPHAGY IS ACTIVATED WITH EXERCISE
A basal level of autophagy is ongoing in all eukaryotic cells, and it aids in cellular housekeeping, as it is responsible for the homeostatic turnover of molecules and organelles (21). During metabolic stress, such as increased oxidative stress, energetic imbalance, or protein misfolding, autophagy can be further upregulated. In this scenario, autophagy acts as an adaptive response system, essential for cell survival and the maintenance of cellular vitality (13, 101). Aberrantly low autophagy contributes to tissue dysfunction and collapse of cellular homeostasis. Indeed, many degenerative diseases, muscle dystrophies, and even aging are characterized by insufficient autophagy (18, 74, 106). On the other hand, excessive autophagy may contribute to protein catabolism during conditions such as prolonged muscle disuse and cancer-related cachexia (57, 79). Various cellular stressors have been documented to induce autophagy in many tissues and cell types. These include metabolic stress in the form of nutrient insufficiency, oxidative stress, calcium imbalance, and general disturbances in cellular homeostasis (65, 71, 73, 85, 91). Coincidently, exercise can elicit many of these stresses, which may contribute to autophagy activation (26, 28, 54).
The first documented evidence for a change in autophagy during exercise was noted back in the 1980s (93, 97). An increase in both the number and size of autophagic vacuoles was observed in the liver and skeletal muscle of exercised animals. However, the importance of this activation and the contribution of autophagy to exercise-induced metabolic benefits were not established until recent years. In 2011, Grumati et al. demonstrated that autophagy was induced in skeletal muscle with an acute bout of exercise, and that animals lacking collagen VI, a model where autophagy is compromised (18), were exercise intolerant (19). Moreover, these mice demonstrated an exacerbation of their dystrophic phenotype with acute and chronic exercise (19), suggesting that functional autophagy is required for the proper response to exercise. An increase in the expression of autophagy genes, as well as induction in autophagy proteins in response to exercise, have since been noted in various tissues in both humans and rodents (9, 35, 36, 52, 62, 111). Indeed, autophagy induction following exercise has been observed in multiple metabolically relevant organs, such muscle, liver, heart, pancreas, and adipose tissue (27). Moreover, autophagy activation after exercise has been reported even in the brain and may account, at least in part, for the neuroprotective effects of chronic exercise (27, 58, 119). The importance of autophagy for chronic exercise-mediated metabolic benefits has also begun to emerge (Fig. 2).
Fig. 2.
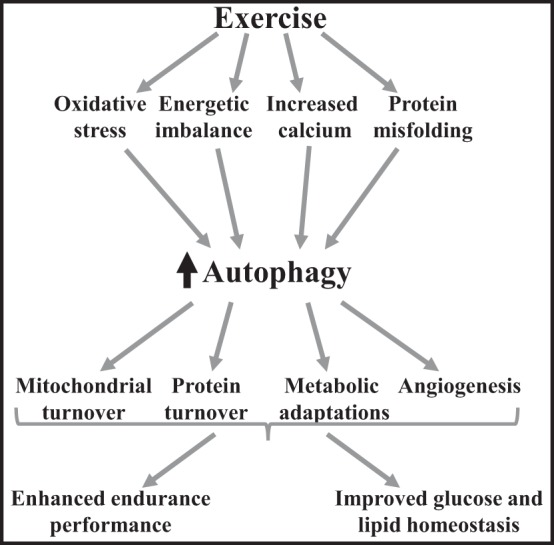
Long-term adaptations to exercise that are potentiated by exercise-induced autophagy. Exercise initiates a cascade of events, which leads to increased oxidative stress, energetic imbalance, increase intracellular calcium, and protein misfolding (28, 115). Oxidative stress (54), energetic imbalance (53), calcium (62, 95), and misfolded proteins (50, 72, 118) can induce autophagy activation. Autophagy then participates in mitochondrial turnover (54, 111), protein turnover (11, 54), metabolic adaptations (10), as well as angiogenesis (52). These adaptations together result in enhanced endurance performance (52), as well as improved glucose and lipid homeostasis (26).
MOLECULAR MECHANISMS OF AUTOPHAGY ACTIVATION
Muscle contraction constitutes a form of energetic stress, much like nutrient deprivation, which leads to alterations in molecular messengers, such as, calcium, AMP, NAD+, and reactive oxygen species (ROS). These messengers activate downstream signaling cascades, culminating in a biphasic autophagic response aimed at restoring homeostasis (Fig. 1).
Under steady-state conditions, when energetic supply and demand are in equilibrium, the metabolic sensors mammalian target of rapamycin (mTOR) and protein kinase A negatively regulate autophagy through the phosphorylation and inhibition of the induction complex (29, 39, 107). When energy demand outweighs supply, the AMP-to-ATP ratio rises, and this activates AMP-dependent kinase (AMPK), while inhibiting mTOR activity. Elevated production of ROS and an increase in NAD+ during exercise also result in the activation of AMPK (7, 24), as well as additional cellular stress responders, such as sirtuin-1 (SIRT1) and p38 MAPK (2, 7, 82). These molecules, in turn, lead to the activation of autophagic machinery (29, 44). The induction phase of phagophore (an immature precursor of the autophagosome) formation is mediated by the unc-51-like kinase 1 (ULK1) complex (61). This induction complex is composed of the core components, including ULK1, which is a serine/threonine protein kinase, autophagy-related gene 13 (Atg13), which is a phosphoprotein, and FIP200 (focal adhesion kinase family-interacting protein of 200 kDa) (23, 29, 30). AMPK can activate ULK1 during exercise by directly phosphorylating it on Ser-555. Moreover, the inhibition of mTOR during exercise also releases the inhibitory phosphorylation of ULK1 on Ser-757, thus further activating the induction complex (66). The activation of AMPK with exercise is well established, and recent evidence indicates that it is associated with autophagy activation during exercise (26, 66). A single bout of exercise increased the activity of autophagy in the skeletal muscle of wild-type mice, but not in AMPKα2−/− mice. This finding was attributed to an increase in AMPK-α2 association with sestrins 2 and 3 following exercise (53). Sestrins can inhibit mTOR function, thus inducing autophagy, and they have been recently documented to facilitate the degradation of autophagy substrates by promoting ULK1 phosphorylation of p62 (87). ULK1 activation has been documented with exercise in both humans and rodents. One hour of cycling at 50% of maximal oxygen consumption increased autophagic signaling through the phosphorylation of ULK1 at Ser-555 in human skeletal muscle (66). This activation was also associated with AMPK-α Thr-172 phosphorylation and was independent of nutritional status, as a 36-h fast before exercise did not result in any further induction of ULK1. The effects of nutrient deprivation preceding exercise on autophagy activation remains controversial, however, as other studies have reported that 24-h starvation before exercise resulted in both enhanced, and diminished, autophagy following an acute bout of exercise (37, 66, 123).
The next stage of autophagy is the nucleation and expansion of the autophagosomal membrane. Autophagosome formation is no simple task, requiring the tethering together of membranes from different donor sources. The exact origin of the autophagosomal membranes is the subject of much debate, but mitochondria (20, 21), endoplasmic reticulum (17, 113), plasma membrane (86), and even the nuclear envelope (84) have all been implicated in membrane donation to the growing phagophore. It is becoming more and more evident that the autophagosome membrane source may vary based on the autophagic stimulus, as well as the cargo destined for degradation. Autophagosome nucleation involves the beclin-1 complex, which is composed of beclin-1, class III phosphatidylinositol 3-kinase, vacuolar protein sorting (VPS) 34, VPS15, AMBRA1 (activating molecule in BECN1 regulated autophagy protein 1), and UVRAG (ultraviolet irradiation resistance-associated gene). Bcl-2 can serve to antagonize this step by sequestering beclin-1 and AMBRA1 (15, 34, 78). Seminal work by He et al. (26) has revealed that exercise-induced autophagy engages the Bcl-2-beclin-1 complex. In this scenario, phosphorylation of Bcl-2 during exercise results in the emancipation of beclin-1, allowing autophagosome nucleation. Mice in which beclin-1 cannot be dissociated from Bcl-2 (Bcl2AAA mice) demonstrate normal basal autophagy, but impaired stimulus-induced autophagy. These animals display compromised exercise performance and impaired exercise-mediated metabolic adaptations, indicating the importance of autophagy activation with exercise. Interestingly, these findings are not limited to Bcl2AAA mice, but are rather ubiquitous among various autophagy-deficient models. Mice heterozygous for beclin-1 (beclin1+/− mice), as well as mice hypomorphic for Atg16L1, an essential autophagy protein vital for autophagosomal membrane synthesis, display defects in exercise performance that are similar to those observed in Bcl2AAA mice. Moreover, these animals also fail to display exercise-mediated improvements in whole body metabolism (26, 52).
Vesicle expansion is a poorly defined event, but it is thought to involve Atg9-mediated membrane recruitment (116). Atg9 levels seem to determine the number of autophagosomes formed. Moreover, there are two Ub-like protein conjugation systems that act to promote the formation of the autophagosome precursor, the phagophore (73). These are involved in the ultimate conjugation of microtubule-associated protein light chain 3 (LC3), and its close relatives GABARAP (GABA receptor-associated protein), and GATE16 (32, 40, 48), to phosphatidylethanolamine. This is an essential step in the autophagic process, and it occurs through a cascade of conjugation reactions performed by several autophagy-related genes [reviewed in detail elsewhere (64)]. Global loss of genes coding for proteins involved in vesicle expansion is often lethal. For instance, systemic loss of Atg7 leads to neonatal lethality, while mice with a conditional knockout of Atg7 in skeletal muscle are viable, but experience impaired muscle function and atrophy (47, 60). An increase in cellular NAD+ level during exercise activates the NAD-dependent deacetylase SIRT1 (7), which has been documented to regulate the acetylation status of Atg7 (49). Surprisingly, exercise performance was not altered in muscle-specific Atg7−/− mice, as it was in animals with global autophagy defects (45, 54). This suggests a cell non-autonomous role for autophagy during exercise. Although autophagy induction following exercise has been documented to occur in various tissues, including liver, heart, pancreas, adipose tissue, and even brain (26, 27), the contribution of this activation to overall exercise-induced metabolic benefits has not been well defined, and this topic requires further investigation. Examining the role of autophagy during exercise in a variety of conditional autophagy null animals will be vital for gaining a better understanding of the cell non-autonomous role of this pathway. However, results from experiments involving long-term autophagic deficiency should be interpreted with some caution due to the confounding effect of aggregated toxic cellular debris and dysfunctional organelles in such models. Indeed, alterations in exercise capacity in autophagy-deficient animals may be a result of secondary aggregates and degeneration, rather than a primary deficiency in autophagic capacity. Interestingly, repeated bouts of damaging downhill running do result in a progressive drop in performance, as well as diminished mitochondrial membrane potential in muscle-specific Atg7−/− mice, a finding that was not evident in wild-type exercised animals (54). These results suggest that the expansion stage of autophagy is vital for proper autophagic turnover of dysfunctional proteins and organelles following exercise.
Generally, a series of conjugation reactions by several autophagy-related genes act to promote phagophore expansion and sealing by LC3 lipidation. LC3 exists in its inactive form, free in the cytosol, and must first be cleaved by the protease Atg4, giving rise to LC3-I (100), which is subsequently conjugated to phosphatidylethanolamine. Lipid conjugation converts LC3-I to the LC3-II form that is attached to either side of the growing phagophore membrane. LC3 lipidation and formation of autophagosomes is an early autophagic event that has been documented to increase progressively with exercise in humans and rodents (19, 26, 35). Aside from its role in autophagosome expansion, LC3-II also plays an essential role in cargo selection. In selective types of autophagy, cargo selection occurs during the expansion step. Substrate specific receptors such as p62 (68), NBR-1 (neighbor of BRCA1 gene 1) (46), and others can interact with ubiquitinated proteins on organelles and recruit the autophagosome. These proteins act as molecular adaptors, as they are equipped with both Ub (Ub) and LC3-interacting regions. The cargo-receptor complex is then selectively encapsulated by the autophagosome through receptor-mediated interaction with LC3-II.
Organelle-specific autophagy receptors that act independently of Ub have also been identified. Mitophagy refers to the selective removal of mitochondria, and it involves specific receptors such as BCL2/adenovirus E1B 19-kDa interacting protein 3 (BNIP3) and BNIP3-like (also known as NIX) that interact directly with LC3-II on the phagophore (22, 67, 98). Mitophagy has also been recently documented to take place following exercise (52, 54, 111, 117). This removal is thought to occur at least in part due to enhanced ROS production with exercise. Indeed, treatment with general or mitochondria-specific ROS scavengers in wild-type mice has been shown to attenuate autophagy induction, resulting in deteriorating exercise performance with repeated bouts (54). Moreover, the stress of exercise can enhance the formation of a protein complex involving REDD1 (regulated in development and DNA damage responses 1) (an mTORC1 inhibitor) and TXNIP (thioredoxin-interacting protein) (a prooxidant protein), which induces ROS production and facilitates autophagosome formation (83). In cells and tissues lacking either REDD1 or TXNIP, ROS is reduced, which results in the enhanced catalytic activity of the redox sensitive Atg4B, thus promoting LC3-II delipidation, and halting autophagy. This reduction in autophagy culminates in the accumulation of defective mitochondria, impaired oxidative phosphorylation, and reduced exercise capacity.
Exercise-induced mitophagy also seems to involve the mitochondrial E3 ligase parkin (54, 111). Although the precise role of parkin during exercise has not yet been dissected, its activation can be the result of an increase in oxidative stress, or a loss of membrane potential, which result in the stabilization of the kinase PINK1 (PTEN-induced putative kinase 1) on the mitochondrial outer membrane. PINK1 then recruits parkin to ubiquitinate targets on the mitochondrial membrane, thus signaling autophagosome formation. Aerobic exercise enhances mitochondrial localization of LC3II, p62, and Ub, further indicating mitophagy induction with exercise (95). The removal of dysfunctional mitochondria through mitophagy following exercise seems vital for muscle health and exercise performance in subsequent bouts, and this effect may be more pronounced in females (54). Studies on the possible sex differences in exercise-mediated autophagy induction are necessary to better understand this phenomenon.
Once the autophagosome is complete, it begins its journey along microtubule tracks to the lysosome where fusion can take place. LC3, Rab7, and FYCO1 (FYVE and coiled-coil domain containing 1) form an adaptor protein complex that promotes the microtubule transport of autophagic vesicles. FYCO1, which contains both an LC3-interacting region and a phosphatidylinositol 3-phosphate-binding domain, is important for autophagosome movement (76). The autophagosome and lysosome then fuse together to form the autolysosome, a process that requires the assistance of lysosomal-associated membrane proteins, as well as the tethering and fusion components, including Rab proteins, and soluble N-ethylmaleimide-sensitive factor attachment protein receptors (64). However, these proteins are not specific to macroautophagy, and very little is known about autophagosome trafficking during exercise.
Autophagosomal contents subsequently undergo degradation within the lysosomal acidic environment, induced by a wide range of hydrolases. The macromolecules released can then be recycled to restore cellular energetic balance, or they can be used for biosynthesis. Moreover, the amino acids that efflux from proteolysis within the lysosomal lumen can act in a retrograde manner to activate mTOR and inhibit autophagy progression, in a negative feedback loop. The activity of lysosomal hydrolases is enhanced following exercise (97), and lysosomal abundance has also been documented to increase with chronic exercise (123).
LONG-TERM POTENTIATION OF AUTOPHAGY VIA TRANSCRIPTIONAL ACTIVATION
A growing body of evidence suggests that the autophagic response to exercise as well as other stresses may be biphasic. Cellular metabolic distress first induces a rapid increase in autophagy flux, which occurs within minutes to hours of exposure to a stressful stimulus, and is mediated by posttranslational protein modifications. This initial phase is generally followed by a long-term potentiation that relies on the activation of a transcriptional program (80). Stress-responsive transcription factors, including the Forkhead box O (FoxO) family of transcription factors, transcription factor EB (TFEB), p53, as well the transcriptional peroxisome proliferator co-activator-1α (PGC-1α), have all been identified to play a major role in the regulation of autophagy gene expression in response to exercise.
The evolutionarily conserved FoxO family of transcription factor was one of the first to be recognized to regulate the expression of autophagy- and lysosome-related genes (57, 120). The function and activity of FoxO proteins are mainly regulated by posttranslational modifications, such as phosphorylation and acetylation, which affect their cellular localization. The FoxO family is negatively regulated by the insulin-Akt axis, which is upstream of mTOR. Both transcription-dependent and -independent functions have been described for the FoxO family in the regulation of autophagy. FoxO3 drives a transcriptional program that enhances the expression of numerous autophagy-related genes in various cell types. For instance, FoxO3 upregulates the transcription of core autophagy machinery, such as LC3, p62, cathepsin L, and BNIP3 in response to cellular stress (63, 112, 120, 121). FoxO1, another family member, also transactivates autophagy genes once in the nuclear compartment (25, 124). However, cytosolic FoxO1 can also regulate autophagy independently of its transcriptional activity (122). Recent evidence indicates that FoxO1 and FoxO3 gene expression is induced following an acute bout of exercise in humans and rodents (35, 36). Moreover, exercise-mediated increases in AMPK, SIRT1, and p38 MAPK can all activate FoxO1 and FoxO3 through posttranslational modifications (7), thus implicating these transcriptional regulators in the long-term potentiation of autophagy with exercise.
TFEB is a basic helix-loop-helix, leucine zipper transcription factor, belonging to the family of microphthalmia-associated transcription factors. TFEB has been termed the master regulator of cellular recycling, as it coordinates the expression of both autophagy and lysosomal genes. A recent study documented an enhanced nuclear localization of TFEB following exercise, an event regulated by the calcium-dependent phosphatase calcineurin (62). Calcium transients are typical during muscle contraction, and the temporary increases in intracellular calcium signal downstream to calcium/calmodulin phosphatase (calcineurin) and kinase (CaMK) (8). Indeed, calcineurin deficiency or its pharmacological inhibition attenuate exercise-induced adaptations (69, 77). TFEB transcriptional activity is also regulated by mTORC1. When mTORC1 is activated by amino acids arising from within the lysosomal lumen, it phosphorylates TFEB on several serine and threonine residues (105). Phosphorylated TFEB is detained in the cytosol by the chaperone 14-3-3. Additionally, TFEB can also be phosphorylated and negatively regulated by ERK. During exercise, TFEB is liberated due to reduced inhibition by mTOR, along with a simultaneous increase in dephosphorylation by calcineurin (62). TFEB can then translocate to the nucleus, where it stimulates the expression of multiple autophagy-lysosome genes that possess a consensus E-box binding motif. TFEB regulates a gene network collectively termed CLEAR (coordinated lysosomal expression and regulation), which includes autophagy-lysosome factors involved in various stages of the pathway (75, 99, 104). Beclin-1, p62, and lysosomal cathepsin D, are all bona fide TFEB targets, just to name a few (75, 105).
The tumor suppressor p53 has been documented to regulate an ever-increasing number of biological processes. Indeed, this protein has been implicated in the transcriptional regulation of the autophagy-lysosome system (42). p53 Activation inhibits mTOR activity and promotes the transcriptional upregulation of autophagy genes in the nucleus (14, 80). A global genomic profiling of the p53 transcriptome revealed a large number of autophagy genes involved in various steps of the process to be direct targets of p53 (42, 56). Moreover, the p53 target genes sestrins-1 and -2 have also been documented to regulate autophagy through the modulation of AMPK-mTORC1 axis (56). Interestingly, p53 also appears important for exercise-induced metabolic benefits, and loss of this transcription factor results in reduced exercise-mediated adaptations (94). However, the regulation of autophagy by p53 is complex and is highly dependent on the subcellular localization of this protein. For instance, cytosolic p53, in direct opposition to its nuclear location, appears to arrest autophagy (5, 89, 109).
The transcriptional co-activator PGC-1α has also been documented time and again to be induced by exercise (2, 3, 96, 111) and to mediate many exercise-induced metabolic benefits (3). AMPK, p38, CaMK, SIRT1, and p53 have all been found to regulate PGC-1α activity and expression following exercise (28, 117). Moreover, PGC-1α has also been documented to exert transcriptional regulation over autophagy in several cell types (102, 108, 110). We have recently documented that AMPK and p38 are both activated following acute exercise, which correlated with the nuclear localization of PGC-1α (111). We also demonstrated that PGC-1α is required for normal autophagy and mitophagy kinetics during exercise, as well as for the induction of the autophagy-related genes LC3 and p62 following exercise.
Contractile activity also results in sarcoplasmic reticulum stress and a PGC-1α-mediated unfolded protein response (43, 115). Certain unfolded protein response factors have been implicated in the activation of autophagy in various cell types (50, 70, 118). Interestingly, TFEB and PGC-1α have been documented to regulate the expression of one another in liver and neuronal cells under different cellular and metabolic stressors, which makes for an interesting metabolic regulatory circuit that is worthy of further study (103, 110).
Altogether, a number of transcriptional regulators of autophagy have emerged, and these appear to potentiate a nuclear-autophagic response to exercise. However, the transcriptional regulation of autophagy and lysosomal components, especially following exercise, has only recently begun to be elucidated, and much more work is necessary to establish the regulatory network of this important pathway.
AUTOPHAGY AND CHRONIC EXERCISE
Autophagy is fast emerging as an important mechanism underlying exercise-induced metabolic benefits (Fig. 2). Functional autophagy was found to be required for exercise-mediated protection against high-fat diet (HFD)-induced metabolic derailment (26). Bcl2AAA mutant mice with defective autophagy were more susceptible to HFD-induced obesity and failed to exhibit chronic exercise-mediated protection against HFD-induced impairments in glucose tolerance. Moreover, repeated bouts of damaging exercise reveal the importance of autophagy for cellular turnover postexercise, thus contributing to exercise-induced adaptations (54). Indeed, chronic voluntary exercise results in increased basal autophagy and mitophagy protein expression (52); however, this increase is compromised in mice that are heterozygous for beclin 1 (beclin1−/+). Muscle of beclin1−/+ mice is also deficient in mitochondrial biogenesis and angiogenesis, which culminate in reduced adaptive endurance capacity. Therefore, increased basal autophagy is required for chronic exercise-induced skeletal muscle adaptations and improvements of physical performance (52). Moreover, since the autophagy-lysosome system is also involved in exosome release and vesicular trafficking (4), it is conceivable that deficient autophagy may alter the release of muscle-derived myokines and miRs. Due to the importance of muscle as a metabolic center, this could dampen whole body exercise-induced benefits. Interestingly, chronic exercise was also demonstrated to restore autophagy flux in skeletal muscle of animals treated with the lysosomal inhibitor chloroquine, as well as increase the abundance of autophagy markers with aging (38, 114). This suggests exercise as potential form of therapy for myopathies characterized by deficient autophagy. However, exercise should be prescribed with caution, as in certain autophagy-deficient conditions such as collagen VI-deficiency, it may be detrimental (19).
EXERCISE AND AUTOPHAGY WITH AGING
The autophagy-lysosome system appears to have a pleiotropic role in tissue turnover and cellular clearance, which are compromised during cellular senescence (31, 90). In accordance with this, the suppression of autophagy causes progressive dysfunction in various organs with age. Interestingly, multiple factors currently recognized to be involved in life extension also impinge on autophagy. For instance, rapamycin-mediated longevity is mediated by mTOR suppression, which leads to the activation of autophagy. Similarly, resveratrol mediated-sirtuin activation also leads to enhanced autophagy. Caloric restriction can inhibit mTOR and activate sirtuins as well as AMPK, all signals that converge on autophagy activation. Moreover, spermidine- and coffee-mediated life-span extension have been attributed to autophagy activation (12, 81). Indeed, life-long exercise, in combination with caloric restriction, improved the decline in autophagy proteins with aging and dampened the age-related increase in oxidative damage and apoptosis (114). Autophagy involvement in longevity and aging is a growing field of interest and has been reviewed extensively (55, 59, 90). However, whether autophagy dysfunction contributes to aging, or whether cellular senescence contributes to autophagic dysfunction, thus leading to a vicious cycle, needs to be further teased out. The compilation of these findings indicates that autophagy may indeed be the common element required for life extension. How autophagy may mediate longevity is not fully elucidated yet, but it could be related to its role in facilitating the disposal of noxious proteins and organelles, thus preventing their accumulation and promoting cellular rejuvenation.
CONCLUSION
A growing body of evidence indicates that autophagy is activated during an acute bout of exercise in various tissues and cell types, both acutely through posttranslational modifications, as well as more chronically by the enlistment of a transcriptional program. It is also becoming increasingly evident that autophagy is involved in exercise-induced adaptations through increased cellular turnover. With this in mind, the study of autophagy and exercise is still in its infancy, and additional research delving into the mechanisms of autophagy activation and contribution to long-term adaptations are required. The involvement of additional types of autophagy, such as chaperone-mediated autophagy and microautophagy, during exercise and exercise-induced adaptations should be investigated, as many metabolic factors are substrates of these pathways. Moreover, the importance of factors released by muscle via the autophagy pathway, such as exosomes and their contribution to intertissue information relay, is another important avenue for exploration (16). This may be fruitful in identifying blood-borne factors and interorgan communication axes that fosters the pleiotropic health benefits of exercise.
GRANTS
This work was supported by funding from the Natural Sciences and Engineering Research Council of Canada (NSERC) to D. A. Hood. D. A. Hood is also the holder of a Canada Research Chair in Cell Physiology. A. Vainshtein was a recipient of NSERC-Canada Graduate Scholarship.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: A.V. and D.A.H. drafted manuscript; A.V. and D.A.H. edited and revised manuscript; A.V. and D.A.H. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address of A. Vainshtein: Dept. of Molecular and Human Genetics at Baylor College of Medicine, and the Jan and Dan Duncan Neurological Research Institute at Texas Children's Hospital, 1250 Moursund St., Houston, TX 77030.
REFERENCES
- 1.Agudelo LZ, Femenía T, Orhan F, Porsmyr-Palmertz M, Goiny M, Martinez-Redondo V, Correia JC, Izadi M, Bhat M, Schuppe-Koistinen I, Pettersson AT, Ferreira DMS, Krook A, Barres R, Zierath JR, Erhardt S, Lindskog M, Ruas JL. Skeletal muscle PGC-1α1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell 159: 33–45, 2014. [DOI] [PubMed] [Google Scholar]
- 2.Akimoto T, Pohnert SC, Li P, Zhang M, Gumbs C, Rosenberg PB, Williams RS, Yan Z. Exercise stimulates Pgc-1alpha transcription in skeletal muscle through activation of the p38 MAPK pathway. J Biol Chem 280: 19587–19593, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J 16: 1879–1886, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Baixauli F, López-Otín C, Mittelbrunn M. Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front Immunol 5: 403, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J 28: 3015–3026, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Booth FW, Roberts CK, Laye MJ. Lack of exercise is a major cause of chronic diseases. Compr Physiol 2: 1143–1211, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cantó C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: 1056–1060, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chin ER. Role of Ca2+/calmodulin-dependent kinases in skeletal muscle plasticity. J Appl Physiol 99: 414–423, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Chua JP, Reddy SL, Merry DE, Adachi H, Katsuno M, Sobue G, Robins DM, Lieberman AP. Transcriptional activation of TFEB/ZKSCAN3 target genes underlies enhanced autophagy in spinobulbar muscular atrophy. Hum Mol Genet 23: 1376–1386, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui M, Yu H, Wang J, Gao J, Li J. Chronic caloric restriction and exercise improve metabolic conditions of dietary-induced obese mice in autophagy correlated manner without involving AMPK. J Diabetes Res 2013: 852754, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dohm GL, Tapscott EB, Kasperek GJ. Protein degradation during endurance exercise and recovery. Med Sci Sports Exerc 19: S166–S171, 1987. [PubMed] [Google Scholar]
- 12.Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, Fussi H, Deszcz L, Hartl R, Schraml E, Criollo A, Megalou E, Weiskopf D, Laun P, Heeren G, Breitenbach M, Grubeck-Loebenstein B, Herker E, Fahrenkrog B, Fröhlich KU, Sinner F, Tavernarakis N, Minois N, Kroemer G, Madeo F. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 11: 1305–1314, 2009. [DOI] [PubMed] [Google Scholar]
- 13.Ezaki J, Matsumoto N, Takeda-Ezaki M, Komatsu M, Takahashi K, Hiraoka Y, Taka H, Fujimura T, Takehana K, Yoshida M, Iwata J, Tanida I, Furuya N, Zheng DM, Tada N, Tanaka K, Kominami E, Ueno T. Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy 7: 727–736, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A 102: 8204–8209, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F. Ambra1 regulates autophagy and development of the nervous system. Nature 447: 1121–1125, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Frühbeis C, Helmig S, Tug S, Simon P, Krämer-Albers EM. Physical exercise induces rapid release of small extracellular vesicles into the circulation. J Extracell Vesicles 4: 28239, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ge L, Melville D, Zhang M, Schekman R. The ER-Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. Elife 2: e00947, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grumati P, Coletto L, Sabatelli P, Cescon M, Angelin A, Bertaggia E, Blaauw B, Urciuolo A, Tiepolo T, Merlini L, Maraldi NM, Bernardi P, Sandri M, Bonaldo P. Autophagy is defective in collagen VI muscular dystrophies, and its reactivation rescues myofiber degeneration. Nat Med 16: 1313–1320, 2010. [DOI] [PubMed] [Google Scholar]
- 19.Grumati P, Coletto L, Schiavinato A, Castagnaro S, Bertaggia E, Sandri M, Bonaldo P. Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles. Autophagy 7: 1415–1423, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141: 656–667, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T. Autophagosomes form at ER-mitochondria contact sites. Nature 495: 389–393, 2013. [DOI] [PubMed] [Google Scholar]
- 22.Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson ÅB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 287: 19094–19104, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hara T, Takamura A, Kishi C, Iemura SI, Natsume T, Guan JL, Mizushima N. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 181: 497–510, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hardie DG. Energy sensing by the AMP-activated protein kinase and its effects on muscle metabolism. Proc Nutr Soc 70: 92–99, 2011. [DOI] [PubMed] [Google Scholar]
- 25.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res 107: 1470–1482, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW, Gilpin C, Xiao G, Bassel-Duby R, Scherer PE, Levine B. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481: 511–515, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He C, Sumpter R, Levine B. Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 8: 1548–1551, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hood DA, Uguccioni G, Vainshtein A, D'souza D. Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle: implications for health and disease. Compr Physiol 1: 1119–1134, 2011. [DOI] [PubMed] [Google Scholar]
- 29.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, Guan JL, Oshiro N, Mizushima N. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 20: 1981–1991, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy 5: 973–979, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Hubbard VM, Valdor R, Macian F, Cuervo AM. Selective autophagy in the maintenance of cellular homeostasis in aging organisms. Biogerontology 13: 21–35, 2012. [DOI] [PubMed] [Google Scholar]
- 32.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, Noda T, Ohsumi Y. A ubiquitin-like system mediates protein lipidation. Nature 408: 488–492, 2000. [DOI] [PubMed] [Google Scholar]
- 33.Irrcher I, Adhihetty PJ, Joseph AM, Ljubicic V, Hood DA. Regulation of mitochondrial biogenesis in muscle by endurance exercise. Sports Med 33: 783–793, 2003. [DOI] [PubMed] [Google Scholar]
- 34.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 19: 5360–5372, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jamart C, Benoit N, Raymackers JM, Kim HJ, Kim CK, Francaux M. Autophagy-related and autophagy-regulatory genes are induced in human muscle after ultraendurance exercise. Eur J Appl Physiol 112: 3173–3177, 2012. [DOI] [PubMed] [Google Scholar]
- 36.Jamart C, Francaux M, Millet GY, Deldicque L, Frère D, Féasson L. Modulation of autophagy and ubiquitin-proteasome pathways during ultra-endurance running. J Appl Physiol 112: 1529–1537, 2012. [DOI] [PubMed] [Google Scholar]
- 37.Jamart C, Naslain D, Gilson H, Francaux M. Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state. Am J Physiol Endocrinol Metab 305: E964–E974, 2013. [DOI] [PubMed] [Google Scholar]
- 38.Jiang D, Chen K, Lu X, Gao H, Qin Z, Lin F. Exercise ameliorates the detrimental effect of chloroquine on skeletal muscles in mice via restoring autophagy flux. Acta Pharmacol Sin 35: 135–142, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joassard OR, Amirouche A, Gallot YS, Desgeorges MM, Castells J, Durieux AC, Berthon P, Freyssenet DG. Regulation of Akt-mTOR, ubiquitin-proteasome and autophagy-lysosome pathways in response to formoterol administration in rat skeletal muscle. Int J Biochem Cell Biol 45: 2444–2455, 2013. [DOI] [PubMed] [Google Scholar]
- 40.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 22: 407–417, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, Attardi LD. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev 27: 1016–1031, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim HJ, Jamart C, Deldicque L, An G, Lee YH, Kim CK, Raymackers J, Francaux M. Endoplasmic reticulum stress markers and ubiquitin-proteasome pathway activity in response to a 200-km run. Med Sci Sports Exerc 43: 18–25, 2011. [DOI] [PubMed] [Google Scholar]
- 44.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim DH, Hur KY, Kim HK, Ko T, Han J, Kim HL, Kim J, Back SH, Komatsu M, Chen H, Chan DC, Konishi M, Itoh N, Choi CS, Lee MS. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med 19: 83–92, 2013. [DOI] [PubMed] [Google Scholar]
- 46.Kirkin V, Lamark T, Sou YS, Bjørkøy G, Nunn JL, Bruun JA, Shvets E, McEwan DG, Clausen TH, Wild P, Bilusic I, Theurillat JP, Øvervatn A, Ishii T, Elazar Z, Komatsu M, Dikic I, Johansen T. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 33: 505–516, 2009. [DOI] [PubMed] [Google Scholar]
- 47.Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, Kominami E, Tanaka K, Chiba T. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 169: 425–434, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuma A, Mizushima N, Ishihara N, Ohsumi Y. Formation of the approximately 350-kDa Apg12-Apg5. Apg16 multimeric complex, mediated by Apg16 oligomerization, is essential for autophagy in yeast. J Biol Chem 277: 18619–18625, 2002. [DOI] [PubMed] [Google Scholar]
- 49.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A 105: 3374–3379, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li J, Ni M, Lee B, Barron E, Hinton DR, Lee AS. The unfolded protein response regulator GRP78/BiP is required for endoplasmic reticulum integrity and stress-induced autophagy in mammalian cells. Cell Death Differ 15: 1460–1471, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li W, Li J, Bao J. Microautophagy: lesser-known self-eating. Cell Mol Life Sci 69: 1125–1136, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lira VA, Okutsu M, Zhang M, Greene NP, Laker RC, Breen DS, Hoehn KL, Yan Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J 27: 4184–4193, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu X, Niu Y, Yuan H, Huang J, Fu L. AMPK binds to Sestrins and mediates the effect of exercise to increase insulin-sensitivity through autophagy. Metabolism 64: 658–665, 2015. [DOI] [PubMed] [Google Scholar]
- 54.LoVerso F, Carnio S, Vainshtein A, Sandri M. Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy 10: 11, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Madeo F, Zimmermann A, Maiuri MC, Kroemer G. Essential role for autophagy in life span extension. J Clin Invest 125: 85–93, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maiuri MC, Malik SA, Morselli E, Kepp O, Criollo A, Mouchel PL, Carnuccio R, Kroemer G. Stimulation of autophagy by the p53 target gene Sestrin2. Cell Cycle 8: 1571–1576, 2009. [DOI] [PubMed] [Google Scholar]
- 57.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6: 458–471, 2007. [DOI] [PubMed] [Google Scholar]
- 58.Marques-Aleixo I, Santos-Alves E, Balça MM, Rizo-Roca D, Moreira PI, Oliveira PJ, Magalhães J, Ascensão A. Physical exercise improves brain cortex and cerebellum mitochondrial bioenergetics and alters apoptotic, dynamic and auto(mito)phagy markers. Neuroscience 301: 480–495, 2015. [DOI] [PubMed] [Google Scholar]
- 59.Martinez-Lopez N, Athonvarangkul D, Singh R. Autophagy and aging. Adv Exp Med Biol 847: 73–87, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M. Autophagy is required to maintain muscle mass. Cell Metab 10: 507–515, 2009. [DOI] [PubMed] [Google Scholar]
- 61.Matsuura A, Tsukada M, Wada Y, Ohsumi Y. Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 192: 245–250, 1997. [DOI] [PubMed] [Google Scholar]
- 62.Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, Xu H, Sandri M, Rizzuto R, De Matteis MA, Ballabio A. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17: 288–299, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, Seydel A, Zhao J, Abraham R, Goldberg AL, Blaauw B, DePinho RA, Sandri M. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 6: 6670, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 147: 728–741, 2011. [DOI] [PubMed] [Google Scholar]
- 65.Mofarrahi M, Guo Y, Haspel JA, Choi AMK, Davis EC, Gouspillou G, Hepple RT, Godin R, Burelle Y, Hussain SNA. Autophagic flux and oxidative capacity of skeletal muscles during acute starvation. Autophagy 9: 1604–1620, 2013. [DOI] [PubMed] [Google Scholar]
- 66.Møller AB, Vendelbo MH, Christensen B, Clasen BFF, Bak AM, Jørgensen JOL, Møller N, Jessen N. Physical exercise increases autophagic signaling through ULK1 in human skeletal muscle. J Appl Physiol (1985) 118: 971–979, 2015. [DOI] [PubMed] [Google Scholar]
- 67.Motley AM, Nuttall JM, Hettema EH. Atg36: the Saccharomyces cerevisiae receptor for pexophagy. Autophagy 8: 1680–1681, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6: 1090–1106, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Naya FJ, Mercer B, Shelton J, Richardson JA, Williams RS, Olson EN. Stimulation of slow skeletal muscle fiber gene expression by calcineurin in vivo. J Biol Chem 275: 4545–4548, 2000. [DOI] [PubMed] [Google Scholar]
- 70.Ngo JK, Pomatto LCD, Davies KJA. Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol 1: 258–264, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O'Leary MFN, Hood DA. Denervation-induced oxidative stress and autophagy signaling in muscle. Autophagy 5: 230–231, 2009. [DOI] [PubMed] [Google Scholar]
- 72.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback a J, Urano F, Imaizumi K. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol 26: 9220–9231, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ohsumi Y, Mizushima N. Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol 15: 231–236, 2004. [DOI] [PubMed] [Google Scholar]
- 74.Palikaras K, Tavernarakis N. Mitophagy in neurodegeneration and aging. Front Genet 3: 297, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 20: 3852–3866, 2011. [DOI] [PubMed] [Google Scholar]
- 76.Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, Bjørkøy G, Johansen T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol 188: 253–269, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Parsons SA, Wilkins BJ, Bueno OF, Molkentin JD. Altered skeletal muscle phenotypes in calcineurin Aalpha and Abeta gene-targeted mice. Mol Cell Biol 23: 4331–4343, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122: 927–939, 2005. [DOI] [PubMed] [Google Scholar]
- 79.Penna F, Costamagna D, Pin F, Camperi A, Fanzani A, Chiarpotto EM, Cavallini G, Bonelli G, Baccino FM, Costelli P. Autophagic degradation contributes to muscle wasting in cancer cachexia. Am J Pathol 182: 1367–1378, 2013. [DOI] [PubMed] [Google Scholar]
- 80.Pietrocola F, Izzo V, Niso-Santano M, Vacchelli E, Galluzzi L, Maiuri MC, Kroemer G. Regulation of autophagy by stress-responsive transcription factors. Semin Cancer Biol 23: 310–322, 2013. [DOI] [PubMed] [Google Scholar]
- 81.Pietrocola F, Malik SA, Mariño G, Vacchelli E, Senovilla L, Chaba K, Niso-Santano M, Maiuri MC, Madeo F, Kroemer G. Coffee induces autophagy in vivo. Cell Cycle 13: 1987–1994, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pogozelski AR, Geng T, Li P, Yin X, Lira VA, Zhang M, Chi JT, Yan Z. p38gamma mitogen-activated protein kinase is a key regulator in skeletal muscle metabolic adaptation in mice. PLoS One 4: e7934, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qiao S, Dennis M, Song X, Vadysirisack DD, Salunke D, Nash Z, Yang Z, Liesa M, Yoshioka J, Matsuzawa SI, Shirihai OS, Lee RT, Reed JC, Ellisen LW. A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat Commun 6: 7014, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Radtke K, English L, Rondeau C, Leib D, Lippé R, Desjardins M. Inhibition of the host translation shutoff response by herpes simplex virus 1 triggers nuclear envelope-derived autophagy. J Virol 87: 3990–3997, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rahman M, Mofarrahi M, Kristof AS, Nkengtac B, Harel S, Hussain SN. Reactive oxygen species regulation of autophagy in skeletal muscles. Antioxid Redox Signal 20: 443–459, 2014. [DOI] [PubMed] [Google Scholar]
- 86.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 12: 747–757, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ro SH, Semple IA, Park H, Park H, Park HW, Kim M, Kim JS, Lee JH. Sestrin2 promotes Unc-51-like kinase 1 mediated phosphorylation of p62/sequestosome-1. FEBS J 281: 3816–3827, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rogers MA, Evans WJ. Changes in skeletal muscle with aging: effects of exercise training. Exerc Sport Sci Rev 21: 65–102, 1993. [PubMed] [Google Scholar]
- 89.Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, Adams PD, Anderson KI, Gottlieb E, Sansom OJ, Ryan KM. p53 status determines the role of autophagy in pancreatic tumour development. Nature 504: 296–300, 2013. [DOI] [PubMed] [Google Scholar]
- 90.Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell 146: 682–695, 2011. [DOI] [PubMed] [Google Scholar]
- 91.Russ DW, Krause J, Wills A, Arreguin R. “SR stress” in mixed hindlimb muscles of aging male rats. Biogerontology 13: 547–555, 2012. [DOI] [PubMed] [Google Scholar]
- 92.Safdar A, Little JP, Stokl AJ, Hettinga BP, Akhtar M, Tarnopolsky MA. Exercise increases mitochondrial PGC-1alpha content and promotes nuclear-mitochondrial cross-talk to coordinate mitochondrial biogenesis. J Biol Chem 286: 10605–10617, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 93.Salas M, Tuchweber B, Kourounakis P. Liver ultrastructure during acute stress. Pathol Res Pract 167: 217–233, 1980. [DOI] [PubMed] [Google Scholar]
- 94.Saleem A, Adhihetty PJ, Hood DA. Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol Genomics 37: 58–66, 2009. [DOI] [PubMed] [Google Scholar]
- 95.Saleem A, Carter HN, Hood DA. p53 is necessary for the adaptive changes in cellular milieu subsequent to an acute bout of endurance exercise. Am J Physiol Cell Physiol 306: C241–C249, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Saleem A, Hood DA. Acute exercise induces tumour suppressor protein p53 translocation to the mitochondria and promotes a p53-Tfam-mitochondrial DNA complex in skeletal muscle. J Physiol 591: 3625–3636, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Salminen A, Vihko V. Autophagic response to strenuous exercise in mouse skeletal muscle fibers. Virchows Arch B Cell Pathol Incl Mol Pathol 45: 97–106, 1984. [DOI] [PubMed] [Google Scholar]
- 98.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 454: 232–235, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A. A gene network regulating lysosomal biogenesis and function. Science 325: 473–477, 2009. [DOI] [PubMed] [Google Scholar]
- 100.Satoo K, Noda NN, Kumeta H, Fujioka Y, Mizushima N, Ohsumi Y, Inagaki F. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J 28: 1341–1350, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schworer CM, Shiffer KA, Mortimore GE. Quantitative relationship between autophagy and proteolysis during graded amino acid deprivation in perfused rat liver. J Biol Chem 256: 7652–7658, 1981. [PubMed] [Google Scholar]
- 102.Scott I, Webster BR, Chan CK, Okonkwo JU, Han K, Sack MN. GCN5-like protein 1 (GCN5L1) controls mitochondrial content through coordinated regulation of mitochondrial biogenesis and mitophagy. J Biol Chem 289: 2864–2872, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, Wollenberg AC, Di Bernardo D, Chan L, Irazoqui JE, Ballabio A. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol 15: 647–658, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A. TFEB links autophagy to lysosomal biogenesis. Science 332: 1429–1433, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31: 1095–1108, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Spitali P, Grumati P, Hiller M, Chrisam M, Aartsma-Rus A, Bonaldo P. Autophagy is impaired in the tibialis anterior of dystrophin null mice. PLoS Curr 2013: 5, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stephan JS, Yeh YY, Ramachandran V, Deminoff SJ, Herman PK. The Tor and PKA signaling pathways independently target the Atg1/Atg13 protein kinase complex to control autophagy. Proc Natl Acad Sci U S A 106: 17049–17054, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Takikita S, Schreiner C, Baum R, Xie T, Ralston E, Plotz PH, Raben N. Fiber type conversion by PGC-1α activates lysosomal and autophagosomal biogenesis in both unaffected and Pompe skeletal muscle. PLoS One 5: e15239, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tasdemir E, Chiara Maiuri M, Morselli E, Criollo A, D'Amelio M, Djavaheri-Mergny M, Cecconi F, Tavernarakis N, Kroemer G. A dual role of p53 in the control of autophagy. Autophagy 4: 810–814, 2008. [DOI] [PubMed] [Google Scholar]
- 110.Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RAV, Lazarowski ER, Damian VA, Masliah E, La Spada AR. PGC-1α rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med 4: 142ra97, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Vainshtein A, Tryon LD, Pauly M, Hood DA. Role of PGC-1α during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am J Physiol Cell Physiol 308: C710–C719, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Van der Vos KE, Eliasson P, Proikas-Cezanne T, Vervoort SJ, van Boxtel R, Putker M, van Zutphen IJ, Mauthe M, Zellmer S, Pals C, Verhagen LP, Groot Koerkamp MJA, Braat AK, Dansen TB, Holstege FC, Gebhardt R, Burgering BM, Coffer PJ. Modulation of glutamine metabolism by the PI(3)K-PKB-FOXO network regulates autophagy. Nat Cell Biol 14: 829–837, 2012. [DOI] [PubMed] [Google Scholar]
- 113.Wang J, Tan D, Cai Y, Reinisch KM, Walz T, Ferro-Novick S. A requirement for ER-derived COPII vesicles in phagophore initiation. Autophagy 10: 708–709, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wohlgemuth SE, Seo AY, Marzetti E, Lees HA, Leeuwenburgh C. Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life-long exercise. Exp Gerontol 45: 138–148, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wu J, Ruas JL, Estall JL, Rasbach KA, Choi JH, Ye L, Boström P, Tyra HM, Crawford RW, Campbell KP, Rutkowski DT, Kaufman RJ, Spiegelman BM. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1α/ATF6α complex. Cell Metab 13: 160–169, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo-Kakuta C, Ichikawa R, Kinjo M, Ohsumi Y. Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol 198: 219–233, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yan Z, Lira VA, Greene NP. Exercise training-induced regulation of mitochondrial quality. Exerc Sport Sci Rev 40: 159–164, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yu Z, Wang AM, Adachi H, Katsuno M, Sobue G, Yue Z, Robins DM, Lieberman AP. Macroautophagy is regulated by the UPR-mediator CHOP and accentuates the phenotype of SBMA mice. PLoS Genet 7: e1002321, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang L, Hu X, Luo J, Li L, Chen X, Huang R, Pei Z. Physical exercise improves functional recovery through mitigation of autophagy, attenuation of apoptosis and enhancement of neurogenesis after MCAO in rats. BMC Neurosci 14: 46, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 6: 472–483, 2007. [DOI] [PubMed] [Google Scholar]
- 121.Zhao J, Brault JJ, Schild A, Goldberg AL. Coordinate activation of autophagy and the proteasome pathway by FoxO transcription factor. Autophagy 4: 378–380, 2008. [DOI] [PubMed] [Google Scholar]
- 122.Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, Zhu WG. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat Cell Biol 12: 665–675, 2010. [DOI] [PubMed] [Google Scholar]
- 123.Zheng DM, Bian Z, Furuya N, Oliva Trejo JA, Takeda-Ezaki M, Takahashi K, Hiraoka Y, Mineki R, Taka H, Ikeda SI, Komatsu M, Fujimura T, Ueno T, Ezaki J. A treadmill exercise reactivates the signaling of the mammalian target of rapamycin (mTor) in the skeletal muscles of starved mice. Biochem Biophys Res Commun 456: 519–526, 2015. [DOI] [PubMed] [Google Scholar]
- 124.Zhou J, Liao W, Yang J, Ma K, Li X, Wang Y, Wang D, Wang L, Zhang Y, Yin Y, Zhao Y, Zhu WG. FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy 8: 1712–1723, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]