

Abstract
Interleukin-18 (IL-18), a proinflammatory cytokine, has been implicated in pathologic left ventricular hypertrophy and is elevated in plasma of heart failure patients. However, IL-18 blockade strategies have been conflicting. The purpose of these experiments was to determine whether genetic ablation of IL-18 would protect mice against hypobaric hypoxia (HH)-induced right ventricular (RV) hypertrophy, a condition in which chamber-specific inflammation is prominent. We hypothesized that IL-18 knockout (KO) mice would be protected while wild-type (WT) mice would demonstrate RV hypertrophy in response to HH exposure. KO and WT mice were exposed to HH for 7 wk, and control mice were exposed to normoxic ambient air. Following echocardiography, the RV was dissected and flash-frozen for biochemical analyses. HH exposure increased IL-18 mRNA (P = 0.08) in RV from WT mice. Genetic ablation of IL-18 mildly attenuated RV hypertrophy as assessed by myocyte size. However, IL-18 KO mice were not protected against HH-induced organ-level remodeling, as evidenced by higher RV weights, elevated RV systolic pressure, and increased RV anterior wall thickness compared with normoxic KO mice. These RV changes were similar to those seen in HH-exposed WT mice. Compensatory upregulation of other proinflammatory cytokines IL-2 and stromal cell-derived factor-1 was seen in the HH-KO animals, suggesting that activation of parallel inflammatory pathways might mitigate the effect of IL-18 KO. These data suggest targeted blockade of IL-18 alone is not a viable therapeutic strategy in this model.
Keywords: pulmonary hypertension, right ventricle, cardiac, IL-18
sustained production of inflammatory cytokines is implicated in the development and progression of heart failure (HF) (21). Various studies have assessed the effect of cytokines on cardiac function in vivo, many using tumor necrosis factor-α (TNF-α) as a representative cytokine, and show that infusion of TNF-α results in left ventricular (LV) systolic dysfunction (12). Mechanistically, TNF-α and similar cytokines induce cellular changes responsible for systolic dysfunction, including altered β-adrenergic sensitivity (14), alterations of calcium currents (29), and remodeling of the cardiac extracellular matrix (18). While many of these studies were conducted in the context of LV hypertrophy and failure, the right ventricle (RV) is also sensitive to inflammatory stimuli. The infiltration of immune cells in and around the RV and pulmonary vasculature during exposure to hypobaric hypoxia (HH) suggests an underlying inflammatory component may be equally important in the context of RV hypertrophy (36).
Interleukin-18 (IL-18), a proinflammatory cytokine of the IL-1 family, is chronically induced during various immune and inflammatory conditions (10). IL-18 elicits characteristics of other proinflammatory cytokines of the IL-1 family, such as increases in cell adhesion molecule expression, nitric oxide synthesis, and production of cytokines such as IL-1β, TNF-α, and interferon-γ (24). In the heart, macrophage and endothelial cells most abundantly express IL-18. However, IL-18 has also been detected in cardiomyocytes and cardiac fibroblasts, particularly in areas of inflammation where it stimulates hypertrophic (6) and proapoptotic properties (7) and induces contractile dysfunction and remodeling of the extracellular matrix (13).
An increasing number of both animal and human epidemiological studies indicate IL-18 is highly associated with cardiac disease. Daily administration of IL-18 causes LV hypertrophy, increased collagen content (27), and elevated LV diastolic pressure in mice (39). In addition, overexpression of IL-18 in the lungs results in mild pulmonary hypertension (PH) and RV dilation (15). LV pressure overload, accomplished by transaortic constriction (TAC), induces IL-18 expression and results in fibrotic remodeling of the LV and subsequent LV dysfunction (43). IL-18 protein and mRNA are upregulated in the myocardium of patients with ischemic cardiomyopathy, and plasma IL-18 is significantly elevated in patients with HF compared with controls, and directly correlates with disease severity (20). Together, these data suggest a role for IL-18 in the etiology and progression of ventricular dysfunction and HF.
Despite the well-established role for IL-18 as a maladaptive cardiac cytokine, IL-18 blockade strategies in animal models of heart disease have been conflicting (8, 35). Whether IL-18 blockade strategies are deleterious by inhibiting necessary remodeling or beneficial by limiting pathological maladaptation remains unknown. Therefore, we tested the hypothesis that genetic ablation of IL-18 would protect mice against maladaptive RV remodeling following exposure to HH. This model induces modest PH in the mouse associated with RV hypertrophy and dysfunction and in addition is associated with infiltration of the RV with inflammatory cells (4, 5). We show for the first time in a model of PH and RV pressure overload that genetic ablation of IL-18 does not protect against HH-induced RV remodeling, and thus does not represent a singular therapeutic strategy.
METHODS
Mouse model of RV hypertrophy.
Experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of Colorado Denver. Homozygous IL-18 knockout (KO) mice (33) were purchased from Jackson Laboratories. C57BL/6J mice were used as wild-type (WT) controls. Male and female mice (12–15 wk) were used for the experiments. Mice were housed for 7 wk in HH chambers (equivalent to ∼5,380 m; Po2: 10%). Control mice were housed at Denver, CO, room atmosphere. No morbidity or mortality of the mice occurred during the study period. Mouse cohorts are defined as follows: IL-18 KO HH (n = 6), IL-18 KO control (CO; n = 5), WT HH (n = 6), and WT CO (n = 4). Following the 7-wk exposure, echocardiographic assessments were obtained; the RV was dissected from the atria and LV and flash-frozen for biochemical analyses.
Echocardiography.
Echocardiographic assessments were performed using a Vevo770 (VisualSonics). Animals were anesthetized using 2% isoflurane, and body temperature was maintained at 37°C. Pulse-wave Doppler of pulmonary outflow was recorded in the parasternal short-axis view at the level of the aortic valve. RV systolic pressure was assessed at the end of the 7-wk period and after the last ultrasound analysis. During this assessment, mice were ventilated with 100% oxygen and 2% isoflurane.
Histological analysis.
Histology was performed on a small subset of samples. Pieces of RV were embedded and frozen in optimum cutting temperature buffer. Sections (8 μm) were cut on a precooled −20°C cryostat. Slides were stained with hematoxylin-eosin (H&E) to evaluate cell size. Myocyte size was measured on an Olympus CK40 light microscope under ×20 magnification. Calculations were made using Motic Images Plus 2.0 with repeated measurements on each slide. All measurements were conducted in a blind fashion.
Gel electrophoresis.
RV proteins were separated on 12.5% SDS-PAGE gels. Gels were stained with phosphospecific Pro-Q Diamond Stain to detect phosphorylation, and phosphoproteins were imaged using a Typhoon 9410 Gel Imager. After imaging, the same gels were stained with Coomassie brilliant blue to quantify total protein. Relative phosphorylation was calculated by dividing the Pro-Q Diamond signal of each protein by the total protein signal for myosin light chain 1 (37).
Western blot analysis.
RV and LV lysates were homogenized in RIPA buffer (50 mM Tris·HCl, 150 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Nonidet P-40, and 0.5% sodium deoxycholate, pH 7.4) containing 1 mM dithiothreitol, 2 mM tributylphosphine, and protease inhibitors. Protein concentration was determined using a modified protein assay (Bio-Rad) and prepared in Laemmli sample buffer (Bio-Rad). Proteins were resolved on a gradient SDS-PAGE gel and transferred to nitrocellulose or polyvinyl difluoride. Following blocking in 5% nonfat dry milk or 5% bovine serum albumin for 1 h at room temperature, membranes were incubated with primary antibody overnight at 4°C. The following primary antibodies were used: collagen (sc-8784R, 1:200; Santa Cruz), pan troponin I (TnI; 2010-TNI, 1:5,000; PhosphoSolutions), IL-2 (sc-7896, 1:200; Santa Cruz), stromal cell-derived factor-1 (SDF-1, sc-6193, 1:200; Santa Cruz), phospho-p44/42 mitogen-activated protein kinase (MAPK) (Thr202/Tyr204) (no. 9101, 1:1,000; Cell Signaling), and p44/42 MAPK (no. 9102, 1:1,000; Cell Signaling). Membranes were washed and incubated with secondary antibody for 1 h at room temperature. Protein bands were visualized using a chemiluminescent substrate and autoradiography. Even loading of proteins was verified by pan TnI and Ponceau S staining.
Real-time RT-PCR.
RNA was extracted from RV by the ReliaPrep RNA MiniPrep System (Promega) and was reverse transcribed using the iScript cDNA synthesis kit (Bio-Rad). Real-time RT-PCR was performed with the iCycler My iQ using iQ SYBR Green Supermix (Bio-Rad), normalized to the housekeeping gene 18S ribosomal RNA (18S). Data are expressed as ΔCt values to permit comparison across all four groups. As such, a higher ΔCt value indicates lower expression. Myosin heavy chain-β (MYHCβ), atrial natriuretic factor (ANF), brain natriuretic peptide (BNP), myosin heavy chain-α (MYHCα), sarcoplasmic reticulum Ca2+-ATPase (SERCA), and 18S oligonucleotide sequences were used as previously published (11). IL-18 oligonucleotide sequences were as follows: forward 5′-GCCTCAAACCTTCCAAATCA-3′ and reverse 5′-TGGATCCATTTCCTCAAAGG-3′.
Collagen assay.
Hydroxyproline content was assessed using a commercially available kit (Sigma-Aldrich) following the manufacturer instructions. Briefly, RV lysates were hydrolyzed overnight at 95°C in 12 M HCl. Hydroxyproline concentration was determined spectrophotometrically using known hydroxyproline standards.
Cytokine profile array.
A mouse cytokine array panel was performed following the manufacturer instructions (R&D Systems). This kit allows simultaneous detection of multiple chemokine and cytokine analytes. Briefly, four RV from each cohort were pooled and incubated with the cytokine array antibody cocktail overnight to allow binding to the analyte-containing membrane. Membranes were visualized with streptavidin-horseradish peroxidase, and imaged via autoradiography, with two biological replicates of each group. Within each genotype (WT and KO), a ratio of each cytokine/chemokine was calculated from HH/CO. This ratio was log transformed to allow comparisons of HH response between the two genotypes.
The following cytokines and chemokines were assessed in this profile array: B lymphocyte chemoattractant/CXCL13, C5/C5a/complement component 5a, granulocyte-colony stimulating factor, granulocyte macrophage colony-stimulating factor, I-309/CCL1, eotaxin/CCL11, sICAM-1/CD54, interferon-γ, IL-1α, IL-1β, interleukin-1 receptor antagonist, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-10, IL-13, IL-12 p70, IL-16, IL-17, IL-23, IL-27, IP-10/interferon-γ induced protein 10 (CXCL10), T-cell-α chemoattractant/CXCL11, keratinocyote chemoattractant/CXCL1, macrophage colony-stimulating factor, JE/CCL2/monocyote chemoattractant protein-1, monocyte chemoattractant protein-5/CCL12, MIG/CXCL9, macrophage inflammatory protein (MIP)-1α/CCL3, MIP-1β/CCL4, MIP-2/CXCL2, RANTES/CCL5, stromal cell derived factor-1 (SDF-1)/CXCL12, thymus and activation-regulated chemokine/CCL17, tissue inhibitor of metalloproteinases-1, TNF-α, and triggering receptor expressed on myeloid cells-1.
Statistical analyses.
Significance was set a priori at P < 0.05. Data were analyzed by Student's t-test or 2-way ANOVA using IBM SPSS Statistics Version 22. Data with unequal variance were transformed to meet assumptions for homoscedasticity. Data are expressed as means ± SE. Where statistical analyses trend toward significance (P < 0.1), values are also noted above Figs. 1–8.
Fig. 1.
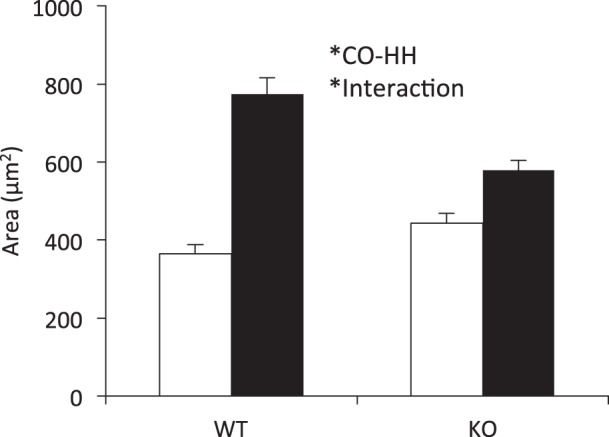
Hypobaric hypoxia (HH) exposure increases right ventricular (RV) myocyte size in both wild-type (WT) and interleukin (IL)-18 knockout (KO) mice compared with control (CO), with a moderate increase in cell size in WT mice compared with KO. RV sections were stained with hematoxylin and eosin (H&E), and cell size was measured in the circumferential plane. Two animals from each cohort were assessed, and cell size was repeatedly measured within each cohort. CO WT (n = 25), HH WT (n = 16), CO KO (n = 25), HH KO (n = 17). Open bars, CO; filled bars, HH.
Fig. 8.

Exposure to HH tends to activate extracellular signal-regulated kinase (ERK) in RV from IL-18 KO mice. Phosphorylation of ERK was assessed by immunoblotting and normalized to total ERK expression. Data are expressed as a phospho (P)-to-total (T) ratio. Membranes were probed for phospho-ERK (Thr202/Tyr204), stripped, and reprobed for total ERK protein. TnI verifies equal loading of protein. Open bars, CO; filled bars, HH. *P < 0.05. All four conditions were run on the same gel, and nonessential lanes were cropped for generation of the representative images.
RESULTS
PH and RV remodeling.
Chronic exposure to HH results in PH and concomitant RV dysfunction. In the current experiment, 7 wk of HH significantly increased lung weight and reduced pulmonary artery acceleration time (PAAT). PAAT, defined as the time from the onset of pulmonary flow to the peak of the ejection flow velocity, provides evidence of pulmonary vascular remodeling (16). Thus, the shorter the acceleration time, the more severe the PH (28). These indexes are in agreement with the pulmonary changes observed in other models of HH and confirm the development of PH. Analyses of RV myocyte size indicated hypertrophy in HH animals under both WT and KO genotypes (Fig. 1). An interaction effect suggested a moderate attenuation of RV cell size in KO mice compared with WT exposed to HH. Additionally, indexes of RV remodeling, including RV weight, RV-to-LV ratio, RV systolic pressure, and RV anterior wall thickness (RVAW), were significantly higher in HH compared with CO. However, none of the echocardiographic variables differed between WT and KO mice, and there were no significant interactions between genotype and environment (Table 1). Therefore, from the level of whole organ remodeling, KO mice did not respond differently to HH compared with WT mice.
Table 1.
Seven weeks of exposure to HH causes mild pulmonary hypertension and RV remodeling in both WT and IL-18 KO mice
| Body Wt, g | RV Wt, g | Lung Wt, g | RV/LV | HR, beats/min | RVSP, mmHg | PAAT, ms | RVAW, mm | |
|---|---|---|---|---|---|---|---|---|
| WT CO | 24.1 ± 1.6 | 0.025 ± 0.004 | 0.144 ± 0.009 | 0.275 ± 0.03 | 518 ± 18 | 29.2 ± 3.3 | 16.6 ± 0.31 | 0.37 ± 0.03 |
| WT HH | 23.9 ± 0.7 | 0.041 ± 0.004 | 0.175 ± 0.005 | 0.555 ± 0.06 | 476 ± 17 | 42.4 ± 2.5 | 13.1 ± 0.38 | 0.62 ± 0.05 |
| KO CO | 22.4 ± 0.8 | 0.027 ± 0.003 | 0.137 ± 0.001 | 0.313 ± 0.03 | 476 ± 27 | 28.7 ± 2.8 | 16.8 ± 0.35 | 0.4 ± 0.02 |
| KO HH | 22.8 ± 0.9 | 0.042 ± 0.002 | 0.172 ± 0.002 | 0.585 ± 0.04 | 416 ± 11 | 48.8 ± 0.5 | 14.2 ± 0.64 | 0.61 ± 0.02 |
| CO-HH | NS | <0.001* | <0.001* | <0.001* | <0.05* | <0.001* | <0.001* | <0.001* |
| KO-WT | NS | NS | NS | NS | <0.05* | NS | NS | NS |
| Interaction | NS | NS | NS | NS | NS | NS | NS | NS |
Values are means ± SE. Exposure to hypobaric hypoxia (HH) (7 wk) causes mild pulmonary hypertension and right ventricular (RV) remodeling in both wild-type (WT) and interleukin (IL)-18 knockout (KO) mice compared with control (CO). BW, body weight; RV/LV, right-to-left ventricle ratio; HR, heart rate; RSVP, right ventricular systolic pressure; PAAT, pulmonary artery acceleration time; RVAW, right ventricular anterior wall thickness.
Statistically significant difference; NS, not significant.
Induction of IL-18 by HH.
IL-18 expression was assessed in all WT RV to determine whether exposure to 7 wk of HH resulted in upregulation of the inflammatory cytokine. HH exposure trended to increase expression of IL-18 (P = 0.08) compared with control conditions (Fig. 2). Consistent with the murine genotype, IL-18 was undetectable in the hearts of the KO mice.
Fig. 2.
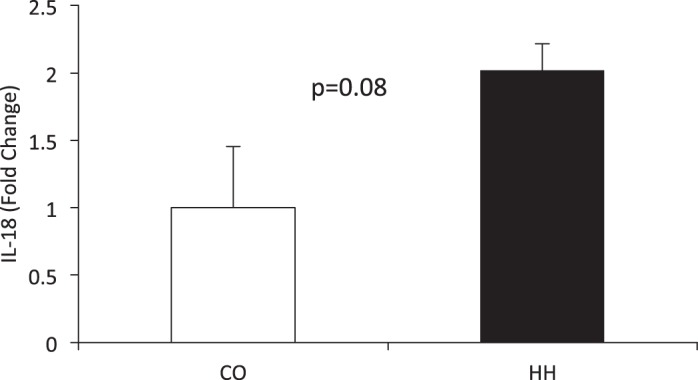
Exposure to HH for 7 wk trends toward a significant induction of IL-18 mRNA in the RV (P = 0.08) compared with CO. IL-18 expression was assessed in all WT RV by real-time RT-PCR and normalized to 18S. Fold change was calculated by 2−ΔΔCt. IL-18 expression was undetectable in the RV from KO mice.
Biochemical responses to RV pressure overload.
LV failure in rodents and humans is characterized by changes in phosphorylation of the myofilament proteins, and our group has previously shown decreases in phosphorylation of troponin T (TnT), TnI, and myosin light chain 2 (MLC2) in a neonatal bovine model of RV pressure overload (38). Therefore, we assessed the phosphorylation status of TnI, TnT, MLC2, and desmin in mice exposed to HH. We found desmin, MLC2, and TnI phosphorylation status to be unchanged by HH, with no differences between KO and WT mice (Fig. 3).
Fig. 3.

HH exposure for 7 wk does not significantly alter phosphorylation status of myofilament proteins, nor is phosphorylation status different between WT and IL-18 KO mice. Phosphorylation status was determined by Pro-Q Diamond staining of phosophoproteins relative to Coomassie brilliant blue staining of total protein. Open bars, CO; filled bars, HH. MLC1, myosin light chain 1; TnI, troponin I; MLC2, myosin light chain 2; TnT, troponin T.
To further quantify biochemical changes in the RV as a result of HH exposure, we assessed the expression of five genes in the hypertrophic gene program. These genes, components of the fetal gene program, are differentially expressed in established pathologic cardiac hypertrophy. We found MYHCβ, atrial natriuretic factor (ANF), brain natriuretic peptide (BNP), and sarcoplasmic reticulum Ca2+-ATPase (SERCA) expression to be unchanged by hypoxia, and did not differ between genotypes (Fig. 4, A–D). MYHCα expression (Fig. 4E) trended toward decreased expression in response to HH (P = 0.08), and decreased expression in KO compared with WT (P = 0.06).
Fig. 4.

Exposure to HH does not significantly induce the hypertrophic gene program, as assessed by expression of atrial natriuretic factor (ANF, A), brain natriuretic peptide (BNP, B), sarcoplasmic reticulum Ca2+-ATPase (SERCA, C), and myosin heavy chain-β (MYHCβ, D). However, 7 wk of HH exposure trended to decrease myosin heavy chain-α (MYHCα) expression in the RV (P = 0.08) (E), but this did not differ between IL-18 KO and WT mice. Furthermore, IL-18 KO animals trended toward decreased expression of MYHCα compared with WT mice (P = 0.06). Hypertrophic gene expression was assessed by real-time RT-PCR and normalized to 18S. Data are presented as ΔCt values. Open bars, CO; filled bars, HH.
RV remodeling in response to pressure overload is often characterized by increased collagen deposition. Therefore, we assessed collagen expression and content in the RV. Collagen (Fig. 5A) and procollagen (Fig. 5B) expression was not changed by 7 wk exposure to HH and did not differ between genotypes. Hydroxyproline content was assessed as a measurement of collagen content and was also unchanged by HH exposure and did not differ between WT and KO animals (Fig. 5C).
Fig. 5.

Collagen remodeling does not occur in the RV in response to 7 wk of HH and is not affected by IL-18 genetic ablation. Collagen (A) and procollagen (B) protein expression did not differ as a result of HH, nor did expression differ between genotypes. Collagen and procollagen expression was assessed by immunoblotting and normalized to TnI. All four conditions were run on the same gel, and nonessential lanes were cropped for generation of the representative images. C: collagen content, as assessed by hydroxyproline concentration, did not differ between CO and HH or between WT and KO mice. Hydroxyproline concentration was assessed spectrophotometrically using a commercially available kit. Open bars, CO; filled bars, HH.
Cytokine array.
Interleukins often display overlapping signaling, where knockdown of one member of the interleukin family results in upregulation of other family members. To assess whether IL-18 KO animals differentially expressed inflammatory cytokines, we assessed the relative expression of an array of cytokines in the RV. Cytokines and chemokines were not consistently upregulated within the KO animals under normoxic conditions. We then assessed the response to HH relative to CO within each genotype by calculating fold change of all cytokines in KO and WT mice. Fourteen of the 40 cytokines and chemokines were expressed with a change of greater than twofold (Fig. 6A), and 7 cytokines were differentially expressed between WT and KO mice (Fig. 6B). Of these, only IL-2 and SDF-1 showed a significant increase in the KO mice. Together, these assessments suggest that either or both of these compounds might compensate for the lack of protection against HH seen in the KO animals.
Fig. 6.

Compensatory upregulation of other proinflammatory cytokines IL-2 and stromal cell-derived factor-1 (SDF-1) in HH KO mice. A commercially available cytokine profile array was used to assess the expression of 40 chemokines and cytokines in all 4 groups. Log ratios of HH/CO were calculated within each genotype, and those with a fold change of >2 are reported in A. Only 14 proteins were induced >2-fold within each genotype, and, of these, only 7 showed differential expression between KO and WT mice (B). Green indicates significantly induced in the HH compared with CO, and proteins in red were significantly repressed in the HH compared with CO; n = 4 mice/group, with biological duplicates. BLC, B lymphocyte chemoattractant; C5a, complement component 5a; G-CSF, granulocyte-colony stimulating factor; GM-CSF, granulocyte macrophage colony-stimulating factor, IFN-γ, interferon-γ; IL, interleukin; KC, keratinocyote chemoattractant; M-CSF, macrophage colony-stimulating factor; TARC, thymus and activation-regulated chemokine; TNF-α, tumor necrosis factor-α.
Chamber-specific expression of IL-2 and SDF-1.
To confirm the induction of IL-2 and SDF-1 in RV from KO mice in response to HH as assessed by cytokine array, we performed immunoblotting in both the RV and the LV. We found a chamber-specific effect of HH on IL-2 expression, with KO mice expressing higher IL-2 compared with WT only in the RV (Fig. 7A), with no effect in the LV (Fig. 7B). Furthermore, 7 wk of HH resulted in a significant interaction between genotype and environment in the RV, with KO animals showing increased expression of IL-2 in response to HH (Fig. 7A). The induction of IL-2 only in the KO mice was confirmed by calculating a ratio of HH/CO, which showed a significant induction of IL-2 expression of >2.5-fold in the KO animals, with no effect in WT mice or in the LV.
Fig. 7.

Chamber-specific upregulation of IL-2 and SDF-1 protein expression in IL-18 KO mice in response to 7 wk of HH. A: in the RV, IL-18 KO mice expressed higher IL-2 than WT mice. IL-2 was significantly induced upon HH in KO mice, whereas no effect was observed in WT (P value for interaction = 0.05). B: no significant differences in IL-2 expression were observed between genotypes or CO and HH within the left ventricle (LV). C: the interaction between genotype and environment trended toward significance in the RV, and calculation of the ratio between HH/CO indicated an induction of SDF-1 expression in KO mice but not in WT. D: SDF-1 expression trended to decrease in response to HH in both genotypes of the LV. IL-2 and SDF-1 expression was assessed by immunoblotting in the RV and LV and normalized to TnI. Open bars, CO; filled bars, HH. *P ≤ 0.05. All four conditions were run on the same gel, and nonessential lanes were cropped for generation of the representative images.
SDF-1 protein expression was also confirmed in the RV and LV of WT and KO mice exposed to HH. A trend toward a significant interaction between genotype and environment was observed in the RV (Fig. 7C), and calculation of the HH-to-CO ratio indicated an induction of 1.5-fold in the KO mice in response to HH exposure. Exposure to HH tended to suppress SDF-1 expression in both WT and KO mice (P = 0.07) in the LV. However, SDF-1 expression was not influenced by genotype, nor did exposure to HH differentially affect WT compared with KO mice, since the interaction effect was not statistically significant (Fig. 7D) and the ratio of HH/CO was unchanged between WT and KO.
Downstream inflammatory signaling.
Cytokines and chemokines utilize a variety of downstream signaling pathways, including MAPK. Specifically, both IL-2 (3) and SDF-1 (9, 41) have been shown to signal through extracellular signal-regulated kinase (ERK) MAPK. Therefore, we assessed activation of ERK by immunoblotting in the RV. We found ERK to be higher in the WT RV compared with KO. Furthermore, we found an interaction effect (P = 0.09) between environment and genotype and a higher ratio of phospho/total ERK in response to HH in the KO mice compared with WT (Fig. 8). These data further support the cytokine array and Western data of compensatory upregulation of IL-2 and SDF-1 in the KO RV in response to HH and suggest other downstream cytokine signaling pathways may be similarly activated in this early response to HH.
DISCUSSION
Sustained production of inflammatory cytokines is implicated in the development and progression of HF (21), with IL-18 implicated in maladaptive cardiac remodeling (6, 7, 13). Despite the established deleterious effects of IL-18 on cardiac function, IL-18 blockade strategies have been discordant in animal models of heart disease (8, 35). Therefore, we assessed whether genetic ablation of IL-18 would protect against HH-induced RV dysfunction. We report that, despite a trend toward significant induction of IL-18 following hypoxia, knockout of IL-18 does not protect against maladaptive RV remodeling.
The stimulus to PH and RV overload employed in the present study has been used by others in a spectrum of models from mouse, rat, and cow (5, 17). However, the severity of HH-induced PH and RV pressure overload greatly varies between species (30), with mice displaying enhanced resistance to HH-induced pulmonary remodeling compared with humans, cows, and rats. Despite their resilient phenotype, the hemodynamic response seen in our mice mirrors that seen by others (4, 25), since chronic exposure to HH induced PH, as evidenced by increased lung weights and reduced pulmonary artery acceleration times. From a cardiac perspective, the mice also showed elevated RV systolic pressures, increased RV anterior wall thickness, and RV hypertrophy. With pressure overload, the degree of hypertrophy is linearly related to RV systolic dysfunction (2). Adaptive remodeling shows minimal increases in RV size and is characterized by a lack of change in RV systolic function. Our model shows significant hypertrophy as evidenced by increased RV weight and higher RVAW, increased myocyte size, and significant RV systolic pressure, none of which are characteristic of adaptive RV remodeling. This maladaptive RV remodeling should be even more apparent in the pressure-overloaded RV, since the RV is even more sensitive to increases in afterload compared with the LV (19). Thus, we believe the current investigation represents a mild early form of RV dysfunction in response to the pressure overload, since it is not yet associated with matrix remodeling. Furthermore, changes in ventricular functional measurements without corresponding biochemical signaling are not without precedence. Previous publications demonstrate that gene expression changes are not seen until the RV has progressed to failure (1) and are not always associated with measures of ventricular dysfunction. One might predict that the early remodeling phase assessed in the current investigation would be most reflective of interleukin activation and thus most responsive to IL-18 KO. Furthermore, this early disease stage provides the most clinical relevance, since this is when pharmaceutical intervention would occur. However, it is also possible that the effects of IL-18 activation target the later phases of cardiac maladaptation, a hypothesis that should be tested in future investigations.
To our knowledge, only two previous studies have examined the effect of IL-18 genetic knockout in response to a pathological stimulus in the heart, and these have yielded discrepant outcomes. IL-18 KO mice displayed reduced LV volume at baseline compared with WT in addition to blunted hypertrophy in TAC-induced LV pressure overload (8), suggesting a key role for IL-18 in cardiac growth and in the hypertrophic response to pressure overload. In this model of compensatory hypertrophy assessed 7 days following TAC, LV contractility increased in the WT TAC hearts but decreased in KO TAC mice compared with sham controls. Therefore, in the context of LV pressure overload, IL-18 was required for the hypertrophic response. However, in another report of IL-18 KO mice subjected to IL-1β-induced systolic dysfunction, IL-18 KO mice were protected against LV systolic dysfunction, as evidenced by preserved LV fractional shortening (34). Furthermore, this study reported no differences in LV dimension and function between IL-18 KO and WT mice at baseline. Our data provide the first evidence for IL-18 in HH-induced RV hypertrophy but do not shed additional light on this apparent discrepancy. Rather, our data suggest that the role of IL-18 in cardiac adaptation may be chamber and stimulus specific, as well as dependent on the timing of the disease process, in early vs. late remodeling.
Cytokine signaling is biologically redundant. Often, knockdown of one member of the interleukin family results in upregulation of other proinflammatory cytokines (22). Therefore, we assessed the expression of 40 chemokines and cytokines to determine whether compensatory upregulation of other inflammatory signals could account for the lack of protection against HH-induced RV hypertrophy in the context of IL-18 KO. We did not find cytokines and chemokines to be preferentially upregulated in the IL-18 KO compared with WT. However, we did identify two cytokines that were preferentially induced in the KO but not in WT under hypoxic conditions: IL-2 and SDF-1, the protein coded by CXCL12. Large-scale epidemiological studies indicate a detrimental role for SDF-1 in the context of HF. In the Framingham Heart Study cohort, plasma SDF-1 was positively associated with HF and all-cause mortality (32). Higher circulating SDF-1 in these patients was associated with a 20% increase in risk of new-onset HF after adjustment for clinical HF risk factors. Furthermore, increased plasma levels of SDF-1 have been observed in patients with idiopathic PH, and these levels significantly correlated with indexes of RV dysfunction (40). Preclinical experimental data support a detrimental role for SDF-1 in RV remodeling, since inhibition of the SDF-1/CXCR4 axis attenuates hypoxia-induced PH and subsequent RV hypertrophy in neonatal mice (42). The cardiotoxic effects of IL-2 have been recognized since the 1980s when IL-2 chemotherapy was observed to impart multiple cardiovascular maladaptive outcomes (26). Patients receiving recombinant IL-2 exhibited reductions in LV stroke work, an index of cardiac contractility, as well as severe capillary leak and increased incidence of myocardial infarction (23). Subsequent studies of the mechanism of IL-2-induced cardiotoxicity revealed atrial and ventricular arrhythmias as well as indirect inhibition of contractility in isolated cardiac tissue (31). Thus it is possible, based on the established maladaptive cardiac remodeling elicited by these two cytokines, that the upregulation of SDF-1 and IL-2 by HH in IL-18 KO mice may partially explain the lack of protection observed in KO HH animals. Analyses of the potential role of these cytokines and their differential expression in IL-18 KO and WT mice are beyond the scope of the current investigation but certainly warrant future investigation.
In conclusion, we show that genetic ablation of IL-18 does not protect mice against maladaptive RV remodeling following exposure to HH. This model of PH represents early adaptation in the RV to elevated pulmonary pressure. From a cardiac perspective, the animals exhibit clear evidence of RV hypertrophy despite a lack of upregulation of hypertrophic and collagen deposition mechanisms. Compensatory upregulation of SDF-1 and IL-2 in the KO animals in response to HH might explain the lack of an IL-18 effect in this model and warrants future investigation. Together, our data suggest that IL-18 blockade is not a holistic therapeutic strategy in this model.
GRANTS
This project is supported by National Heart, Lung, and Blood Institute Training Grant 2T32-HL-007822-16 (D. R. Bruns) and Program Project Grant P01-HL014985-36A1.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.R.B. and L.A.W. conception and design of research; D.R.B. performed experiments; D.R.B. and L.A.W. analyzed data; D.R.B. and L.A.W. interpreted results of experiments; D.R.B. prepared figures; D.R.B. drafted manuscript; P.M.B. and L.A.W. edited and revised manuscript; L.A.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Yanmei Du for technical assistance.
REFERENCES
- 1.Aguero J, Ishikawa K, Hadri L, Santos-Gallego C, Fish K, Hammoudi N, Chaanine A, Torquato S, Naim C, Ibanez B, Pereda D, Garcia-Alvarez A, Fuster V, Sengupta PP, Leopold JA, Hajjar RJ. Characterization of right ventricular remodeling and failure in a chronic pulmonary hypertension model. Am J Physiol Heart Circ Physiol 307: H1204–H1215, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barbera A, Giraud GD, Reller MD, Maylie J, Morton MJ, Thornburg KL. Right ventricular systolic pressure load alters myocyte maturation in fetal sheep. Am J Physiol Regul Integr Comp Physiol 279: R1157–R1164, 2000. [DOI] [PubMed] [Google Scholar]
- 3.Benczik M, Gaffen SL. The interleukin (IL)-2 family cytokines: survival and proliferation signaling pathways in T lymphocytes. Immunol Invest 33: 109–142, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Brown RD, Ambler SK, Li M, Sullivan TM, Henry LN, Crossno JT Jr, Long CS, Garrington TP, Stenmark KR. MAP kinase kinase kinase-2 (MEKK2) regulates hypertrophic remodeling of the right ventricle in hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 304: H269–H281, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cavasin MA, Demos-Davies K, Horn TR, Walker LA, Lemon DD, Birdsey N, Weiser-Evans MC, Harral J, Irwin DC, Anwar A, Yeager ME, Li M, Watson PA, Nemenoff RA, Buttrick PM, Stenmark KR, McKinsey TA. Selective class I histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ Res 110: 739–748, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chandrasekar B, Mummidi S, Claycomb WC, Mestril R, Nemer M. Interleukin-18 is a pro-hypertrophic cytokine that acts through a phosphatidylinositol 3-kinase-phosphoinositide-dependent kinase-1-Akt-GATA4 signaling pathway in cardiomyocytes. J Biol Chem 280: 4553–4567, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Chandrasekar B, Vemula K, Surabhi RM, Li-Weber M, Owen-Schaub LB, Jensen LE, Mummidi S. Activation of intrinsic and extrinsic proapoptotic signaling pathways in interleukin-18-mediated human cardiac endothelial cell death. J Biol Chem 279: 20221–20233, 2004. [DOI] [PubMed] [Google Scholar]
- 8.Colston JT, Boylston WH, Feldman MD, Jenkinson CP, de la Rosa SD, Barton A, Trevino RJ, Freeman GL, Chandrasekar B. Interleukin-18 knockout mice display maladaptive cardiac hypertrophy in response to pressure overload. Biochem Biophys Res Commun 354: 552–558, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delgado-Martin C, Escribano C, Pablos JL, Riol-Blanco L, Rodriguez-Fernandez JL. Chemokine CXCL12 uses CXCR4 and a signaling core formed by bifunctional Akt, extracellular signal-regulated kinase (ERK)1/2, and mammalian target of rapamycin complex 1 (mTORC1) proteins to control chemotaxis and survival simultaneously in mature dendritic cells. J Biol Chem 286: 37222–37236, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dinarello CA, Novick D, Puren AJ, Fantuzzi G, Shapiro L, Muhl H, Yoon DY, Reznikov LL, Kim SH, Rubinstein M. Overview of interleukin-18: more than an interferon-gamma inducing factor. J Leukocyte Biol 63: 658–664, 1998. [PubMed] [Google Scholar]
- 11.Dockstader K, Nunley K, Karimpour-Fard A, Medway A, Nelson P, Port JD, Liggett SB, Bristow MR, Sucharov CC. Temporal analysis of mRNA and miRNA expression in transgenic mice overexpressing Arg- and Gly389 polymorphic variants of the beta1-adrenergic receptor. Physiol Genomics 43: 1294–1306, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eichenholz PW, Eichacker PQ, Hoffman WD, Banks SM, Parrillo JE, Danner RL, Natanson C. Tumor necrosis factor challenges in canines: patterns of cardiovascular dysfunction. Am J Physiol Heart Circ Physiol 263: H668–H675, 1992. [DOI] [PubMed] [Google Scholar]
- 13.Fix C, Bingham K, Carver W. Effects of interleukin-18 on cardiac fibroblast function and gene expression. Cytokine 53: 19–28, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gulick T, Chung MK, Pieper SJ, Lange LG, Schreiner GF. Interleukin 1 and tumor necrosis factor inhibit cardiac myocyte beta-adrenergic responsiveness. Proc Natl Acad Sci USA 86: 6753–6757, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoshino T, Kato S, Oka N, Imaoka H, Kinoshita T, Takei S, Kitasato Y, Kawayama T, Imaizumi T, Yamada K, Young HA, Aizawa H. Pulmonary inflammation and emphysema: role of the cytokines IL-18 and IL-13. Am J Respir Crit Care Med 176: 49–62, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Isobe M, Yazaki Y, Takaku F, Koizumi K, Hara K, Tsuneyoshi H, Yamaguchi T, Machii K. Prediction of pulmonary arterial pressure in adults by pulsed Doppler echocardiography. Am J Cardiol 57: 316–321, 1986. [DOI] [PubMed] [Google Scholar]
- 17.Lemler MS, Bies RD, Frid MG, Sastravaha A, Zisman LS, Bohlmeyer T, Gerdes AM, Reeves JT, Stenmark KR. Myocyte cytoskeletal disorganization and right heart failure in hypoxia-induced neonatal pulmonary hypertension. Am J Physiol Heart Circ Physiol 279: H1365–H1376, 2000. [DOI] [PubMed] [Google Scholar]
- 18.Li YY, McTiernan CF, Feldman AM. Interplay of matrix metalloproteinases, tissue inhibitors of metalloproteinases and their regulators in cardiac matrix remodeling. Cardiovasc Res 46: 214–224, 2000. [DOI] [PubMed] [Google Scholar]
- 19.MacNee W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Part one. Am J Respir Crit Care Med 150: 833–852, 1994. [DOI] [PubMed] [Google Scholar]
- 20.Mallat Z, Heymes C, Corbaz A, Logeart D, Alouani S, Cohen-Solal A, Seidler T, Hasenfuss G, Chvatchko Y, Shah AM, Tedgui A. Evidence for altered interleukin 18 (IL)-18 pathway in human heart failure. FASEB J 18: 1752–1754, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res 91: 988–998, 2002. [DOI] [PubMed] [Google Scholar]
- 22.Mentink-Kane MM, Cheever AW, Wilson MS, Madala SK, Beers LM, Ramalingam TR, Wynn TA. Accelerated and progressive and lethal liver fibrosis in mice that lack interleukin (IL)-10, IL-12p40, and IL-13Ralpha2. Gastroenterology 141: 2200–2209, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nora R, Abrams JS, Tait NS, Hiponia DJ, Silverman HJ. Myocardial toxic effects during recombinant interleukin-2 therapy. J Natl Cancer Inst 81: 59–63, 1989. [DOI] [PubMed] [Google Scholar]
- 24.Novick D, Kim S, Kaplanski G, Dinarello CA. Interleukin-18, more than a Th1 cytokine. Semin Immunol 25: 439–448, 2013. [DOI] [PubMed] [Google Scholar]
- 25.Nozik-Grayck E, Woods C, Taylor JM, Benninger RK, Johnson RD, Villegas LR, Stenmark KR, Harrison DG, Majka SM, Irwin D, Farrow KN. Selective depletion of vascular EC-SOD augments chronic hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 307: L868–L876, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ognibene FP, Rosenberg SA, Lotze M, Skibber J, Parker MM, Shelhamer JH, Parrillo JE. Interleukin-2 administration causes reversible hemodynamic changes and left ventricular dysfunction similar to those seen in septic shock. Chest 94: 750–754, 1988. [DOI] [PubMed] [Google Scholar]
- 27.Platis A, Yu Q, Moore D, Khojeini E, Tsau P, Larson D. The effect of daily administration of IL-18 on cardiac structure and function. Perfusion 23: 237–242, 2008. [DOI] [PubMed] [Google Scholar]
- 28.Ryan JJ, Thenappan T, Luo N, Ha T, Patel AR, Rich S, Archer SL. The WHO classification of pulmonary hypertension: A case-based imaging compendium. Pulmon Circ 2: 107–121, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stamm C, Friehs I, Cowan DB, Moran AM, Cao-Danh H, Duebener LF, del Nido PJ, McGowan FX Jr. Inhibition of tumor necrosis factor-alpha improves postischemic recovery of hypertrophied hearts. Circulation 104: I350–355, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol 297: L1013–L1032, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Sterin-Borda L, Perez Leiros C, Borda ES, de Bracco MM. Effects of IL-2 on the myocardium. Participation of the sympathetic system. J Mol Cell Cardiol 28: 2457–2465, 1996. [DOI] [PubMed] [Google Scholar]
- 32.Subramanian S, Liu C, Aviv A, Ho JE, Courchesne P, Muntendam P, Larson MG, Cheng S, Wang TJ, Mehta NN, Levy D. Stromal cell-derived factor 1 as a biomarker of heart failure and mortality risk. Arteriosclerosis Thromb Vasc Biol 34: 2100–2105, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takeda K, Tsutsui H, Yoshimoto T, Adachi O, Yoshida N, Kishimoto T, Okamura H, Nakanishi K, Akira S. Defective NK cell activity and Th1 response in IL-18-deficient mice. Immunity 8: 383–390, 1998. [DOI] [PubMed] [Google Scholar]
- 34.Toldo S, Mezzaroma E, O'Brien L, Marchetti C, Seropian IM, Voelkel NF, Van Tassell BW, Dinarello CA, Abbate A. Interleukin-18 mediates interleukin-1-induced cardiac dysfunction. Am J Physiol Heart Circ Physiol 306: H1025–H1031, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venkatachalam K, Prabhu SD, Reddy VS, Boylston WH, Valente AJ, Chandrasekar B. Neutralization of interleukin-18 ameliorates ischemia/reperfusion-induced myocardial injury. J Biol Chem 284: 7853–7865, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walker LA, Buttrick PM. The right ventricle: biologic insights and response to disease: updated. Curr Cardiol Rev 9: 73–81, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walker LA, Walker JS, Ambler SK, Buttrick PM. Stage-specific changes in myofilament protein phosphorylation following myocardial infarction in mice. J Mol Cell Cardiol 48: 1180–1186, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walker LA, Walker JS, Glazier A, Brown DR, Stenmark KR, Buttrick PM. Biochemical and myofilament responses of the right ventricle to severe pulmonary hypertension. Am J Physiol Heart Circ Physiol 301: H832–H840, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Woldbaek PR, Sande JB, Stromme TA, Lunde PK, Djurovic S, Lyberg T, Christensen G, Tonnessen T. Daily administration of interleukin-18 causes myocardial dysfunction in healthy mice. Am J Physiol Heart Circ Physiol 289: H708–H714, 2005. [DOI] [PubMed] [Google Scholar]
- 40.Yang T, Li ZN, Chen G, Gu Q, Ni XH, Zhao ZH, Ye J, Meng XM, Liu ZH, Xiong CM, He JG. Increased levels of plasma CXC-Chemokine Ligand 10, 12 and 16 are associated with right ventricular function in patients with idiopathic pulmonary arterial hypertension. Heart Lung 43: 322–327, 2014. [DOI] [PubMed] [Google Scholar]
- 41.Yao C, Li P, Song H, Song F, Qu Y, Ma X, Shi R, Wu J. CXCL12/CXCR4 axis upregulates twist to induce EMT in human glioblastoma. Mol Neurobiol In press. [DOI] [PubMed] [Google Scholar]
- 42.Young KC, Torres E, Hatzistergos KE, Hehre D, Suguihara C, Hare JM. Inhibition of the SDF-1/CXCR4 axis attenuates neonatal hypoxia-induced pulmonary hypertension. Circ Res 104: 1293–1301, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu Q, Vazquez R, Khojeini EV, Patel C, Venkataramani R, Larson DF. IL-18 induction of osteopontin mediates cardiac fibrosis and diastolic dysfunction in mice. Am J Physiol Heart Circ Physiol 297: H76–H85, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]