

Abstract
Accumulation of oxidized proteins, and especially β-amyloid (Aβ), is thought to be one of the common causes of Alzheimer's disease (AD). The current studies determine the effect of an in vivo methionine sulfoxidation of Aβ through ablation of the methionine sulfoxide reductase A (MsrA) in a mouse model of AD, a mouse that overexpresses amyloid precursor protein (APP) and Aβ in neurons. Lack of MsrA fosters the formation of methionine sulfoxide in proteins, and thus its ablation in the AD-mouse model will increase the formation of methionine sulfoxide in Aβ. Indeed, the novel MsrA-deficient APP mice (APP+/MsrAKO) exhibited higher levels of soluble Aβ in brain compared with APP+ mice. Furthermore, mitochondrial respiration and the activity of cytochrome c oxidase were compromised in the APP+/MsrAKO compared with control mice. These results suggest that lower MsrA activity modifies Aβ solubility properties and causes mitochondrial dysfunction, and augmenting its activity may be beneficial in delaying AD progression.
Keywords: posttranslational modification, Alzheimer's disease, mitochondria, oxidative stress
oxidative stress occurs in biological systems when generation of reactive oxygen species and reactive nitrogen species exceeds the system's capacity to eliminate these species. Oxidative stress is a major deleterious mechanism in Alzheimer's disease (AD) (8, 35, 42), other neurodegenerative diseases (11), and normal aging (43). In AD, oxidative damage markers, including lipid peroxidation and nitration, nucleic acid oxidation, and protein carbonylation, are increased in vulnerable brain areas relative to age-matched healthy individuals (26).
AD is characterized pathologically by extracellular amyloid plaques comprised predominantly of fibrillar β-amyloid (Aβ) peptide and intracellular neurofibrillary tangles made of hyperphosphorylated tau (38). Amyloid plaques are surrounded by inflammation, including activated microglia and astrocytes, which contribute to creation and maintenance of oxidative stress (13). Although historically amyloid plaques were thought to cause AD (36), current evidence indicates that the pathological process leading to AD begins with synaptic injury by neurotoxic Aβ oligomers, whereas formation of plaques and tangles are downstream events (36). Oxidative stress is one of the earliest consequences of toxic insults mediated by soluble Aβ oligomers (27). Mitochondria are particularly sensitive to oxidative stress, and reduced metabolic activity resulting from oxidative damage to vital mitochondrial components has been demonstrated in AD (14). Consequently, antioxidant therapy has been associated with reduced risk for AD (7, 35).
Methionine (Met) is highly susceptible to oxidation in vivo, particularly under conditions of oxidative stress. For example, the sulfoxide form has been found to comprise 10–50% of Aβ in amyloid plaques of AD brain (2, 6, 16, 25), although it is difficult to determine whether its existence in the plaques contributes to AD etiology or results from the highly oxidative environment around the amyloid plaques, where Aβ may be trapped for long periods. Oxidation of Met to Met(O) is reversible, and the reverse reaction is catalyzed in vivo by the methionine sulfoxide reductase (Msr) system, comprising peptide-methionine (S)-S-oxide reductase (MsrA) and peptide-methionine (R)-S-oxide reductase (MsrB), which reduce the S and R enantiomers, respectively, of the sulfoxide group. Thus, these enzymes provide both an efficient repair mechanism for oxidative damage to Met residues and general protection against oxidative stress by scavenging reactive oxygen species through the recycling of Met (28).
Mammalian MsrA is encoded by a single gene (19) and is found in both the cytosol and mitochondria (12). Studies in Msr-knockout (MsrAKO) mice have shown increased vulnerability to oxidative stress (21) and oxidative pathology similar to that associated with AD (32) and Parkinson's disease (PD) (29). Conversely, overexpression of MsrA in various organisms has been shown to provide enhanced protection against oxidative stress and extend survival rate (5, 18, 20).
A cell culture study relevant to AD showed elevated MsrA activity and mRNA levels in human neuroblastoma (IMR-32) cells in response to treatment with the sulfoxide form of Aβ42, suggesting that the cells sensed the presence of Met(O) in Aβ and upregulated MsrA to provide enhanced cellular protection (17). Recently, we reported similar findings in primary rat hippocampal and cortical neurons, in which we observed increased total Msr activity ascribed to both MsrA and MsrB in correlation with protection from cell death induced by the sulfoxide forms of Aβ40 or Aβ42 (22). Furthermore, exposure of wild-type (WT) and MsrAKO mouse cortical neurons to Aβ42 and Met(O)-Aβ showed that lack of MsrA abolishes the protective effect induced by Met(O)-Aβ (22). A different study demonstrated that overexpression of MsrA in differentiated rat pheochromocytoma (PC-12) cells increased significantly the resistance of the cells to Aβ42-induced toxicity (15). These cell culture studies suggest that neurons protect themselves against oxidative stress in AD by elevated expression of the Msr system.
Studies using WT rats have shown age-related decline in Msr activity unrelated to disease (33, 40, 44), which correlated with a decrease in the mRNA levels of MsrA (10). Overall, the combination of human and animal data suggests that the low Msr levels observed in AD brain (10) result both from aging-related decreased transcription and disease-related translational or posttranslational defects. The decline in Msr activity would be expected to lead to gradual accumulation of protein-Met(O). Thus, it is proposed that both compromised Msr activity and the resulting accumulation of protein-Met(O) [including specifically Met(O)-Aβ] will increase the levels of soluble-Aβ while compromising mitochondrial function. Accordingly, to facilitate obtaining relevant data to support our hypothesis, we have created a novel mouse model in which the MsrA gene is deleted in an AD mouse model that overexpresses amyloid precursor protein (APP) and Aβ in neurons.
MATERIALS AND METHODS
Creation of APP+/MsrAKO mice and validation of their high Aβ levels.
The transgenic mice overexpressing an isoform of human Alzheimer's Aβ precursor protein (APP) exhibit AD-like phenotypes at mature to older ages. These mice produce high levels of Aβ in the brain following cleavage of the overexpressed human APP protein. The APP transgenic mice we used for our studies are Tg (PDGFB-APPSwInd, or “J20”) mice that express a mutant form of human APP bearing both the Swedish (K670N/M671L) and the Indiana (V717F) mutations (APPSwInd) and were purchased from Jackson's Laboratory (Sacramento, CA) and will be denoted as APP+ mice. This AD mouse model is well characterized in terms of mitochondrial function, oxidative stress, Aβ accumulation, and synaptic function (23). Crossing these APP+ mice with MsrAKO mice will result in a hybrid mouse model that overexpresses APP and lacks the MsrA gene. Briefly, female MsrAKO mice on a C57BL6/129sv background (21) were back-crossed 10 times into a C57BL6 background and mated with males of PDGFB-APPSwInd on a C57BL6 background (APP+ mice). Double-heterozygous F1s were selected by PCR typing for the human APP gene and intercrossed with MsrA+/− from F1 to produce F2 progenies that included mice that are homozygous for the MsrA−/− allele in combination with heterozygous APP+ transgenic gene (MsrAKO/PDGFB-APPSwInd, named shortly as “APP+/MsrAKO mice”). Littermates that were MsrA+/+/PDGFB-APPSwInd served as the APP+ control mice for the APP+/MsrAKO cohorts. Littermates that were non-APP+ transgenic MsrA+/+ mice served as the WT control for the non-APP+/MsrAKO cohorts (MsrA−/− mice). All mouse-related experimental protocols were submitted to and approved by the Animal Care Unit Committee of the University of Kansas.
Brain tissue processing.
The following indicated brain extractions were performed based on a previously published method (39). Briefly, AD-relevant brain regions (a mixture of hippocampus and cortex) were dissected out of one hemisphere of all mouse brains at euthanasia (n = 5/mouse strain, 7 mo of age). Ice-cold Tris-buffered saline (TBS) consisting of 20 mM Tris·HCl and 150 mM NaCl, pH 7.4, was added to the frozen regions (4:1 vol/wt) in the presence of protease inhibitors mixture (Roche and Life Technologies) and homogenized with a mechanical Dounce homogenizer. The homogenate was spun down at 157,000 g for 15 min at 4°C, and the supernates were aliquoted and stored at −80°C (denoted as the “Tris”-soluble fractions). The pellets were rehomogenized (4:1 vol/wt) in TBS + 1% Triton X-100 and spun as described above. The resultant supernates (denoted as “Triton”-soluble fractions) were aliquoted and stored at −80°C, and the pellet was rehomogenized in TBS + 5 M guanidine HCl. Following the same centrifugation indicated above, the resultant supernates (denoted as “guanidine”-soluble fractions) were aliquoted and stored as well at −80°C until use. The Tris-soluble fraction contains mainly a mixture of both monomeric and oligomeric Aβ that is representative of the most soluble species of Aβ. The quinidine soluble fraction contains mainly aggregative Aβ species that are representative of the least soluble species of Aβ. The Triton soluble fraction represents intermediate fraction between the Tris and quinidine soluble fractions that contains mainly a mixture of both oligomeric and aggregative Aβ species. Having these fractions will facilitate the search for possible correlation between types of Aβ and their toxicity in brain.
Determination of Aβ levels.
First, to determine that indeed total Aβ levels were similar in both APP+ and APP+/MsrAKO brains and higher compared with the non-APP+ carrier brains (WT and MsrAKO), a dot blot analyses were performed. Equal protein amounts of the evenly combined three soluble fractions (Tris, Triton, and guanidine) from each brain sample were applied to a dot blot analyses (in a range of 1–10 μg protein·dot−1·strain−1). The MOAB-2 antibodies were used as the primary antibody, followed by the secondary antibody horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Santa Cruz Biotechnology, Dallas, TX). The MOAB-2 antibodies recognize human as well as mouse Aβ, unaggregated, oligomeric, and fibrilar forms of synthetic Aβ42 and unaggregated Aβ40, without recognizing the APP protein (Ref. 45 and personal communication from Dr. Mary Jo LaDu, University of Illinois). The densities of the resulting signals, following exposure of the blot to an X-ray film, were quantified by the National Institutes of Health ImageJ program.
Detection and measurement of Met(O)-Aβ distribution in brain.
We used our anti-Met(O) antibodies to detect Met(O)-Aβ in the hippocampal and cortex, as it was shown to detect Met(O) in other brain proteins associated with neurodegenerative diseases (3, 30, 31, 41). For this analysis, we have developed a novel sandwich ELISA for the detection of sulfoxidized Aβ [Aβ-Met(O)]. The assay worked in the following format: extracted or synthetic Aβ was captured by an anti-Aβ antibody (MOAB-2, which is bound to the ELISA 96-well plate), and the rabbit anti-Met(O) antibody is used for the detection of Met(O) of Aβ. Then, fluorescence visualization is mediated by HRP-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology). For calibrating the ELISA, synthetic Aβ (1–40/42) and Aβ-Met(O) (1–40/42) (a gift from Dr. Gal Bitan, University of California Los Angeles) were used in various concentrations in this ELISA format. The Tris, Triton, and guanidine-soluble fractions of the four mouse strains were loaded into the 96-well plate, and the ELISA assay was performed according to the format described above. Briefly, MOAB-2 antibodies (1:1,000) were added into coating buffer [50 mM carbonate-bicarbonate (pH 9.6)] at ∼1.0 μg/ml, and then they were added to a Nunc-Immuno 96-well plate (Maxisorp Surface Treatment; Thermo-Fisher Scientific, Waltham, MA). The plate was incubated overnight at 4°C and washed with 0.05% (vol/vol) Tween-20 in PBS (PBST). Blocking buffer [0.5% (wt/vol) BSA in PBS] was added to each well and incubated for 1 h at room temperature. Then, the wells were washed with PBST. Equal amounts of proteins from each brain fraction were added to the plate in various dilutions (from 1:20 to 1:100 to eliminate the effect of the Triton-guanidine) and were incubated for overnight at 4°C. The plate was washed with PBST, and anti-Met(O) antibodies were added in PBST (1:1,000) and incubated at room temperature for 2 h. Following washes with PBST, HRP goat anti-rabbit IgG (Santa Cruz Biotechnology) was added to each well (1:5,000) and incubated at room temperature for 2 h. Finally, the plate was washed with PBST, and a mixture of peroxidase substrate solutions was added to each well (Substrate Reagent Pack; R & D Systems-Tocris, Minneapolis, MN). Then, a stopper solution (1 N sulfuric acid) was added, and the plate was read at 450 nm.
Mitochondrial respiration and cytochrome c oxidase expression and activity.
Mouse brain regions (a mixture of hippocampus and cortex) were dissected out of one hemisphere of all mouse brains at euthanasia (n = 5/mouse strain). These brain regions were used as a source for the isolated mitochondrial preparations. Oxygen consumption of each preparation was determined by following mitochondrial respiration assay, using the OROBOROS Oxygraph-2K (OROBOROS Instruments, Innsbruck, Austria), and as described previously (4). The tissue homogenates or isolated mitochondria were resuspended in KCl medium (80 mM KCl, 10 mM Tris·HCl, 3 mM MgCl2, 1 mM EDTA, and 5 mM potassium phosphate, pH 7.4). Various substrates and inhibitors for mitochondrial respiratory chain complexes were used as described in Figs. 3 and 4. OROBOROS DatLab software was used to calculate the oxygen consumption. The cytochrome c oxidase activities of mitochondrial fractions were measured with a cytochrome c oxidase kit (Sigma-Aldrich, St. Louis, MO). Briefly, mitochondrial fraction and enzyme solution were added to the assay buffer. The reaction was initiated by the addition of ferrocytochrome c substrate solution. Enzyme kinetics was determined by following changes in OD values at 550 nm at 10-s intervals (using a Synergy H1 monochromator-microplate reader; Biotek, Winooski, VT). The expression levels of cytochrome c oxidase and heat shock protein 60 (HSP60; mitochondrial marker) were determined by a Western blot analysis performed on mitochondrial protein fractions made from the four mouse types (primary antibody: anti cytochrome c oxidase subunit VIb and anti-HSP60 antibodies; Abcam, Cambridge, MA).
Fig. 3.
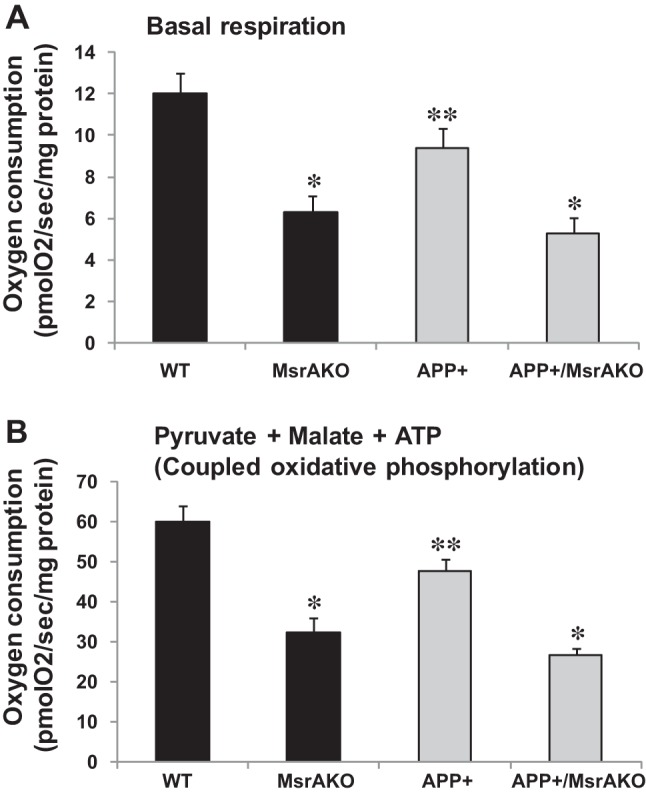
Mitochondrial respiratory chain activity is impaired in freshly isolated mitochondria in brains of WT, MsrAKO, APP+, and APP+/MsrAKO mice (7 mo of age). Basal respiration (A) and pyruvate + malate + ADP (coupled oxidative phosphorylation) (B). The respiratory values were measured using OROBOROS Oxygraph-2K as described, in materials and methods. Values are means ± SD; n = 5. *P < 0.001 vs. APP+ and WT groups in A and B; **P < 0.05 vs. WT (1-way ANOVA with Tukey's post hoc comparison).
Fig. 4.
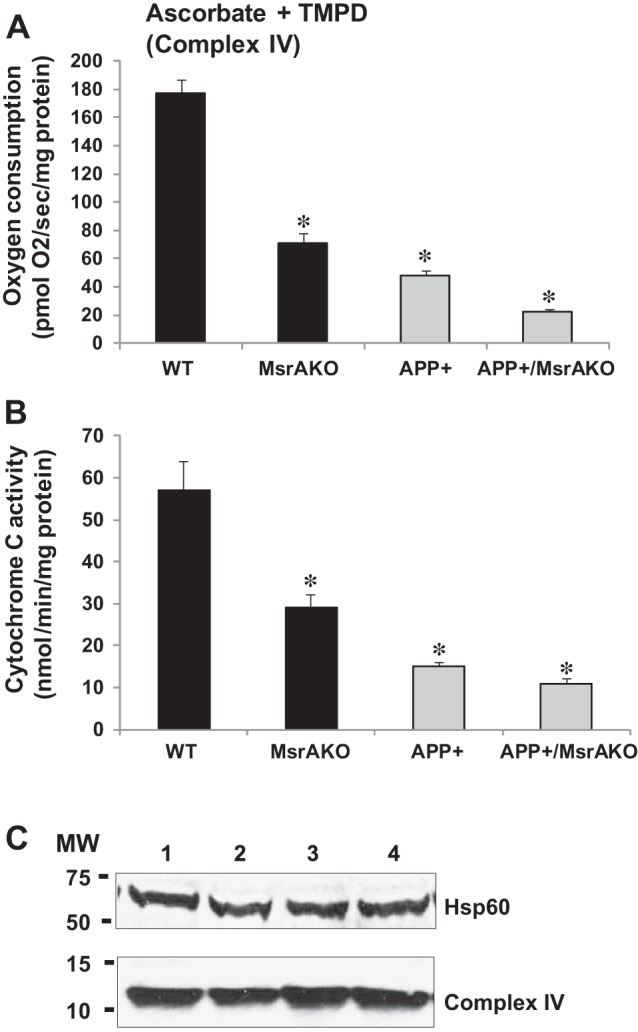
Cytochrome c oxidase (complex IV) mitochondrial function and activity decline in mitochondria of MsrAKO, APP+, and APP+/MsrAKO brains (7 mo of age). A: complex IV mediated oxygen consumption in mitochondria, which was measured using OROBOROS Oxygraph-2K, as described in materials and methods. Complex IV respiration was calculated as the portion sensitive to potassium cyanide (KCN), a specific inhibitor of cytochrome c oxidase (first, the ascorbate-TMPD respiratory rates in the presence and absence of KCN were obtained, and then the final respiratory rate was obtained by subtracting the KCN-insensitive respiration). B: cytochrome c oxidase activities of mitochondrial fractions were measured with a cytochrome c oxidase kit (Sigma). Briefly, mitochondrial fraction and enzyme solution were added to the assay buffer. The reaction was initiated by the addition of ferrocytochrome c substrate solution. Enzyme kinetics was determined by following changes in optical density values at 550 nm at 10-s intervals (using a Synergy H1 monochromator-microplate reader; Biotek). C: protein expression levels of cytochrome c oxidase and heat shock protein 60 (HSP60; loading control as a mitochondrial marker) were determined by Western blot analyses (a represented blot is shown, in which each lane contains a pool of equal mitochondrial protein extracts originating from 5 animals/mouse group). Values are means ± SD; n = 5. *P < 0.001 vs. WT (1-way ANOVA). MW, molecular weight markers.
Statistical analyses.
All data values in the presented figures are displayed as means ± SD. All analyses were run by two-way analysis of variance (ANOVA), followed by Newman-Keuls test for post hoc comparisons.
RESULTS
We have created the APP+/MsrAKO mice as described in materials and methods. It is worth noting that the rate of success of having APP+/MsrAKO pups was below the expectations. Only 1% of all resulting littermates contained the desired genotype (we have obtained only 5 out of 500 resulting pups from the F2 progenies, as described in materials and methods). This phenomenon may indicate that complete absence of MsrA causes enhanced toxicity of Aβ, which negatively affects the survival of the newborn pups. The obtained APP+/MsrAKO pups were grown until they reached ≥7 mo of age before any analyses was performed, since the APP+ mice start to exhibit AD-associated phenotypes only at that age. Accordingly, we have first determined the levels of soluble Aβ in the four mouse strains. As predicted, a significantly high increase in Aβ levels was observed in the brain fractions of the APP+ carriers compared with the non-APP+ carrier brains (Fig. 1). Additionally, the levels of total Aβ were compatible in both the APP+ carrier and the non-APP+ carrier, suggesting that ablation of MsrA in either WT or APP+ mice did not have an effect on the expression of Aβ levels. Then, using our novel ELISA assay, we determined the relative ratio of Met(O) levels in the Tris, Triton, and guanidine-soluble fractions containing mainly the monomeric and oligomeric Aβ, oligomeric and aggregative Aβ, and aggregative Aβ species, respectively. The added signal values obtained from the respective three fractions of each mouse represented its 100% level of Aβ-Met(O). Accordingly, the distribution of Aβ-Met(O) levels in all of the fractions in each mouse strain is provided as percent of total Aβ-Met(O). The ability of the anti-Met(O) antibody to specifically recognize Aβ-Met(O) was further confirmed by dot blot and our ELISA analyses using synthetic Aβ and Aβ-Met(O). As shown in Fig. 2, the pattern of Aβ-Met(O) distribution exhibited in MsrA carriers (WT and APP+ mouse strains) is similar to and different from the Aβ-Met(O) distribution exhibited in the MsrA−/− carriers (MsrAKO and APP+/MsrAKO mouse strains). Compared with the mouse strains having an intact MsrA gene, the lack of the MsrA gene caused a reduction in the levels of the Aβ-Met(O) in the guanidine- and Triton-soluble fractions and an increase in the Aβ-Met(O) levels in the Tris-soluble fraction (Fig. 2). This increased presence of Aβ-Met(O) in the Tris fraction of the MsrAKO and APP+/MsrAKO brain extracts supports the hypothesis that there is a shift from aggregates to the soluble oligomers of MetO-Aβ in mice lacking MsrA. These soluble oligomers are thought to possess more toxic elements to neurons and synapses than aggregative Aβ forms alone (37, 39). Furthermore, since MsrA is also transported into mitochondria (12), we examined the effect of MsrA ablation on mitochondrial function in the presence or absence of Aβ overexpression. Oxygen consumption of each preparation was determined by following mitochondrial respiration assay, using the OROBOROS Oxygraph-2K. In addition, various substrates and inhibitors for mitochondrial respiratory chain complexes were used as described in Figs. 3 and 4. Basal respiration was significantly reduced in the MsrA KO mice and APP+/MsrAKO mice compared with WT control and APP+ control, respectively. The mouse values pattern presented in Fig. 3A was reproduced in the coupled oxidative phosphorylation experiments, as shown in Fig. 3B, with the expected higher oxygen consumption values. Ascorbate and N,N,N′,N′-tetramethyl-p-phenylenediamine (TMPD) were added to the respiratory mixture to determine the capacity of cytochrome c oxidase (complex IV). TMPD is an artificial redox mediator that assists the transfer of electrons from ascorbate to cytochrome c. Complex IV respiration was calculated as the portion sensitive to potassium cyanide (KCN), a specific inhibitor of cytochrome c oxidase (first, the ascorbate-TMPD respiratory rates in the presence and absence of KCN were obtained, and then the final respiratory rate was obtained by subtracting the KCN-insensitive respiration). Respiration rates with ascorbate plus TMPD were decreased by ∼60% in the MsrAKO compared with WT mice, and a similar reduction was observed in the APP+/MsrAKO compared with APP+ mice (Fig. 4A). These data show that the lack of MsrA caused a similar negative impact on mitochondrial respiration (mediated by compromising complex IV function) of both WT and APP+ mice. Furthermore, under inhibitory conditions of complex IV by KCN, the APP+/MsrAKO mice were the most affected. These data strongly suggest that the mitochondrial MsrA activity contributes for maintaining complex IV function, which may be of most importance especially when the mitochondrial function of complex IV in the APP+ mice is already inhibited (Fig. 4A). Thus, it is suggested that the mitochondrial MsrA's ability to reduce Met(O) residues of either complex IV or other related proteins may contribute to the complex IV-mediated respiration. Indeed, monitoring the enzymatic activity of cytochrome c oxidase in these mice showed that the activity paralleled complex IV-dependent respiration changes (Fig. 4, A vs. B). In addition, no significant changes in the expression levels of either cytochrome c oxidase HSP60 (a marker of mitochondria) were observed as determined by a Western blot analyses performed on mitochondrial protein fractions made from the four mouse types (Fig. 4C). Accordingly, it is suggested that the observed declined complex IV respiration (Fig. 4A) is associated with Met oxidation of cytochrome c oxidase and/or other related mitochondrial proteins to which the APP+/MsrAKO mice are mostly vulnerable.
Fig. 1.
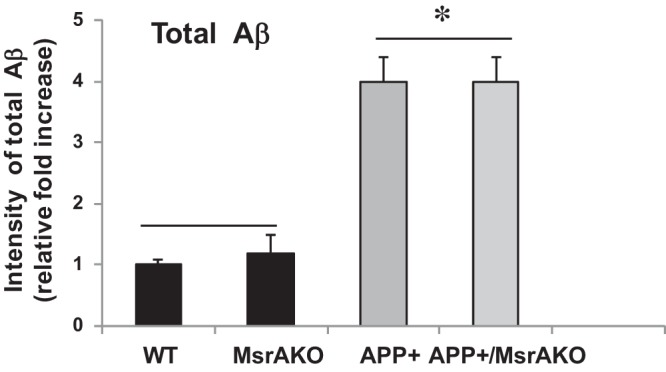
Levels of total β-amyloid (Aβ) in brains of wild-type (WT), peptide-methionine (S)-S-oxide reductase (MsrAKO), amyloid precursor protein (APP)+, and APP+/MsrAKO mice. Mouse brains (hippocampal and cortical regions) were homogenized and processed as described in materials and methods. Equal protein amounts of the evenly combined 3 soluble fractions (Tris, Triton, and guanidine) from each brain sample were applied to a dot blot analyses (in a range of 1–10 μg protein·dot−1·strain−1). The MOAB-2 antibodies were used as the primary antibody, followed by secondary antibody horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG antibodies. The densities of the resulting signals, following exposure of the blot to an X-ray film, were quantified by the NIH ImageJ program. MsrA−/− is denoted as MsrAKO. *Statistically significant differences between the APP+ and APP+/MsrAKO group and the WT and MsrAKO group (P < 0.001).
Fig. 2.

Levels of Aβ in brains of WT, MsrAKO, APP+, and APP+/MsrAKO mice (7 mo of age). Mouse brains (hippocampal and cortical regions) were homogenized and processed as described in materials and methods, resulting in Tris-, Triton-, and guanidine-soluble fractions. A sandwich ELISA for the detection of sulfoxidized Aβ [Aβ-Met(O)] was performed using anti-Aβ antibody (MOAB-2) as the capture antibody, rabbit anti-Met(O) antibody for the detection of Met(O) of Aβ, and the HRP-conjugated goat anti-rabbit IgG as the signaling probe. MsrA−/− is denoted as MsrAKO. Statistically significant differences: for WT, *P < 0.001 between guanidine and Tris and Triton fractions and **P < 0.02 between Tris and Triton fractions; for APP+, *P < 0.001 between guanidine and Tris and Triton fractions; for MsrAKO, *P < 0.001 between Triton and Tris and Guanidine fractions and **P < 0.04 between Tris and guanidine fractions; for APP+/MsrAKO, *P < 0.001 between Triton and Tris and guanidine fractions and **P < 0.01 between Tris and guanidine fractions.
DISCUSSION
Modification of proteins by methionine oxidation can change their biochemical function and/or structure. Accordingly, the oxidation of the sole methionine residue of Aβ causes it to lose its fibrillation, and overexpression of MsrA leads to a protective effect against neuronal cell death in culture in the presence of either Aβ or Aβ-Met(O) (15, 17, 22). In the current studies, we show that lack of MsrA promotes a shift of aggregated forms of Aβ toward soluble oligomers in vivo (Fig. 2). Since soluble oligomers are thought to possess more toxic elements to neurons and synapses than aggregative Aβ forms alone (37, 39), it is suggested that enhancing MsrA activity by compounds that can upregulate its transcription may have therapeutic application. For example, we have showed that pergolide, pergolide sulfoxide, and S-adenosyl methionine can upregulate MsrA transcription and activity in cultured neuronal cell (9). Alternatively, immunization of the APP+ type of mice with methionine sulfoxide-rich protein caused a reduction in amyloid plaque burden in the hippocampus, presumably through clearance of protein-Met(O) [including Aβ-Met(O)] from brain (22). The role of MsrA in cellular protection against oxidative stress is also evident from its presence in mitochondria, although the regulatory process of its transport into the mitochondria is not clear yet. Evidence for the link between mitochondrial dysfunction and AD has been accumulated in various studies on the role of mitochondria in normal aging and neurodegenerative diseases. For example, reduced cytochrome c oxidase activities have been reported in both platelets and brains of mild cognitive impermanent and AD patients (1, 24), and this activity reduction was attributed to an enhanced mitochondrial oxidative stress occurring especially at an advanced age. As shown in Figs. 3 and 4, lack of MsrA exacerbates mitochondrial malfunction that is observed in the APP+ mice. Thus, this situation may mimic the reduction of MsrA expression with age (33, 40, 44), which is suggested to be a risk factor for AD-related mitochondrial malfunction when Aβ levels are elevated. Further studies are needed to elucidate the interaction between Aβ-Met(O), mitochondrial function, and neuronal cell death.
GRANTS
This research was supported by the Hedwig Miller Fund for Aging Research, by the Institutional Development Award (IDeA) from the National Institute of General Medical Sciences (P20 GM-103418) of the National Institutes of Health, and by KU ADC P30-AG-035982 and the Landon Center on Aging provided to J. Moskovitz; and by the National Institute on Aging (R37AG037319) of the National Institutes of Health to S. S. Yan.
DISCLOSURES
All authors declare that they do not have any potential conflicts of interest, financial or otherwise.
AUTHOR CONTRIBUTIONS
J.M., F.D., and S.Y. conception and design of research; J.M., F.D., and C.F.B. performed experiments; J.M., F.D., C.F.B., and S.Y. analyzed data; J.M., F.D., and S.Y. interpreted results of experiments; J.M. prepared figures; J.M. drafted manuscript; J.M. and S.Y. edited and revised manuscript; J.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Rick T. Dobrowsky for consulting on mitochondrial function.
REFERENCES
- 1.Bosetti F, Brizzi F, Barogi S, Mancuso M, Siciliano G, Tendi EA, Murri L, Rapoport SI, Solaini G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer's disease. Neurobiol Aging 23: 371–376, 2002. [DOI] [PubMed] [Google Scholar]
- 2.Boutte AM, Woltjer RL, Zimmerman LJ, Stamer SL, Montine KS, Manno MV, Cimino PJ, Liebler DC, Montine TJ. Selectively increased oxidative modifications mapped to detergent-insoluble forms of Aβ and β-III tubulin in Alzheimer's disease. FASEB J 20: 1473–1483, 2006. [DOI] [PubMed] [Google Scholar]
- 3.Canello T, Frid K, Gabizon R, Lisa S, Friedler A, Moskovitz J, Gasset M, Gabizon R. Oxidation of Helix-3 methionines precedes the formation of PK resistant PrP. PLoS Pathog 6: e1000977, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chowdhury SK, Zherebitskaya E, Smith DR, Akude E, Chattopadhyay S, Jolivalt CG, Calcutt NA, Fernyhough P. Mitochondrial respiratory chain dysfunction in dorsal root ganglia of streptozotocin-induced diabetic rats and its correction by insulin treatment. Diabetes 59: 1082–1091, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung H, Kim AK, Jung SA, Kim SW, Yu K, Lee JH. The Drosophila homolog of methionine sulfoxide reductase A extends lifespan and increases nuclear localization of FOXO. FEBS Lett 584: 3609–36014, 2010. [DOI] [PubMed] [Google Scholar]
- 6.Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA, Perry G, Carey PR. Metal binding and oxidation of amyloid-beta within isolated senile plaque cores: Raman microscopic evidence. Biochemistry 42: 2768–2773, 2003. [DOI] [PubMed] [Google Scholar]
- 7.Dumont M, Lin MT, Beal MF. Mitochondria and antioxidant targeted therapeutic strategies for Alzheimer's disease. J Alzheimer's Dis 20: S633–S643, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Floyd RA, Hensley K. Oxidative stress in brain aging. Implications for therapeutics of neurodegenerative diseases. Neurobiol Aging 23: 795–807, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Franklin JM, Carrasco GA, Moskovitz J. Induction of methionine sulfoxide reductase activity by pergolide, pergolide sulfoxide, and S-adenosyl-methionine in neuronal cells. Neurosci Lett 533: 86–89, 2003. [DOI] [PubMed] [Google Scholar]
- 10.Gabbita SP, Aksenov MY, Lovell MA, Markesbery WR. Decrease in peptide methionine sulfoxide reductase in Alzheimer's disease brain. J Neurochem 73: 1660–1666, 1999. [DOI] [PubMed] [Google Scholar]
- 11.Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging 18: 685–716, 2001. [DOI] [PubMed] [Google Scholar]
- 12.Hansel A, Kuschel L, Hehl S, Lemke C, Agricola HJ, Hoshi T, Heinemann SH. Mitochondrial targeting of the human peptide methionine sulfoxide reductase (MSRA), an enzyme involved in the repair of oxidized proteins. FASEB J 16: 911–913, 2002. [DOI] [PubMed] [Google Scholar]
- 13.Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science 256: 184–185, 1992. [DOI] [PubMed] [Google Scholar]
- 14.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci 21: 3017–3023, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung B, Lee EH, Chung WS, Lee SJ, Shin SH, Joo SH, Kim SK, Lee JH. Increased viability of PC12 cells exposed to amyloid-beta peptide by transduction with human TAT-methionine sulfoxide reductase. Neuroreport 14: 2349–2353, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Kuo YM, Kokjohn TA, Beach TG, Sue LI, Brune D, Lopez JC, Kalback WM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Roher AE. Comparative analysis of amyloid-beta chemical structure and amyloid plaque morphology of transgenic mouse and Alzheimer's disease brains. J Biol Chem 276: 12991–12998, 2001. [DOI] [PubMed] [Google Scholar]
- 17.Misiti F, Clementi ME, Giardina B. Oxidation of methionine 35 reduces toxicity of the amyloid beta-peptide(1–42) in neuroblastoma cells (IMR-32) via enzyme methionine sulfoxide reductase A expression and function. Neurochem Int 56: 597–602, 2010. [DOI] [PubMed] [Google Scholar]
- 18.Moskovitz J, Rahman MA, Strassman J, Yancey SO, Kushner SR, Brot N, weissbach H. Escherichia coli peptide methionine sulfoxide reductase gene: regulation of expression and role in protecting against oxidative damage. J Bacteriol 177: 502–507, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moskovitz J, Weissbach H, Brot N. Cloning the expression of a mammalian gene involved in the reduction of methionine sulfoxide residues in proteins. Proc Natl Acad Sci USA 93: 2095–2099, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moskovitz J, Flescher E, Berlett BS, Azare J, Poston JM, Stadtman ER. Overexpression of peptide-methionine sulfoxide reductase in Saccharomyces cerevisiae and human T cells provides them with high resistance to oxidative stress. Proc Natl Acad Sci USA 95: 14071–14075, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moskovitz J, Bar-Noy S, Williams WM, Requena J, Berlett BS, Stadtman ER. Methionine sulfoxide reductase (MsrA) is a regulator of antioxidant defense and lifespan in mammals. Proc Natl Acad Sci USA 98: 12920–12925, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moskovitz J, Maiti P, Lopes DH, Oien DB, Attar A, Liu T, Mittal S, Hayes J, Bitan G. Induction of methionine-sulfoxide reductases protects neurons from amyloid β-protein insults in vitro and in vivo. Biochemistry 50: 10687–10697, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 20: 4050–4058, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muirhead KE, Borger E, Aitken L, Conway SJ, Gunn-Moore FJ. The consequences of mitochondrial amyloid beta-peptide in Alzheimer's disease. Biochem J 426: 255–270, 2010. [DOI] [PubMed] [Google Scholar]
- 25.Näslund J, Schierhorn A, Hellman U, Lannfelt L, Roses AD, Tjernberg LO, Silberring J, Gandy SE, Winblad B, Greengard P. Relative abundance of Alzheimer A beta amyloid peptide variants in Alzheimer disease and normal aging. Proc Natl Acad Sci USA 91: 8378–8382, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neurosci 19: 1959–1964, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60: 759–767, 2001. [DOI] [PubMed] [Google Scholar]
- 28.Oien DB, Moskovitz J. Substrates of the methionine sulfoxide reductase system and their physiological relevance. Curr Top Dev Biol 80: 93–133, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Oien DB, Osterhaus GL, Latif SA, Pinkston JW, Fulks J, Johnson M, Fowler SC, Moskovitz J. MsrA knockout mouse exhibits abnormal behavior and brain dopamine levels. Free Radic Biol Med 45: 193–200, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oien DB, Canello T, Gabizon R, Gasset M, Lundquist BL, Burns JM, Moskovitz J. Detection of oxidized methionine in selected proteins, cellular extracts and blood serums by novel anti-methionine sulfoxide antibodies. Arch Biochem Biophys 485: 35–40, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oien DB, Carrasco GA, Moskovitz J. Decreased Phosphorylation and Increased Methionine Oxidation of α-Synuclein in the Methionine Sulfoxide Reductase A Knockout Mouse. J Amino Acids 2011: 721094 and Erratum 2012: 415713, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pal R, Oien DB, Ersen FY, Moskovitz J. Elevated levels of brain-pathologies associated with neurodegenerative diseases in the methionine sulfoxide reductase A knockout mouse. Exp Brain Res 180: 765–774, 2007. [DOI] [PubMed] [Google Scholar]
- 33.Petropoulos I, Mary J, Perichon M, Friguet B. Rat peptide methionine sulphoxide reductase: cloning of the cDNA, and down-regulation of gene expression and enzyme activity during aging. Biochem J 355: 819–825, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pratico D. Evidence of oxidative stress in Alzheimer's disease brain and antioxidant therapy: lights and shadows. Ann NY Acad Sci 1147: 70–78, 2008. [DOI] [PubMed] [Google Scholar]
- 35.Resende R, Moreira PI, Proenca T, Deshpande A, Busciglio J, Pereira C, Oliveira CR. Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease. Free Radic Biol Med 44: 2051–2057, 2008. [DOI] [PubMed] [Google Scholar]
- 36.Roychaudhuri R, Yang M, Hoshi MM, Teplow DB. Amyloid beta-protein assembly and Alzheimer disease. J Biol Chem 284: 4749–4753, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ryan TM, Roberts BR, McColl G, Hare DJ, Doble PA, Li QX, Lind M, Roberts AM, Mertens HD, Kirby N, Pham CL, Hinds MG, Adlard PA, Barnham KJ, Curtain CC, Masters CL. Stabilization of nontoxic Aβ-oligomers: insights into the mechanism of action of hydroxyquinolines in Alzheimer's disease. J Neurosci 35: 2871–2884, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81: 741–766, 2001. [DOI] [PubMed] [Google Scholar]
- 39.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med 14: 837–842, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stadtman ER, Moskovitz J, Berlett BS, Levine RL. Cyclic oxidation and reduction of protein methionine residues is an important antioxidant mechanism. Mol Cell Biochem 234: 3–9, 2002. [PubMed] [Google Scholar]
- 41.Sideri TC, Koloteva-Levine N, Tuite MF, Grant CM. Methionine oxidation of Sup35 protein induces formation of the [PSI+] prion in a yeast peroxiredoxin mutant. J Biol Chem 286: 38924–38931, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sultana R, Butterfield DA. Role of oxidative stress in the progression of Alzheimer's disease. J Alzheimers Dis 19: 341–353, 2010. [DOI] [PubMed] [Google Scholar]
- 43.Terman A, Brunk UT. Oxidative stress, accumulation of biological ‘garbage’, and aging. Antioxid Redox Signal 8: 197–204, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Vinokur V, Grinberg L, Berenshtein E, Gross M, Moskovitz J, Reznick AZ, Chevion M, Eliashar R. Methionine-centered redox cycle in organs of the aero-digestive tract of young and old rats. Biogerontology 10: 43–52, 2009. [DOI] [PubMed] [Google Scholar]
- 45.Youmans KL, Tai LM, Kanekiyo T, Stine WB Jr, Michon SC, Nwabuisi-Heath E, Manelli AM, Fu Y, Riordan S, Eimer WA, Binder L, Bu G, Yu C, Hartley DM, LaDu MJ. Intraneuronal Aβ detection in 5xFAD mice by a new Aβ-specific antibody. Mol Neurodegener 7: 8, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]