

Abstract
The arteriovenous fistula (AVF) is the preferred hemodialysis vascular access, but it is complicated by high failure rates and attendant morbidity. This study provides the first description of a murine AVF model that recapitulates two salient features of hemodialysis AVFs, namely, anastomosis of end-vein to side-artery to create the AVF and the presence of chronic kidney disease (CKD). CKD reduced AVF blood flow, observed as early as 3 days after AVF creation, and increased neointimal hyperplasia, venous wall thickness, thrombus formation, and vasculopathic gene expression in the AVF. These adverse effects of CKD could not be ascribed to preexisting alterations in blood pressure or vascular reactivity in this CKD model. In addition to vasculopathic genes, CKD induced potentially vasoprotective genes in the AVF such as heme oxygenase-1 (HO-1) and HO-2. To determine whether prior HO-1 upregulation may protect in this model, we upregulated HO-1 by adeno-associated viral gene delivery, achieving marked venous induction of the HO-1 protein and HO activity. Such HO-1 upregulation improved AVF blood flow and decreased venous wall thickness in the AVF. Finally, we demonstrate that the administration of carbon monoxide, a product of HO, acutely increased AVF blood flow. This study thus demonstrates: 1) the feasibility of a clinically relevant murine AVF model created in the presence of CKD and involving an end-vein to side-artery anastomosis; 2) the exacerbatory effect of CKD on clinically relevant features of this model; and 3) the beneficial effects in this model conferred by HO-1 upregulation by adeno-associated viral gene delivery.
Keywords: arteriovenous fistula, murine model, chronic kidney disease, heme oxygenase-1
hemodialysis vascular access dysfunction is a common and challenging problem in patients with end-stage renal disease (ESRD) maintained on chronic hemodialysis, and, indeed, such dysfunction is the single most influential determinant of the rates of hospitalization, morbidity, and mortality in this patient population (15, 31, 42, 43). Of the three types of vascular accesses used for maintenance hemodialysis, the arteriovenous fistula (AVF) is widely preferred. While broadly accepted as the vascular access of choice, the AVF exhibits relatively low functionality and longevity: more than half of all AVFs created for use as a dialysis vascular access may be unusable because of maturational failure, and the cumulative patency for functional AVFs, placed as the first vascular access, is ∼5 yr (15, 31, 42, 43).
At least three fundamental pathobiologic processes contribute to AVF dysfunction and failure, and these include: 1) an impaired maturational process whereby an adaptive increase in blood flow and outward remodeling do not adequately occur in the afferent artery; 2) neointimal hyperplasia that is prone to occur at the juxta-anastomotic site; and 3) thrombosis occurring in the venous limb of the AVF (2, 15, 28, 31, 42, 43).
To investigate these pathogenetic pathways, AVFs have been created in rats and mice so as to model what occurs in clinical hemodialysis AVFs, and several such models have been described previously (17, 23, 29, 32, 39, 45, 47). Murine models, while technically more challenging than models in rats, are of particular interest as they can be imposed on genetically altered mice, thereby affording the assessment of the functional significance of a given gene or protein.
The present study provides the first description of a murine AVF model that recapitulates two essential features of a clinical hemodialysis AVF. First, this AVF model was created in mice already subjected to chronic kidney disease (CKD), the latter representing a disease invariantly present in patients in whom a hemodialysis AVF is created. Second, in the present study, the AVF was established by anastomosing the end-vein to the side-artery. Other types of anastomoses described in the literature on murine AVFs involve surgical connections that are end-artery to side-vein or end-artery to end-vein. While technically easier, these anastomoses do not broadly recapitulate the general topography and configuration of what is performed clinically; the latter is an important consideration as configuration of the AVF determines the pathologic shear stress imposed on an AVF, and such stress is regarded as a major determinant of AVF dysfunction and failure (30, 41).
In addition to describing and characterizing this particular murine model, the present study explored a new approach to the prevention of AVF dysfunction and failure. Our prior studies of an end-artery to side-vein murine AVF model, in the absence of CKD, demonstrated that the deficiency of heme oxygenase-1 (HO-1) markedly impaired the AVF (24). With the use of the present more clinically relevant model, the current studies questioned whether upregulation of HO-1 by adeno-associated viral (AAV) gene delivery exerts salutary effects in this model.
MATERIALS AND METHODS
Studies of the Murine AVF (End-Vein to Side-Artery) in CKD
All studies presented here were approved by the Institutional Animal Care and Use Committee of Mayo Clinic and were done according to National Institutes of Health guidelines. Male C57BL/6J mice (10–15 wk of age; Jackson Laboratory, Bar Harbor, ME) were used for these studies. The surgical procedures described below were performed under pentobarbital anesthesia (50 mg/kg ip) with postsurgical analgesia provided by the administration of buprenorphine (0.1 mg/kg sc).
Murine model of CKD.
Mice were subjected to subtotal nephrectomy as described previously (18, 19, 25) in two surgical sessions. First, through a flank incision, renal tissue was ablated by electrocoagulation using a surgical cautery (Cardinal Health, Waukegan, IL) from the upper and lower poles of the right kidney, leaving one-third of the kidney intact. One week later, left nephrectomy was performed via a flank incision. Sham operations for control mice (Sham CKD) included anesthesia and flank incisions with kidney exposure. Seven days after left nephrectomy, plasma blood urea nitrogen (BUN) levels were measured using a commercially available kit (Pointe Scientific, Canton, MI).
Murine end-vein to side-artery AVF model.
Two weeks after left nephrectomy, an AVF was created by an end-vein to side-artery anastomosis of the left external jugular vein and the left common carotid artery. After administration of heparin (1 IU/g) via tail vein, the left common carotid artery and left external jugular vein were carefully dissected through a ventral neck incision. Blood flow in the carotid artery was interrupted with microvascular clamps distal to the clavicle and at the bifurcation. A nontraumatic microvascular clamp was also placed on the external jugular vein immediately distal to the clavicle, and a ligature was placed at the branching of this vein with 6-0 silk suture. The jugular vein was then transected immediately proximal to the ligature, and a small longitudinal incision was made in the carotid artery. An anastomosis was created between the vein and artery with eight evenly spaced interrupted sutures (11-0 Ethilon; Ethicon, Somerville, NJ). Clamps were then released and flow through the fistula was confirmed. At 3 days and at 2 wk after AVF creation, blood flow in the carotid artery at points proximal (afferent) and distal (efferent) to the anastomosis and in the right (contralateral), intact carotid artery was assessed using a perivascular flow probe (catalog no. 0.5 PSB; Transonic Systems, Ithaca, NY), as described for our prior study (27). At 1 and 2 wk after placement of the AVF, the venous limb of the AVF and the right (contralateral) jugular vein were harvested for analysis of gene expression and histologic examination; in the AVF, venous cross sections immediately adjacent to the anastomosis were examined. Survival rates for our studies involving the placement of the AVF were comparable to a previously described murine AVF model (47): for studies at the 2-wk time point after AVF surgery, 47 of the 80 mice (58.8%) survived. The survival rate after the placement of the AVF was similar in mice previously subjected to CKD (33 of 56, and 58.9%) or Sham CKD (14 of 24, and 58.3%).
Studies of the effect of HO-1 upregulation by AAV9 vector.
Upregulation of HO-1 expression was mediated by an AAV serotype 9 [AAV9(HO-1)] vector constructed and produced as previously described (21, 26). A negative control vector, AAV9(revHO-1), was made by reversing the orientation of the HO-1 transgene sequence. AAV9(HO-1) vector or control AAV9(revHO-1) vector was intravenously administered (1 × 1011 vg/mouse) at 1 wk after left nephrectomy (1 wk before placement of the AVF) (26). At 2 wk after placement of the AVF, functional studies of blood flow through the AVF were undertaken as well as histologic studies of the venous segment of the AVF.
Studies of the effect of carbon monoxide delivered via CO-releasing molecule-3.
In other studies carbon monoxide (CO) was administered using a CO-releasing molecule {CORM-3; tricarbonylchloro[glycinato] ruthenium [II]; Sigma-Aldrich, St. Louis, MO} (11, 26). CORM-3 (40 mg/kg ip) was administered 13 days after left nephrectomy and 1 day before AVF surgery; additional doses were administered on the day of AVF placement and 1 day thereafter. Inactivated CORM-3 (iCORM-3), prepared as described in our previous study (26), was used as a control. One day following the creation of the AVF, blood flow was measured 45–60 min after the administration of the final dose of CORM-3 or iCORM-3.
Renal function and blood pressure studies.
In separate, additional studies, renal function, and systolic blood pressure were evaluated in response to the placement of the AVF in the setting of CKD and in response to AAV9(HO-1) vector administration to CKD mice. In the first set of experiments, mice were subjected to the two-step surgical induction of CKD (or Sham CKD), as outlined above. One week after CKD or Sham CKD, baseline BUN levels were determined and systolic blood pressures measured by tail cuff plethysmography (catalog no. SC-1000; Hatteras Instruments, Cary, NC). One week later the AVF was created in all mice, and BUN and systolic blood pressure were measured at 1 and 2 wk thereafter. In the second set of experiments, mice were subjected to the two-step surgical induction of CKD, and at 1 wk after left nephrectomy, baseline BUN and systolic blood pressure were measured. Mice were then matched and stratified into two experimental groups according to plasma BUN levels; one group was administered AAV9(HO-1) vector, the other group received control AAV9(revHO-1) vector. Two and three weeks later, BUN and systolic blood pressure were again measured. In a third set of studies, the effect of CKD on mean arterial pressure was assessed in mice with intact vasculature. For these studies, mice were subjected to the two-step surgical induction of CKD (or Sham CKD), as outlined above, and 1 wk later all mice were subjected to sham AVF surgery. Two weeks after the sham AVF, all mice were anesthetized (pentobarbital, 50 mg/kg ip) and mean arterial pressure in the left carotid artery was determined via an intra-arterial catheter (PE-10 tubing), monitored over 3 min after a 3-min equilibration period.
Assessment of Carotid Artery Vascular Reactivity
Vascular reactivity was assessed in mouse carotid arteries as described in our prior studies using small vessel chambers (Living Systems Instrumentation, St. Albans, VT) (13, 25). Briefly, before relaxation assessment, submaximal contraction was stabilized with the thromboxane analog U-46619 (3 × 10−8 to 10−7 M). Responses to 10−9 to 10−5 M acetylcholine or diethylammonium(Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate (DEA NONOate) were measured to characterize endothelium-dependent and endothelium-independent relaxation, respectively. Relaxation responses are calculated and expressed as a percentage of maximal relaxation in response to 3 × 10−4 M papaverine.
mRNA Expression by Quantitative Real-Time RT-PCR
Gene expression was assessed in venous segments of the AVF and in the contralateral jugular veins using quantitative real-time RT-PCR as described in our prior studies (26, 40). Briefly, total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA) with additional purification using an RNeasy kit (Qiagen, Valencia, CA). Reverse transcription was performed with a cDNA synthesis kit using random hexamers (catalog no. 4896866001; Roche Applied Science, Indianapolis, IN). Probes and primers used for quantitative PCR were purchased as assay sets (TaqMan Gene Expression Assays; Applied Biosystems, Foster City, CA). Target gene expression was standardized to the expression of 18S rRNA.
Western Analysis
Western analysis of HO-1 protein expression in jugular veins was undertaken as described in our prior studies (26, 37) with two veins combined for each lane. A polyclonal HO-1 antibody (catalog no. ADI-SPA-895; Enzo Life Sciences, Farmingdale, NY) was used as a primary antibody along with a horseradish peroxidase conjugated secondary antibody (catalog no. 170–6515; Bio-Rad, Hercules, CA). Protein loading was assessed by immunoblotting for GAPDH (catalog no. 2118; Cell signaling Technology, Danvers, MA).
Morphometric Assessment of Venous Wall Thickening And Semiquantitative Assessment of Thrombosis
Morphometric measurements were performed on hematoxylin and eosin-stained 5-μm sections of the venous limb of the AVF and the contralateral jugular vein, as previously described, to determine venous wall thickness (12, 24); wall thickness was determined at 12 sagittal sections around the venous wall, and the mean value for each vein was determined. In studies examining the effect of CKD on thrombosis in the venous limb of the AVF, the number of veins with thrombus occupying an area ≥50% of the venous cross-sectional area, was determined in hematoxylin and eosin-stained sections of the AVF.
Measurement of Venous HO Activity
As described in our prior studies, HO activity was assessed by measuring the rate of bilirubin production from hemin (26, 38). HO activity in the vena cava was expressed as picomoles of bilirubin produced per hour per milligram lysate protein, with the latter being measured by the Lowry method.
Statistical Analysis
Results are expressed as means ± SE and considered statistically significant for P < 0.05. The Student's t-test was used for parametric data and the Mann-Whitney U-test was employed for nonparametric data. Fisher's exact test was used to evaluate differences in thrombosis in the AVF in Sham CKD and CKD groups.
RESULTS
Characterization of the Murine AVF Model at 2 wk After Creation of the AVF
The AVF model employed in this study involved the transected end of the jugular vein anastomosed to the side of the carotid artery (end-vein to side-artery as is performed clinically). Figure 1 shows the afferent carotid artery segment, the jugular vein fed by the afferent carotid artery, and the efferent carotid artery segment. Two weeks after AVF creation in mice with intact kidney function, the carotid blood flow, measured in the afferent carotid artery of the AVF, was markedly increased compared with blood flow in the contralateral, intact carotid artery (2.36 ± 0.19 vs. 0.78 ± 0.02 ml/min, n = 8 in each group, P < 0.01). Thus, in this AVF model, the expected increase in carotid artery blood flow occurs when renal function is intact. Flow through the jugular vein of the AVF, determined as the difference in blood flow between the afferent carotid artery segment feeding the fistula and the efferent carotid segment leaving and distal to the AVF, 2.10 ± 0.18 ml/min, accounted for the bulk of the flow through the AVF.
Fig. 1.
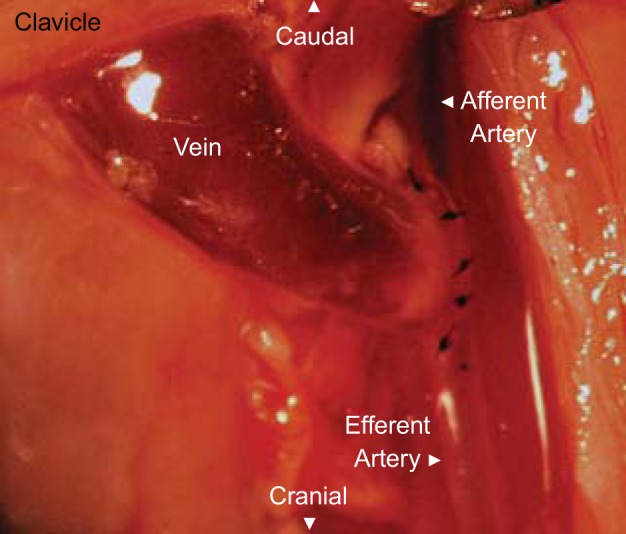
The murine end-vein to side-artery arteriovenous fistula (AVF) model. Appearance of the murine AVF employed in these studies, shown here immediately after it was fashioned by an end-vein to side-artery anastomosis of the left external jugular vein and left common carotid artery.
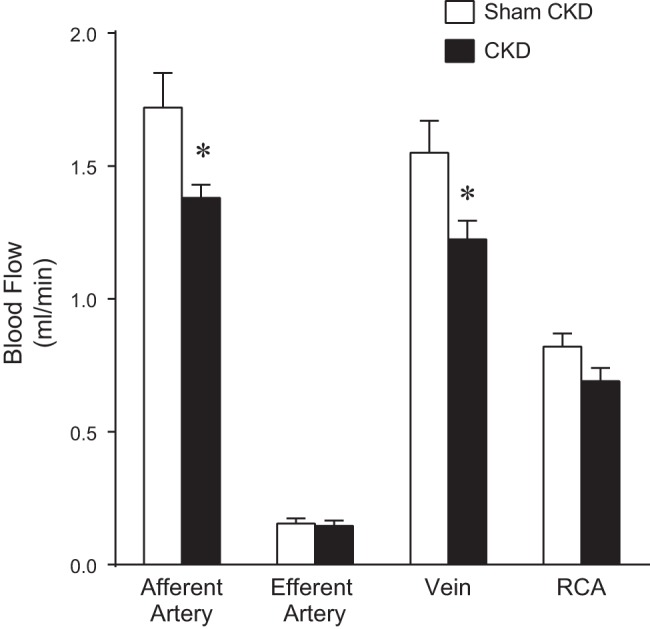
Figure 2 shows the effect of CKD on blood flow through the AVF. As demonstrated, CKD significantly reduced the flow through the afferent carotid artery without influencing flow through the efferent carotid artery. Blood flow through the jugular vein in the AVF was significantly reduced in mice with CKD compared with venous AVF blood flow in mice with intact kidney function. Figure 2 also demonstrates that CKD reduced blood flow through the intact, contralateral, carotid artery.
Fig. 2.

Effect of chronic kidney disease (CKD) on AVF blood flow determined 2 wk after creation of the AVF. Measurements of blood flow in the AVF and contralateral carotid artery were made using a perivascular flow probe in sham-operated control (Sham CKD) and CKD mice. Blood flow was measured in the left carotid artery at a point proximal (afferent) and a point distal (efferent) to the anastomosis, as well as in the right carotid artery (RCA; contralateral). Blood flow through the venous limb of the AVF (vein) was calculated as the difference in blood flow between the afferent and the efferent segments of the carotid artery of the AVF; n = 7 and n = 9 in Sham CKD and CKD groups, respectively. *P < 0.05 vs. corresponding Sham CKD.
In addition to impairing flow through the AVF, CKD enhanced neointimal hyperplasia and venous wall thickening in the AVF due to increased cellular infiltration and matrix deposition (Figure 3). In the AVF, CKD, compared with Sham CKD, also enhanced luminal occlusion by thrombosis, as demonstrated by semiquantitative analysis of histologic sections immediately adjacent to the anastomosis. In the venous segment of all seven mice with an AVF in the presence of CKD, there were thrombi that occluded 50% or greater of the venous cross sectional area, whereas only one of seven mice with an AVF without CKD exhibited such occlusion (100 vs. 14%, P < 0.01). Morphometric analyses demonstrated that CKD, compared with Sham CKD, induced a twofold increase in venous wall thickness in the AVF (Fig. 4).
Fig. 3.

Histologic effects of CKD on the venous limb of the AVF, assessed 2 wk after creation of the AVF. Representative views of the venous limb of the AVF in control mice (Sham CKD; A) and CKD mice (CKD; B). In addition to demonstrating increased wall thickness, the section of the AVF with CKD (B) shows organized and more recent thrombus, both of which were uncommon in the AVF with Sham CKD (A). All sections were stained with hematoxylin and eosin. Scale bars = 200 μm.
Fig. 4.
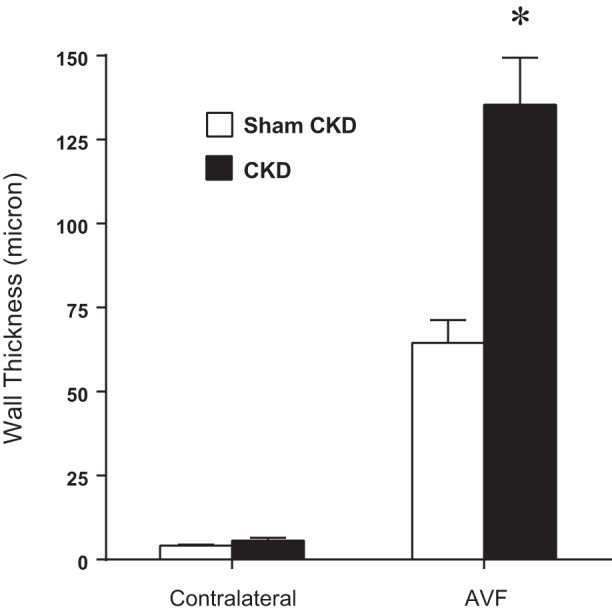
Effect of CKD on wall thickness of the venous limb of the AVF, determined 2 wk after creation of the AVF. Morphometric measurements of wall thickness were performed on the venous limb of the AVF and on the contralateral, intact, jugular vein in mice subjected to Sham CKD or CKD (n = 4 for each of the contralateral groups and n = 7 for each of the AVF groups). *P < 0.05, AVF-CKD vs. AVF-Sham CKD.
Expression of Vasculopathic Genes at 1 wk After Creation of the AVF
In addition to impairing flow through the AVF and increasing venous wall thickness of the AVF, the presence of CKD promoted the upregulation of vasculopathic genes implicated in hemodialysis vascular access dysfunction. In the AVF, CKD, compared with Sham CKD, increased expression of monocyte chemoattractant protein-1 (MCP-1), TNF-α, transforming growth factor-β1 (TGF-β1), and matrix metalloproteinase-9 (MMP-9) (Figs. 5 and 6). In the presence of CKD, compared with Sham CKD, the increments in AVF mRNA expression above expression in contralateral (control) veins were all significantly increased for MCP-1 (37.0 ± 4.6 vs. 14.9 ± 3.1, P < 0.05), TNF-α (43.0 ± 7.7 vs. 13.6 ± 4.9, P < 0.05), TGF-β1 (3.4 ± 1.2 vs. 0.7 ± 0.6, P < 0.05), and MMP-9 (2.2 ± 0.7 vs. −0.9 ± 0.2, P < 0.05). Thus, at least with regards to these representative genes, CKD drives the expression of genes that may promote venous injury in the AVF.
Fig. 5.

Effect of CKD on monocyte chemoattractant protein-1 (MCP-1) and TNF-α mRNA expression in the venous limb of the murine AVF, determined 1 wk after creation of the AVF. MCP-1 (A) and TNF-α (B) mRNA expression was measured by quantitative real-time RT-PCR in the venous limb of the AVF and in the contralateral, intact jugular vein in mice subjected to either Sham CKD or CKD; n = 14 and n = 12 for Sham CKD and CKD groups, respectively. *P < 0.05, AVF-CKD vs. AVF-Sham CKD.
Fig. 6.

Effect of CKD on transforming growth factor-β1 (TGF-β1) and matrix metalloproteinase-9 (MMP-9) mRNA expression in the venous limb of the murine AVF, determined 1 wk after creation of the AVF. TGF-β1 (A) and MMP-9 (B) mRNA expression was measured by quantitative real-time RT-PCR in the venous limb of the AVF and in the contralateral, intact, jugular vein in mice subjected to either Sham CKD or CKD; n = 14 and n = 12 for Sham CKD and CKD groups, respectively. *P < 0.05, AVF-CKD vs. AVF-Sham CKD.
Measurement of Murine AVF Blood Flow at 3 Days After Creation of the AVF
The marked suppression of AVF blood flow observed in AVF-CKD mice at 2 wk (Fig. 2) raised the question whether such effects of CKD were evinced at a relatively early time point after AVF creation. Studies were thus undertaken 3 days after AVF creation to determine if this inhibitory effect of CKD on AVF blood flow was present in the relatively early phase after the creation of the AVF (Fig. 7). While augmented in both groups compared with the contralateral (control) flow, AVF blood flow in mice with CKD, compared with mice with AVFs and Sham CKD, was significantly decreased at 3 days after AVF creation. However, the reduction in AVF blood flow induced by CKD was less at 3 days compared with the reduction observed at 2 wk, when assessed either by percent reduction in AVF blood flow (21% vs. 69%, respectively) or absolute reduction in AVF blood flow (0.33 vs. 2.23 ml/min, respectively); please compare Fig. 7 with Fig. 2, the blood flow rates on the y-axis being differently scaled. Thus the markedly blunted AVF blood flow rate observed in mice with CKD at 2 wk indicates that from days 3 to 14 a progressive rise in AVF blood flow occurred in the absence of CKD but this largely failed to occur in the presence of CKD (Figs. 2 and 7).
Fig. 7.
Effect of CKD on AVF blood flow determined 3 days after creation of the AVF. Measurements of blood flow in the AVF and contralateral carotid artery were made using a perivascular flow probe in Sham CKD and CKD mice, as described in Fig. 2. Blood flow was measured in the left carotid artery at a point proximal (afferent) and a point distal (efferent) to the anastomosis, as well as in the RCA (contralateral). Blood flow through the venous limb of the AVF (vein) was calculated as the difference in blood flow between the afferent and the efferent segments of the carotid artery of the AVF; n = 5 in each group. *P < 0.05 vs. corresponding Sham CKD.
Characterization of the Murine CKD Model
The adverse effects of CKD on the AVF may reflect, among other processes, the instigation of endothelial dysfunction (thus impairing vasorelaxant responses), alterations in blood pressure, and the effects of uremia. We thus assessed the extent to which such changes occurred in the employed model of CKD. Studies of vascular reactivity ex vivo demonstrated no evidence in this CKD model of impairment in either endothelium-dependent or endothelium-independent vascular reactivity (Fig. 8). This model was not associated with alterations in systolic blood pressure, either before or after the creation of the AVF (Fig. 8). Mean arterial pressure in mice with CKD was not significantly different from mean arterial pressure in mice with Sham CKD, when measured during anesthesia (84 ± 2 vs. 85 ± 1 mmHg, P = NS, n = 5 in each group). Thus the reduction in AVF blood flow and in right carotid artery blood flow observed in mice with CKD, when measured during anesthesia, may not be ascribed to a lowering effect of CKD, per se, on mean arterial pressure during conditions of anesthesia.
Fig. 8.

Effects of CKD on vascular reactivity, blood pressure, and renal function. A: endothelium-dependent relaxation of the carotid artery in Sham CKD and CKD mice; relaxation is plotted in response to acetylcholine. Values are means ± SE; n = 5 in each group. There were no significant differences in these responses in the carotid artery between Sham CKD and CKD groups. B: endothelium-independent relaxation of the carotid artery in Sham CKD and CKD mice; relaxation is plotted in response to DEA NONOate. Values are means ± SE; n = 7 in each group. There were no significant differences in these responses between Sham CKD and CKD groups. C and D: systolic blood pressure (C) and plasma blood urea nitrogen (BUN) levels (D) in Sham CKD and CKD mice before (baseline) and after AVF creation. In C and D, all mice in both groups were subjected to the placement of an AVF. Measurements were performed before (baseline) and at 1 wk and 2 wk after AVF placement; n = 7 and n = 10 in Sham CKD and CKD groups, respectively (1 wk); and n = 6 and n = 9 in Sham CKD and CKD groups, respectively (2 wk). *P < 0.05 vs. Sham CKD at that time point.
BUN was increased twofold in this CKD model, and this persisted after the creation of the AVF in Sham CKD and CKD mice (Fig. 8). Anemia was observed in this model as the hematocrit in mice with CKD was lower than in mice with Sham CKD before AVF creation (41 ± 1 vs. 49 ± 1, n = 10 and n = 7, respectively, P < 0.05), and this difference was still observed 2 wk after the creation of the AVF (40 ± 1 vs. 48 ± 1, n = 9 and n = 6, respectively, P < 0.05). This CKD model is thus relatively mild as it is not attended by an increase in systolic blood pressure or intrinsic abnormalities in vascular reactivity as studied ex vivo.
Expression of Vasoprotective Genes
Our prior studies in an AVF model (end-artery to side-vein in the setting of normal kidney function) demonstrated that HO-1 and HO-2 are vasoprotective genes, as the deficiency of either gene exerted adverse effects in this previously employed AVF model. We thus examined the effect of CKD on the expression of HO-1 and HO-2 in the current AVF model. As demonstrated in Fig. 9, CKD, compared with Sham CKD, significantly increased expression of HO-1 and, to a lesser extent, HO-2 mRNA, in the AVF. In the presence of CKD compared with Sham CKD, the increments in AVF mRNA expression above expression in contralateral (control) veins were both significantly increased for HO-1 (25.7 ± 5.2 vs. 6.8 ± 2.1, P < 0.05) and HO-2 (3.0 ± 1.4 vs. −1.4 ± 0.9, P < 0.05). Thus CKD induced not only potentially vasculopathic genes but also putatively vasoprotective ones.
Fig. 9.

Effect of CKD on heme oxygenase-1 (HO-1) and HO-2 mRNA expression in the venous limb of the murine AVF, determined 1 wk after creation of the AVF. HO-1 (A) and HO-2 (B) mRNA expression was measured by quantitative real-time RT-PCR in the venous limb of the AVF and in the contralateral, intact jugular vein in mice subjected to either Sham CKD or CKD; n = 14 and n = 12 for Sham CKD and CKD groups, respectively. *P < 0.05, AVF-CKD vs. AVF-Sham CKD.
Effect of Upregulating HO-1 in the AVF
In view of our prior findings demonstrating that HO-1 deficiency impairs the function of the AVF, and the recent interest in upregulating HO-1 as a vasoprotective strategy, we questioned whether upregulation of HO-1 would confer protective effects in the AVF created in the setting of CKD. To upregulate HO-1, we employed an adeno-associated gene delivery vector, AAV9, delivering HO-1. We confirmed that this method achieved its intended objective: as demonstrated in Fig. 10, HO-1 protein expression and, notably, HO activity, were both increased threefold.
Fig. 10.
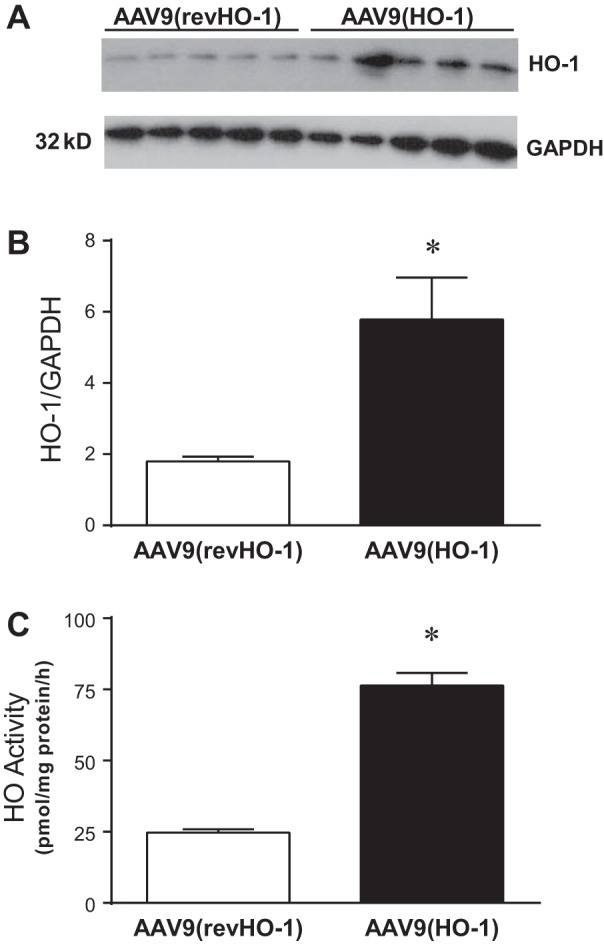
HO-1 expression and HO activity in central veins following the administration of AAV9(HO-1). A: Western analysis of HO-1 protein expression in the jugular vein of control AAV9(revHO-1) vector-treated mice and AAV9(HO-1) vector-treated mice 3 wk after administration. B: densitometric assessment of HO-1 Western analysis displayed in A; n = 5 in each group. *P < 0.05 vs. AAV9(revHO-1). C: HO activity measured in the vena cava of control AAV9(revHO-1) vector-treated mice and AAV9(HO-1) vector-treated mice 3 wk after administration; n = 5 in each group. *P < 0.05 vs. AAV9(revHO-1).
In mice with CKD-AVF, the administration of AAV9(HO-1) vector, compared with control AAV9(revHO-1) vector, markedly and significantly improved blood flow through the venous segment of the AVF (Fig. 11); in addition, such administration of AAV9(HO-1) vector significantly increased flow through the contralateral, intact carotid artery.
Fig. 11.
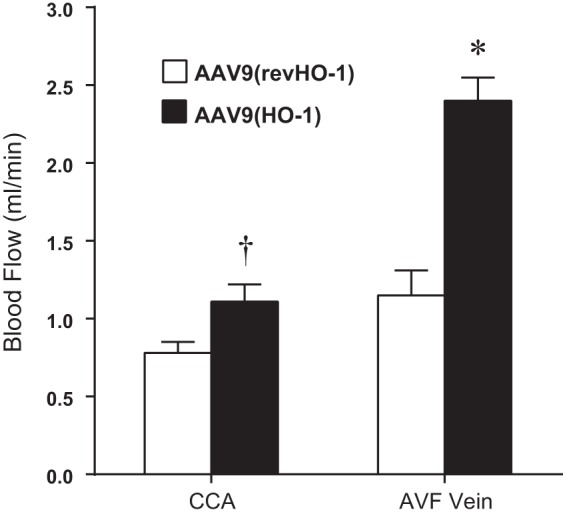
Effect of the administration of AAV9(HO-1) on blood flow in the combined CKD-AVF model, assessed 2 wk after creation of the AVF. Assessment of blood flow through the AVF (AVF vein) and the contralateral carotid artery (CCA) was performed using a perivascular flow probe in mice, all of which were subjected to CKD and AVF and treated with either control AAV9(revHO-1) vector or AAV9(HO-1) vector. Determination of blood flow in the AVF vein and measurement of blood flow in the CCA were made as described in Fig. 2. n = 6 and n = 7 in AAV9(revHO-1) and AAV9(HO-1), respectively. *P < 0.05, AVF-AAV9(HO-1) vs. AVF-AAV9(revHO-1); †P < 0.05, CCA-AAV9(HO-1) vs. CCA-AAV9(revHO-1).
This improvement in AVF blood flow by AAV9(HO-1) vector was accompanied by less neointimal hyperplasia and venous histologic injury in the AVF, as shown by representative profiles in Fig. 12, A and B. Morphometric measurements of venous wall thickness demonstrated that this was reduced by ∼50% in mice with CKD-AVF treated with AAV9(HO-1) vector compared with mice with CKD-AVF treated with AAV9(revHO-1) vector (Fig. 12C). Thus the upregulation of HO-1 by AAV9 not only augments AVF blood flow but also reduces neointimal hyperplasia, venous injury and inflammation, and venous wall thickness as determined by morphometric assessment.
Fig. 12.

Effect of the administration of AAV9(HO-1) on venous histology in the AVF in the combined CKD-AVF model, assessed 2 wk after creation of the AVF. A and B: representative views of the venous limb of the AVF in mice, all of which were subjected to the combined CKD and AVF and treated with either control vector [AAV9(revHO-1); A] or AAV9(HO-1) (B). All sections were stained with hematoxylin and eosin. Scale bars = 200 microns. C: morphometric measurements of wall thickness were performed on the venous limb of the AVF in mice, all of which were subjected to CKD and AVF and treated with either control vector [AAV9(revHO-1)] or AAV9(HO-1); n = 10 in each group. *P < 0.05 vs. AAV9(revHO-1).
To determine whether the beneficial effects of AAV9(HO-1) vector in the AVF reflected alterations in blood pressure or renal function incurred by HO-1 in the CKD model, we assessed these indexes in mice with CKD and treated with either AAV9(HO-1) vector or control AAV9(revHO-1) vector. As demonstrated in Fig. 13, such administration altered neither systolic blood pressure nor BUN in mice with CKD.
Fig. 13.

Evaluation of renal function and blood pressure in the murine CKD model before and after AAV9(HO-1) administration. A: plasma BUN levels were measured in mice, all of which were previously subjected to CKD and then administered either control AAV9(revHO-1) vector or AAV9(HO-1) vector. B: systolic blood pressure was measured in mice, all of which were previously subjected to CKD, and then administered either control AAV9(revHO-1) vector or AAV9(HO-1) vector. Measurements were performed in CKD mice, before (baseline) and 2 wk and 3 wk after the administration of control AAV9(revHO-1) vector or AAV9(HO-1) vector; n = 5 and n = 6 in AAV9(revHO-1) and AAV9(HO-1), respectively. No significant difference in either of these indexes was detected in response to AAV9(HO-1) at any time point.
Effect of CORM-3 on AVF Blood Flow
We also questioned whether a product of HO-1 may exert vasorelaxant effects in the AVF. We selected CO as the product of HO for examination, and we employed a CO-releasing molecule (CORM-3) as a method for delivering CO. Mice with combined CKD and an AVF were accordingly administered CORM-3 or, as a control, iCORM-3, and blood flow in the AVF and in the contralateral, intact carotid artery determined. As shown in Fig. 14, blood flow in the AVF as well as blood flow in the contralateral, intact carotid artery were both increased following the administration of CORM-3 compared with such blood flow rates after the administration of iCORM-3.
Fig. 14.

Effect of the administration of the CO donor CORM-3 on blood flow in the combined CKD-AVF model, assessed 1 day after creation of the AVF. Assessment of blood flow through the AVF (AVF vein) and the contralateral carotid artery was performed using a perivascular flow probe in mice, all of which were previously subjected to CKD and AVF and then administered either iCORM-3 or CORM-3. Determination of blood flow in the AVF vein and measurement of blood flow in the CCA were made as described in Fig. 2; n = 6 in each group. *P < 0.05, AVF-CORM-3 vs. AVF-iCORM-3; †P < 0.05, CCA-CORM-3 vs. CCA-iCORM-3.
DISCUSSION
A number of experimental models have been used to study function and failure of the clinical hemodialysis AVF. These approaches include anastomosis of the end-artery to side-vein in the presence of normal kidney function; end-artery to side-vein in the presence of CKD; end-vein to end-artery in the absence and presence of CKD; and end-vein to side-artery in the presence of normal kidney function (17, 23, 29, 32, 39, 47). The present study is the first to describe the anastomosis of the end-vein to side-artery in the setting of CKD, thereby introducing a model that recapitulates these two salient features of the clinical hemodialysis AVF.
In the presence of normal kidney function, the end-vein to side-artery anastomosis in the present model was attended by a markedly increased afferent carotid artery blood flow compared with blood flow through the contralateral, intact carotid artery. Thus, in the absence of CKD, this end-vein to side-artery AVF model evinces the expected maturational blood flow response in the afferent artery to the AVF. However, in the setting of CKD, and at the 2-wk time point, this increase in afferent arterial AVF blood flow failed to occur, and the blood flow through the venous limb of the AVF was markedly reduced. A reduction in AVF blood flow in the presence of CKD was also noted at a relatively early time point after AVF creation (3 days thereafter), and these data, in conjunction with AVF blood flow at 2 wk, suggest that CKD significantly and progressively limits the maturational increase in AVF blood flow following AVF creation. The presence of CKD not only impaired venous flow in the AVF but also led to substantial thickening of the venous wall and increased venous thrombus formation at 2 wk after the creation of the AVF; in the AVF, CKD also enhanced the upregulation of genes with recognized proliferative, proinflammatory, and procoagulant properties, as assessed 1 wk after creation of the AVF. This CKD model did not exhibit an elevation in blood pressure or intrinsic abnormalities either in endothelium-dependent or endothelium-independent vasorelaxation. The absence of these findings suggests that it was the systemic uremic milieu of the employed CKD model that was largely responsible for these adverse effects of CKD.
These adverse effects of CKD and attendant uremia on AVF flow, histology, and vasculopathic gene expression are clinically relevant because of the following consideration. Hemodialysis AVF dysfunction and failure are usually ascribed to a number of mechanisms/predisposing factors acting singly or, more likely, in combination, and include the following: 1) the adverse effects of pathologic shear stress following the anastomosis of the low pressure, high capacitance vein to the high pressure, low capacitance artery; 2) an impaired maturational response in the artery of the AVF; 3) loss of venous patency due to juxta-anastomotic neointimal hyperplasia and attendant thrombosis; 4) venous tributaries that siphon off blood from the main venous trunk of the AVF; 5) repeated venous injury due to intermittent puncturing of the vein for hemodialysis; 6) relatively small vessel size; and 7) an increased risk that is imposed for largely unclear reasons by age, gender, and ethnicity (15, 31, 42, 43). Hemodialysis AVFs, however, are always created in patients with CKD, and thus AVFs are expected to mature and acquire functionality in a uremic environment. The contribution of uremia per se to AVF failure has received relatively little attention and, until quite recently, has been largely overlooked. Examining this issue clinically, however, is quite challenging, because of the invariant presence of CKD when a hemodialysis AVF is created; it is thus difficult to evaluate the contribution of uremia relative to other putative pathogenetic factors in the evolution of clinical hemodialysis AVF failure. In this regard, manipulations that are possible in disease models may reveal insights not readily gleaned from clinical studies. In the present experimental studies that created an AVF in the presence of CKD, the systemic uremic milieu exerted prominent adverse effects on the AVF, a finding underscored by the relative absence of such adverse effects on the intact vessels (and not subjected to AVF creation). We thus suggest that the contribution of the systemic uremic milieu merits more attention in current constructs regarding, and studies centered on, the pathogenesis of hemodialysis AVF dysfunction and failure. In particular, recent clinical evidence has demonstrated that when an AVF is in place for an extended period before the need to initiate dialysis, interventions are increasingly needed simply to maintain AVF patency and function (3, 8, 14, 22, 44). We thus speculate that chronic exposure to the systemic uremic milieu, in synergy with other pathogenetic processes such as pathologic shear stress, contributes to AVF dysfunction and loss of patency.
Clinical hemodialysis AVFs are usually created in CKD of sufficient severity so as to be attended by systemic hypertension and impaired vasorelaxant responses, both of which may adversely affect the functionality of the AVF (7, 16, 35). Impaired vasorelaxant responses are of special concern for the following reason. For a created AVF to achieve sufficient functionality to support hemodialysis, a maturational response must occur in the artery that starts with, and absolutely requires, arterial vasodilation and increased arterial blood flow. Endothelial function, including flow-mediated dilatation, is impaired in CKD. Such impairment may thus compromise the maturational response in an AVF, and thus the extent to which the AVF is eventually usable for dialysis. Interestingly, in our studies, the employed model of CKD was relatively mild such that neither endothelium-dependent nor endothelium-independent vasorelaxation was impaired. These findings suggest that compromise in AVF blood flow in this model reflects adverse effects on the vasculature of the systemic uremic milieu (for example, assorted uremic “toxins,” vasoconstrictive species, proinflammatory mediators, cytokines, and growth factors), rather than the presence of preexisting intrinsic defects in vascular reactivity or systemic hypertension.
As shown in both experimental and clinical studies, AVFs are attended by upregulation of recognized vasculopathic species. In the present studies, the employed model of CKD/uremia exaggerated the expression of MCP-1, TNF-α, TGF-β1, and MMP-9, genes that are widely regarded as contributors to AVF dysfunction (9, 23, 37, 46). Interestingly, such accentuating effects of CKD/uremia on vasculopathic gene expression in the AVF were not observed in intact veins. Thus the exposure of veins to heightened pressures and pathologic shear stress following AVF creation alters venous responses to uremia, eliciting from such stressed veins increased expression of potentially vasculopathic genes. Such expression may contribute to the venous wall thickening observed in our morphometric and histologic studies of the AVF.
The CKD/uremia model also increased the expression of HO-1 and HO-2 in the AVF. Our prior studies in an end-artery to side-vein murine AVF model in the presence of intact kidney function demonstrated that HO-1 and, to lesser extent, HO-2, are induced in the venous limb; the vasoprotective nature of these responses was demonstrated by the finding that genetic deficiency of either HO-1 or HO-2 exaggerated AVF dysfunction and failure. In the present studies, we evaluated the effects of prior upregulation of HO-1 in the combined CKD-AVF model by examining the clinically relevant, quantitative assessment of AVF blood flow and venous histologic injury; these studies demonstrate that prior HO-1 upregulation substantially preserved AVF blood flow and decreased venous wall thickness. We suggest that these findings are clinically relevant for at least three reasons. First, AVF outcomes are inferior in patients exhibiting genetic HO-1 polymorphisms that are characterized by long GT repeats in the HO-1 gene compared with patients with short GT repeats; long GT repeats are associated with less inducibility of HO-1 gene and protein and less HO activity (34). Second, a modality such as far infrared therapy, which may improve AVF function and longevity, also induces HO-1 (6, 33). Third, enhanced inflammatory processes, regionally at the AVF as well as systemically (5), predict poor AVF outcomes, and among the properties of HO-1 are potent anti-inflammatory actions (1, 36).
As induction of HO-1 and/or administration of its products may provide a therapeutic strategy in improving AVF outcomes, we examined whether upregulating HO-1 may be beneficial in this model. To achieve such upregulation, we utilized an approach based on AAV gene delivery, which we have recently demonstrated to effectively upregulate HO-1 in carotid arteries and improve carotid blood flow in the partial carotid artery ligation model (26). Enthusiasm and support for AAV as a vector for gene delivery are high, in part, because of the lack of immunogenicity of AAV (10, 21); numerous clinical trials using AAV for gene delivery have already been conducted in humans (4, 20). Our studies are the first to demonstrate effective, indeed quite robust, upregulation of HO-1 protein and increased HO activity in veins by this AAV gene delivery approach. Such upregulation was attended by markedly improved flow in the AVF as well as less neointimal hyperplasia and venous wall inflammation and injury in the AVF. Studies examining the effect of HO-1 upregulation on vasculopathic gene expression in the AVF would also be of interest. We also demonstrated that CO, a product of HO activity, can acutely increase flow through the venous limb of the AVF. While these effects of CO/CORMs were studied acutely and in the short term, it is possible that strategies that increase AVF blood flow acutely may promote the maturational response and thus the long-term increase in AVF blood flow. Studies that examine the effects of CORM-3 on gene expression and venous histologic injury at more delayed time points after AVF creation would thus be of interest. Our present studies provide the first demonstration of the protective effects of HO-1 in a clinically relevant model and support therapeutic strategies for the AVF based on upregulating HO-1 and/or administering its products.
The finding that prior upregulation of HO-1 is protective in this AVF model merits comment, especially in light of the fact that in this AVF model, as in prior rodent AVF models, marked induction of HO-1 occurs following the creation of the AVF. HO-1 is quite weakly expressed in healthy veins and in veins exposed to the uremic milieu (and before their use in AVF creation); the induction of HO-1 that ensues in these venous segments following their use in AVF creation requires several hours to occur. The benefit of upregulating venous HO-1 before AVF formation (achieved in this study by AAV9) is that these veins now express increased amounts of HO-1 protein and increased HO activity before, at the time of, and during the early phase following AVF creation, and before the endogenous, inducible HO-1 system (HO-1 mRNA and protein and HO activity) is recruited in the vein now employed in the AVF. The time periods during the surgical creation of the AVF and during the early phase following AVF formation are quite critical ones in determining vasculopathic responses attendant upon the creation of an AVF. Thus we speculate that the beneficial effect of upregulating HO-1 by AAV9 before the creation of the AVF reflects, at least in part, the prominent presence and ready availability of the vasoprotective effects of the HO-1 system during these critical periods wherein the endogenous HO-1 system is yet to be recruited.
In summary, we demonstrate the feasibility of a clinically relevant murine AVF model created in the presence of CKD and involving an end-vein to side-artery anastomosis. We also demonstrate that clinically relevant features of this model are exacerbated by CKD, and that the upregulation of HO-1 by AAV vector-mediated gene delivery confers beneficial effects in this model.
GRANTS
This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-70124 and DK-47060.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: L.K., M.L.H., A.J.C., M.A.B., Z.S.K., and K.A.N. conception and design of research; L.K., M.L.H., and A.J.C. performed experiments; L.K., J.P.G., M.L.H., A.J.C., M.A.B., Z.S.K., and K.A.N. analyzed data; L.K., J.P.G., A.J.C., M.A.B., Z.S.K., and K.A.N. interpreted results of experiments; L.K., A.J.C., and K.A.N. prepared figures; L.K., M.L.H., A.J.C., and K.A.N. drafted manuscript; L.K., J.P.G., M.L.H., A.J.C., M.A.B., Z.S.K., and K.A.N. edited and revised manuscript; L.K., J.P.G., M.L.H., A.J.C., M.A.B., Z.S.K., and K.A.N. approved final version of manuscript.
ACKNOWLEDGMENTS
We gratefully acknowledge the technical expertise of Allan Ackerman and the secretarial expertise of Kara Zelinske in the preparation of this work.
REFERENCES
- 1.Agarwal A, Bolisetty S. Adaptive responses to tissue injury: role of heme oxygenase-1. Trans Am Clin Climatol Assoc 124: 111–122, 2013. [PMC free article] [PubMed] [Google Scholar]
- 2.Agarwal A, Segal MS. Intimal exuberance: veins in jeopardy. Am J Pathol 162: 1759–1761, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aitken E, Jackson A, Kong C, Coats P, Kingsmore D. Renal function, uraemia and early arteriovenous fistula failure. BMC Nephrol 15: 179, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asokan A, Schaffer DV, Samulski RJ. The AAV vector toolkit: poised at the clinical crossroads. Mol Ther 20: 699–708, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banerjee T, Kim SJ, Astor B, Shafi T, Coresh J, Powe NR. Vascular access type, inflammatory markers, and mortality in incident hemodialysis patients: the Choices for Healthy Outcomes in Caring for End-Stage Renal Disease (CHOICE) Study. Am J Kidney Dis 64: 954–961, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bashar K, Healy D, Browne LD, Kheirelseid EA, Walsh MT, Clarke-Moloney M, Burke PE, Kavanagh EG, Walsh SR. Role of far infra-red therapy in dialysis arterio-venous fistula maturation and survival: systematic review and meta-analysis. PLoS One 9: e104931, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Briet M, Burns KD. Chronic kidney disease and vascular remodelling: molecular mechanisms and clinical implications. Clin Sci (Lond) 123: 399–416, 2012. [DOI] [PubMed] [Google Scholar]
- 8.Carney EF. Dialysis: optimal timing of arteriovenous fistula placement in elderly patients. Nat Rev Nephrol 10: 613, 2014. [DOI] [PubMed] [Google Scholar]
- 9.Chang CJ, Ko YS, Ko PJ, Hsu LA, Chen CF, Yang CW, Hsu TS, Pang JH. Thrombosed arteriovenous fistula for hemodialysis access is characterized by a marked inflammatory activity. Kidney Int 68: 1312–1319, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Chen S, Kapturczak MH, Wasserfall C, Glushakova OY, Campbell-Thompson M, Deshane JS, Joseph R, Cruz PE, Hauswirth WW, Madsen KM, Croker BP, Berns KI, Atkinson MA, Flotte TR, Tisher CC, Agarwal A. Interleukin 10 attenuates neointimal proliferation and inflammation in aortic allografts by a heme oxygenase-dependent pathway. Proc Natl Acad Sci USA 102: 7251–7256, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clark JE, Naughton P, Shurey S, Green CJ, Johnson TR, Mann BE, Foresti R, Motterlini R. Cardioprotective actions by a water-soluble carbon monoxide-releasing molecule. Circ Res 93: e2–8, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Croatt AJ, Grande JP, Hernandez MC, Ackerman AW, Katusic ZS, Nath KA. Characterization of a model of an arteriovenous fistula in the rat: the effect of l-NAME. Am J Pathol 176: 2530–2541, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.d'Uscio LV, Smith LA, Santhanam AV, Richardson D, Nath KA, Katusic ZS. Essential role of endothelial nitric oxide synthase in vascular effects of erythropoietin. Hypertension 49: 1142–1148, 2007. [DOI] [PubMed] [Google Scholar]
- 14.Dixon BS. Timing of arteriovenous fistula placement: keeping it in perspective. J Am Soc Nephrol 26: 241–243, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dixon BS. Why don't fistulas mature? Kidney Int 70: 1413–1422, 2006. [DOI] [PubMed] [Google Scholar]
- 16.Drueke TB, Massy ZA. Atherosclerosis in CKD: differences from the general population. Nat Rev Nephrol 6: 723–735, 2010. [DOI] [PubMed] [Google Scholar]
- 17.Feng W, Chumley P, Allon M, George J, Scott DW, Patel RP, Litovsky S, Jaimes EA. The transcription factor E26 transformation-specific sequence-1 mediates neointima formation in arteriovenous fistula. J Am Soc Nephrol 25: 475–487, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gagnon RF, Duguid WP. A reproducible model for chronic renal failure in the mouse. Urol Res 11: 11–14, 1983. [DOI] [PubMed] [Google Scholar]
- 19.Gagnon RF, Gallimore B. Characterization of a mouse model of chronic uremia. Urol Res 16: 119–126, 1988. [DOI] [PubMed] [Google Scholar]
- 20.High KA. The gene therapy journey for hemophilia: are we there yet? Blood 120: 4482–4487, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hillestad ML, Guenzel AJ, Nath KA, Barry MA. A vector-host system to fingerprint virus tropism. Hum Gene Ther 23: 1116–1126, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hod T, Patibandla BK, Vin Y, Brown RS, Goldfarb-Rumyantzev AS. Arteriovenous fistula placement in the elderly: when is the optimal time? J Am Soc Nephrol 26: 448–456, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Juncos JP, Grande JP, Kang L, Ackerman AW, Croatt AJ, Katusic ZS, Nath KA. MCP-1 contributes to arteriovenous fistula failure. J Am Soc Nephrol 22: 43–48, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Juncos JP, Tracz MJ, Croatt AJ, Grande JP, Ackerman AW, Katusic ZS, Nath KA. Genetic deficiency of heme oxygenase-1 impairs functionality and form of an arteriovenous fistula in the mouse. Kidney Int 74: 47–51, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kang L, Grande JP, Farrugia G, Croatt AJ, Katusic ZS, Nath KA. Functioning of an arteriovenous fistula requires heme oxygenase-2. Am J Physiol Renal Physiol 305: F545–F552, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang L, Hillestad ML, Grande JP, Croatt AJ, Barry MA, Farrugia G, Katusic ZS, Nath KA. Induction and functional significance of the heme oxygenase system in pathological shear stress in vivo. Am J Physiol Heart Circ Physiol 308: H1402–H1413, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang L, Yamada S, Hernandez MC, Croatt AJ, Grande JP, Juncos JP, Vercellotti GM, Hebbel RP, Katusic ZS, Terzic A, Nath KA. Regional and systemic hemodynamic responses following the creation of a murine arteriovenous fistula. Am J Physiol Renal Physiol 301: F845–F851, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanwar YS. Functional duality of progenitor cells influxing into arteriovenous fistula during its neoangiogenesis. Am J Physiol Renal Physiol 293: F468–F469, 2007. [DOI] [PubMed] [Google Scholar]
- 29.Kokubo T, Ishikawa N, Uchida H, Chasnoff SE, Xie X, Mathew S, Hruska KA, Choi ET. CKD accelerates development of neointimal hyperplasia in arteriovenous fistulas. J Am Soc Nephrol 20: 1236–1245, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krishnamoorthy MK, Banerjee RK, Wang Y, Choe AK, Rigger D, Roy-Chaudhury P. Anatomic configuration affects the flow rate and diameter of porcine arteriovenous fistulae. Kidney Int 81: 745–750, 2012. [DOI] [PubMed] [Google Scholar]
- 31.Lee T. Novel paradigms for dialysis vascular access: downstream vascular biology–is there a final common pathway? Clin J Am Soc Nephrol 8: 2194–2201, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang A, Wang Y, Han G, Truong L, Cheng J. Chronic kidney disease accelerates endothelial barrier dysfunction in a mouse model of an arteriovenous fistula. Am J Physiol Renal Physiol 304: F1413–F1420, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin CC, Yang WC, Chen MC, Liu WS, Yang CY, Lee PC. Effect of far infrared therapy on arteriovenous fistula maturation: an open-label randomized controlled trial. Am J Kidney Dis 62: 304–311, 2013. [DOI] [PubMed] [Google Scholar]
- 34.Lin CC, Yang WC, Lin SJ, Chen TW, Lee WS, Chang CF, Lee PC, Lee SD, Su TS, Fann CS, Chung MY. Length polymorphism in heme oxygenase-1 is associated with arteriovenous fistula patency in hemodialysis patients. Kidney Int 69: 165–172, 2006. [DOI] [PubMed] [Google Scholar]
- 35.Moody WE, Edwards NC, Madhani M, Chue CD, Steeds RP, Ferro CJ, Townend JN. Endothelial dysfunction and cardiovascular disease in early-stage chronic kidney disease: cause or association? Atherosclerosis 223: 86–94, 2012. [DOI] [PubMed] [Google Scholar]
- 36.Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int 70: 432–443, 2006. [DOI] [PubMed] [Google Scholar]
- 37.Nath KA, Grande JP, Kang L, Juncos JP, Ackerman AW, Croatt AJ, Katusic ZS. β-Catenin is markedly induced in a murine model of an arteriovenous fistula: the effect of metalloproteinase inhibition. Am J Physiol Renal Physiol 299: F1270–F1277, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nath KA, Hernandez MC, Croatt AJ, Katusic ZS, Juncos LA. Heme oxygenase activity as a determinant of the renal hemodynamic response to low-dose ANG II. Am J Physiol Regul Integr Comp Physiol 299: R1183–R1191, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nieves Torres EC, Yang B, Janardhanan R, Brahmbhatt A, Leof E, Mukhopadhyay D, Misra S. Adventitial delivery of lentivirus-shRNA-ADAMTS-1 reduces venous stenosis formation in arteriovenous fistula. PLoS One 9: e94510, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pittock ST, Norby SM, Grande JP, Croatt AJ, Bren GD, Badley AD, Caplice NM, Griffin MD, Nath KA. MCP-1 is up-regulated in unstressed and stressed HO-1 knockout mice: Pathophysiologic correlates. Kidney Int 68: 611–622, 2005. [DOI] [PubMed] [Google Scholar]
- 41.Rajabi-Jagahrgh E, Krishnamoorthy MK, Roy-Chaudhury P, Succop P, Wang Y, Choe A, Banerjee RK. Longitudinal assessment of hemodynamic endpoints in predicting arteriovenous fistula maturation. Semin Dial 26: 208–215, 2013. [DOI] [PubMed] [Google Scholar]
- 42.Riella MC, Roy-Chaudhury P. Vascular access in haemodialysis: strengthening the Achilles' heel. Nat Rev Nephrol 9: 348–357, 2013. [DOI] [PubMed] [Google Scholar]
- 43.Roy-Chaudhury P, Sukhatme VP, Cheung AK. Hemodialysis vascular access dysfunction: a cellular and molecular viewpoint. J Am Soc Nephrol 17: 1112–1127, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Shechter SM, Skandari MR, Zalunardo N. Timing of arteriovenous fistula creation in patients with CKD: a decision analysis. Am J Kidney Dis 63: 95–103, 2014. [DOI] [PubMed] [Google Scholar]
- 45.Tsapenko MV, d'Uscio LV, Grande JP, Croatt AJ, Hernandez MC, Ackerman AW, Katusic ZS, Nath KA. Increased production of superoxide anion contributes to dysfunction of the arteriovenous fistula. Am J Physiol Renal Physiol 303: F1601–F1607, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang Y, Liang A, Luo J, Liang M, Han G, Mitch WE, Cheng J. Blocking Notch in endothelial cells prevents arteriovenous fistula failure despite CKD. J Am Soc Nephrol 25: 773–783, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong CY, de Vries MR, Wang Y, van der Vorst JR, Vahrmeijer AL, van Zonneveld AJ, Roy-Chaudhury P, Rabelink TJ, Quax PH, Rotmans JI. Vascular remodeling and intimal hyperplasia in a novel murine model of arteriovenous fistula failure. J Vasc Surg 59: 192–201.e1, 2014. [DOI] [PubMed] [Google Scholar]