

Abstract
The hypoxia-inducible factor (HIF)-1 and β-catenin protective pathways represent the two most significant cellular responses that are activated in response to acute kidney injury. We previously reported that murine mucin (Muc)1 protects kidney function and morphology in a mouse model of ischemia-reperfusion injury (IRI) by stabilizing HIF-1α, enhancing HIF-1 downstream signaling, and thereby preventing metabolic stress (Pastor-Soler et al. Muc1 is protective during kidney ischemia-reperfusion injury. Am J Physiol Renal Physiol 308: F1452–F1462, 2015). We asked if Muc1 regulates the β-catenin protective pathway during IRI as 1) β-catenin nuclear targeting is MUC1 dependent in cultured human cells, 2) β-catenin is found in coimmunoprecipitates with human MUC1 in extracts of both cultured cells and tissues, and 3) MUC1 prevents β-catenin phosphorylation by glycogen synthase kinase (GSK)3β and thereby β-catenin degradation. Using the same mouse model of IRI, we found that levels of active GSK3β were significantly lower in kidneys of control mice compared with Muc1 knockout (KO) mice. Consequently, β-catenin was significantly upregulated at 24 and 72 h of recovery and appeared in the nuclear fraction at 72 h in control mouse kidneys. Both β-catenin induction and nuclear targeting were absent in Muc1 KO mice. We also found downstream induction of β-catenin prosurvival factors (activated Akt, survivin, transcription factor T cell factor 4 (TCF4), and its downstream target cyclin D1) and repression of proapoptotic factors (p53, active Bax, and cleaved caspase-3) in control mouse kidneys that were absent or aberrant in kidneys of Muc1 KO mice. Altogether, the data clearly indicate that Muc1 protection during acute kidney injury proceeds by enhancing both the HIF-1 and β-catenin protective pathways.
Keywords: acute kidney injury, ischemia, mucin 1, β-catenin
over the past two decades, dramatic increases in the incidence of acute kidney injury (AKI) have been reported, particularly within the United States (36, 48, 54). In addition to a significantly increased mortality, length of stay, and high health care expenditures associated with AKI (8, 48), several studies have linked AKI with the subsequent development of chronic kidney disease (CKD), end-stage kidney disease, and a higher risk of cardiovascular mortality (9, 11, 14, 35). Hypotensive, ischemic, or septic insults are the most common causes of AKI (25, 31), particularly insults in the tubular segments situated within the outer stripe of the outer medulla of the kidney (6, 52). Tubular cells in the S3 segment of the proximal tubule, located in the outer stripe of the outer medulla, likely suffer the most severe injury after an ischemic or toxic insult because these cells are characterized by high metabolic activity with baseline hypoxia even under normal physiological conditions (6, 13).
In the absence of an effective treatment for AKI (4, 35), great efforts have been made to identify the key regulatory mechanisms of renal adaptive responses that may limit the extent of kidney injury and restore homeostasis after AKI (47, 60). The hypoxia-inducible factor (HIF)-1 and β-catenin protective pathways represent two of the most significant cellular responses that are activated during AKI (17, 56, 60). We have also reported that the glycoprotein mucin (Muc)1 stabilizes and enhances the HIF-1 protective pathway in a mouse model of kidney ischemia-reperfusion injury (IRI) (38). We found that levels of HIF-1α were reduced and that expression of HIF-1 target genes associated with a shift in glucose metabolism to glycolysis were aberrant in Muc1 knockout (KO) mice during IRI. Consequently, Muc1 KO mice had reduced kidney function and more tubule damage with increased metabolic stress compared with control mice, consistent with a key role for Muc1 in protection against tubular cell injury during AKI (38). Most notably, Muc1 expression was induced during IRI, with the protein appearing in the cytoplasm and nucleus of all epithelial cells including the S3 segment of the recovering proximal tubule, where the most severe damage occurred (38).
Muc1 (MUC1 in humans) is a transmembrane glycoprotein expressed on the apical surface of normal secretory epithelia including ductal epithelial cells of the mammary gland, liver, pancreas, and kidney (1, 15). In the normal mouse kidney, we found the highest expression of Muc1 on the apical surface of the thick ascending limb, distal convoluted tubule, and collecting ducts (38). The MUC1/Muc1 precursor undergoes autocatalytic cleavage soon after its synthesis, generating a stable dimer of the small transmembrane subunit (∼25 kDa) and a larger mucin-like subunit with a variable number of tandem repeats (TRs; >250 kDa) (23, 27). MUC1 is involved in intracellular signaling via its cytoplasmic domain, which can be phosphorylated by receptor tyrosine kinases like the EGF receptor (EGFR), and nonreceptor tyrosine kinases like glycogen synthase kinase (GSK)3β (24, 40, 55, 61). This phosphorylation of MUC1 promotes differential interactions with protein ligands, thereby generating signal transduction cascades, which, in turn, modify gene expression (33, 45, 59). Interestingly, it has already been shown that the cytoplasmic tail of MUC1 coimmunoprecipitates with transcription factors including HIF-1, p53, and β-catenin (7, 15, 22, 49), all associated with protective pathways that are activated during AKI.
β-Catenin is a unique dual function protein that plays a pivotal role in regulating the coordination of cell-cell adhesion and gene transcription (3, 28, 53). β-Catenin was first characterized as a central component of the adherens junction complex, where it connects E-cadherin to the actin cytoskeleton, thereby stabilizing the cell adherens junction and maintaining epithelial integrity (28, 29, 32). Different stimuli including metabolic stress induce the release of β-catenin from the adherens junction into the cytoplasm (39), where its abundance is controlled by a multiprotein complex called the “β-catenin destruction complex,” which includes GSK3β (34, 42, 44). GSK3β phosphorylates β-catenin on several NH2-terminal serine/threonine residues in unstimulated cells, leading to ubiquitin-mediated proteosomal degradation of β-catenin (26, 37). However, canonical Wnt signaling increases cytoplasmic accumulation of β-catenin by blocking its phosphorylation by GSK3β (5, 12). Consequently, stabilized β-catenin is translocated into the nucleus, where it modulates gene expression through direct binding to transcription factors including members of the T cell factor (TCF)/lymphoid enhancer-binding factor (LEF) family of transcription factors (TCF/LEF proteins) (41). Under normal conditions, β-catenin, through Wnt signaling, is involved in cell proliferation and differentiation; during metabolic stress conditions, Wnt/β-catenin signaling promotes epithelial cell survival through activation of several target genes that inhibit apoptosis, including Akt, an antiapoptotic protein, and survivin, which blocks caspase-3 and caspase-9 activation. Furthermore, Wnt/β-catenin signaling supports cell growth and recovery after exposure to stress by modulating the activity of transcription factors including p53 and TCF/LEF (18, 41, 60).
The results of studies in tumor cell lines have shown that β-catenin binding to a canonical site in the MUC1 cytoplasmic tail blocks β-catenin phosphorylation by GSK3β and thereby stabilizes β-catenin levels (15, 20). Published data now show that β-catenin is induced in all mouse kidney tubular segments including the proximal tubule after either IRI or folic acid injury, whereas either overexpression of β-catenin or Wnt activation of β-catenin is sufficient to protect renal human epithelial cells (HKC-8) against staurosporine-induced apoptosis (56, 60), indicating a protective role for β-catenin during AKI. However, additional studies are needed to fully elucidate how the β-catenin protective pathway is regulated in kidneys under metabolic stress conditions like IRI. As we already established that Muc1 is induced in the kidney during IRI (38), and published data have indicated that β-catenin is upregulated in the kidney after acute injury (19, 58, 60), we tested the hypothesis that Muc1 is protective during IRI through transactivation of the β-catenin protective pathway using Muc1 global KO mice and congenic control mice (see Fig. 1). We found that the induction of β-catenin and its nuclear targeting were absent in Muc1 KO mice in response to IRI. In accord with this observation, we found aberrant modulation of β-catenin targets in kidneys of Muc1 KO mice compared with control mice, including reduced levels of activated Akt (phosphorylated), survivin, transcription factor TCF4 and its target cyclin D1, and increased levels of p53, active Bax, and cleaved caspase-3, consistent with increased apoptosis. Altogether, our data are consistent with Muc1 enhancement of the β-catenin protective pathway during IRI.
Fig. 1.

Mucin (MUC)1 transactivation of the β-catenin pathway. Human MUC1 blocks glycogen synthase kinase (GSK)3β-dependent degradation of β-catenin in tumor cell lines and promotes cell survival and growth by 1) activating the prosurvival factor Akt [phosphorylated (p)Akt], which, in turn, mediates inhibition of proapoptotic factors (p53 and Bax); 2) enhancing transcriptional activity T cell factor (TCF)4, as evidenced by the induction of cyclin D1; and 3) increasing survivin, a member of the inhibitor of apoptosis protein family, as evidence by reduced cleaved caspase-3. We tested the hypothesis that Muc1 also enhances β-catenin activity in the mouse kidney during ischemia-reperfusion injury (IRI).
MATERIALS AND METHODS
Antibodies.
The Armenian hamster monoclonal anti-MUC1 cytoplasmic tail (CT2) antibody has been previously described (46). Mouse monoclonal antibodies against p53, GSK3β, and phospho-GSK3β, rabbit polyclonal antibody against Bax, and rabbit monoclonal antibodies against Akt, phospho-Thr473 Akt, TCF4, cyclin D1, survivin, and cleaved caspase-3 were purchased from Cell Signaling Technology (Danvers, MA). Mouse monoclonal antibodies against active Bax were purchased from Enzo Life Sciences (Farmingdale, NY). Mouse monoclonal antibodies against β-catenin were from BD Biosciences (Franklin Lakes, NJ). Mouse monoclonal antibody B27.29 against MUC1 TR antibody was purchased from Fujirebio Diagnostics (Malvern, PA). Mouse monoclonal antibodies against β-actin were purchased from Sigma (St. Louis, MO).
Mouse renal ischemia-reperfusion model.
All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Indiana University School of Medicine Animal Care and Use Committee. All experiments were conducted using 12-wk-old male C57BL/6J mice. Muc1 global KO mice on a C57BL/6J background were as previously described and are unremarkable under normal physiological conditions (50). AKI was induced as previously described using ischemia (19 min) followed by reperfusion (0, 24, or 72 h) (38, 51). Kidneys were harvested at the time of death after 0–72 h of recovery and subsequently processed for immunoblot and microscopy experiments as described below.
Immunohistochemical analysis.
One kidney from each mouse was flash frozen in liquid nitrogen, and one kidney was sliced lengthwise and then cut in half, yielding a quarter kidney (or one-half of a coronal section), and placed in 4% paraformaldehyde for analysis by microscopy. Fixed kidney samples were then embedded in paraffin to prepare slides (4 μm thick) and subjected to immunohistochemical staining. All images were collected using epifluorescence microscopy.
Immunoblot analysis of kidney extracts.
These experiments were performed according to previously published methods (38) and adapted as follows. The snap-frozen kidney quarter (∼0.4 mg) was homogenized and subjected to immunoblot analysis as previously described (2, 38). Bands were identified either with a Bio-Rad Versadoc or by exposure to Kodak BioMax MR film, and bands were quantified using Bio-Rad Quantity One software. Blots of samples from Muc1 KO and wild-type mouse kidneys were developed side by side for the same time period.
Isolation of nuclear and postnuclear fractions.
A quarter of a kidney was minced with a razor blade on ice and washed three times with ice-cold Dulbecco's PBS with calcium and magenesium (Corning Cellgro) containing Protease Inhibitor Cocktail Set III (Calbiochem). The tissue pellet was resuspended in 0.25 ml of the same buffer and homogenized by 20 strokes with a Dounce homogenizer on ice. The homogenate was centrifuged for 10 min at 1,000 g in the cold, and the nuclear pellet was resuspended in lysis buffer (1% SDS, 10 mM EDTA, and 50 mM Tris·HCl, pH 8) and sonicated in a Bioruptor to shear DNA. Aliquots (60 μg) of the nuclear fraction (1.7% total) and the postnuclear supernatant (3% total) were subjected to immunoblot analysis for β-catenin. Immunoblots of aliquots from control C57BL/6 mouse kidneys were developed with Perkin-Elmer Western Lightning Plus ECL. As levels of β-catenin in Muc1 KO mouse kidneys were significantly lower than control C57BL/6 mouse kidneys (refer to data shown in Fig. 2A), the immunoblots of fractions from Muc1 KO mouse kidneys were developed with ThermoScientific SuperSignal West Femto to improve any chance of finding β-catenin in the nuclear fraction.
Fig. 2.
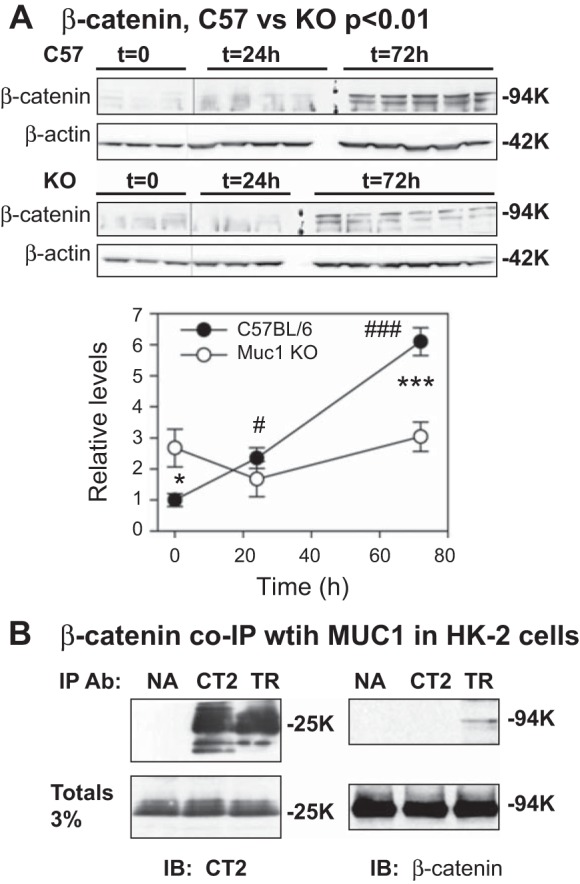
Induction of β-catenin during IRI is Muc1 dependent. Kidneys of Muc1 knockout (KO) mice and control C57BL/6 mice were subjected to 19 min of ischemia and recovery for 0, 24, or 72 h (n = 3–6 mice). A: extracts of kidney homogenates were analyzed by immunoblot analysis for β-catenin, and data were normalized to β-actin loading controls. The level for control mice (C57BL/6) at time (t) = 0 was set at 1, and data are presented as means ± SE. Levels of β-catenin were significantly increased in control mice at 24 h (#P < 0.05) and 72 h (###P < 0.001) of recovery, whereas levels in KO mice were unchanged with time and different from levels in control mice at t = 0 (*P < 0.05) and 72 h of recovery (***P < 0.001). Data analyzed by two-way ANOVA indicate that there was a significant difference in β-cateinin profiles between KO and control mice (P < 0.01). B: β-catenin coimmunoprecipitates (IP) with MUC1 in extracts of cultured human kidney cells (HK-2 cells). Extracts were incubated with protein G-conjugated beads and either no antibody (NA), anti-MUC1 cytoplasmic tail (CT2) antibody, or anti-MUC1 tandem repeat (TR) antibody B27.29. IPs or an aliquot of total extract were immunoblotted (IB) for MUC1 (CT2) or β-catenin.
Cell culture.
HK-2 cells, a proximal tubular cell line derived from the normal kidney, were obtained from the American Type Culture Collection (www.atcc.org). Cells were used at passages 25–29 and maintained at 37°C in a humidified 5% CO2-95% air incubator in DMEM-F-12 medium (Sigma) supplemented with 5 μg/ml insulin, 0.02 μg/ml dexamethasone, 0.01 μg/ml selenium, 0.05 μg/ml transferrin, 2 mM l-glutamine, 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Coimmunoprecipitation experiments.
Extracts of cultured HK-2 cells were incubated with protein G-conjugated beads and either anti-MUC1 CT2 antibody or anti-MUC1 TR antibody (TR antibody was B27.29). Immunoprecipitation in the absence of the antibody (NA) was also performed as a control. After SDS-PAGE and transfer to a nitrocellulose membrane, immunoblot analysis for the expression of β-catenin was first performed using mouse anti-β-catenin antibody. The membrane was then stripped and reprobed using anti-MUC1 CT2 antibody to confirm similar protein expression and loading of the gel for the different conditions. An aliquot of the total extract was also immunoblotted for MUC1 (CT2) or β-catenin.
Statistical analyses.
Data were analyzed for statistical significance with SigmaStat software using two-way ANOVA with a Tukey post hoc test, with P < 0.05 considered significantly different between mouse strains (Muc1 KO vs. C57BL/6 control mice) or between individual time points. All data are reported as means ± SE.
RESULTS
β-Catenin induction during renal IRI is Muc1 dependent.
We have already established that Muc1 is induced in the kidney and plays a protective role in a mouse model of IRI through stabilization of HIF-1α, one of the major adaptive responses to AKI (38). In addition, previously published data have indicated that β-catenin, part of another major adaptive response, is also upregulated in the kidney after acute injury (19, 58, 60). Using the same tissues from our previous study, we assessed β-catenin levels in kidneys of Muc1 KO mice and congenic control mice in response to IRI. Both renal pedicles of mice were clamped for 19 min before a recovery period of 0–72 h, when mice were euthanized and kidneys were harvested to assess protein levels of β-catenin. We have previously reported that serum creatinine was significantly increased at 24 h in control mice and returned to normal at 72 h, whereas serum creatinine did not return to normal in Muc1 KO mice at 72 h (38). Immunoblots of kidney homogenates revealed that β-catenin was significantly increased 2.4-fold in control mice by 24 h (P < 0.05) and 6.1-fold by 72 h (P < 0.001) during IRI, but β-catenin levels in Muc1 KO mice were not significantly changed during IRI (0–72 h of recovery; Fig. 2A). β-Catenin profiles were significantly different between Muc1 KO and control mice, as indicated by two-way ANOVA (P < 0.01), consistent with a role for Muc1 in the induction of β-catenin during IRI (Fig. 2A).
To directly assess the interaction of Muc1 with β-catenin in the kidney, we tested whether β-catenin coimmunoprecipitates with MUC1 using an established human kidney cell line (HK-2 cells) (43). MUC1 coimmunoprecipitation with β-catenin has been previously described in a pancreatic tumor cell line (57). We used antibodies that recognize either the MUC1 cytoplasmic tail (CT2 antibody) or MUC1 TRs (TR antibody). Although the MUC1 small subunit (25K) was similarly immunoprecipitated with either CT2 or TR antibody, β-catenin was found only in the anti-TR immunoprecipitate, indicating that β-catenin binding likely blocked access to the cytoplasmic tail epitope near the β-catenin-binding site (Fig. 2B). The CT2 antibody was prepared against a peptide representing the last 17 residues of the MUC1 cytoplasmic tail (72 residues), including the SSLS residues in the β-catenin canonical binding site (SxxxxxSSLS59) (46).
As previously published data have shown that MUC1 prevents β-catenin phosphorylation by GSK3β and thereby β-catenin degradation in tumor cell lines (15, 20), we asked if Muc1 might stabilize β-catenin during IRI by modulating GSK3β activity. We immunoblotted kidney homogenates with an antibody that detects either phosphorylated GSK3β (inactive) or total GSK3β. We found that while levels of total GSK3β did not change during IRI, levels of inactive GSK3β were significantly increased in both control mice (P < 0.05) and Muc1 KO mice (P < 0.01) by 72 h. However, the proportion of GSK3β that was inactive in control mice (the ratio of inactive phosphorylated to total GSK3β) was significantly higher than the profile in Muc1 KO mice during IRI (P < 0.01 by two-way ANOVA; Fig. 3). These data are consistent with Muc1-dependent stabilization of β-catenin during IRI by regulation of GSK3β activity.
Fig. 3.
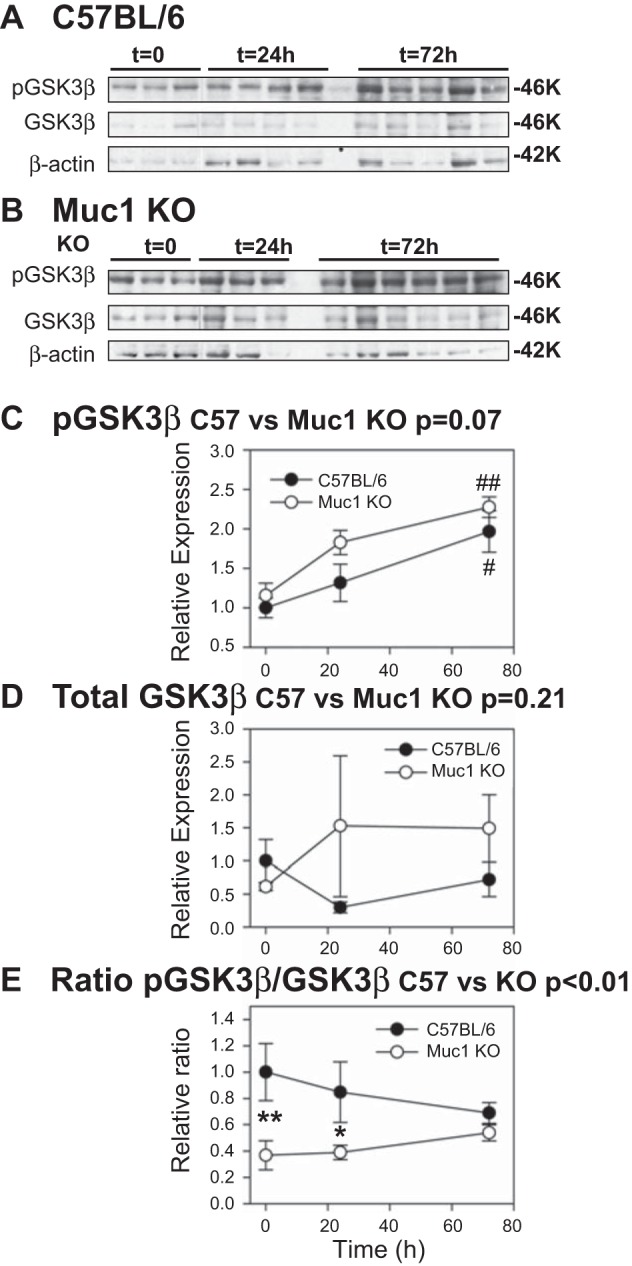
Activation of GSK3β is blocked by Muc1 during IRI. IRI was carried out as described in Fig 2. Kidney extracts from control C57BL/6 (A) and Muc1 KO mice (B) were subjected to immunoblot analysis with anti-pGSK3β antibodies to measure inactive enzyme, then anti-GSK3β antibodies to measure total enzyme, and finally with anti-β-actin antibodies as a loading control. Levels of pGSK3β normalized to β-actin (C) increased significantly for both C57BL/6 control mice (#P < 0.05) and Muc1 KO mice (##P < 0.01) by 72 h, whereas levels of total GSK3β normalized to β-actin (D) were not significantly different when analyzed by two-way ANOVA. However, the ratio of inactive to total enzyme (E), analyzed by two-way ANOVA, indicated that there was a significant difference in the profiles between Muc1 KO and control mice (P < 0.01) and that there were significant differences in the ratio between control and Muc1 KO mice at t = 0 (**P < 0.01) and t = 24 h (*P < 0.05). The level for control mice (C57BL/6) at t = 0 was set at 1 in all cases, and data are presented as means ± SE.
Nuclear localization of β-catenin during renal IRI is Muc1 dependent.
To modulate transcription, stabilized β-catenin needs to translocate into the nucleus, where it controls gene expression through direct binding to transcription factors including members of TCF/LEF factor family (41). To determine the subcellular localization of β-catenin during IRI in our mouse model, we carried out immunohistochemical staining of paraffin-embedded paraformaldehyde-fixed kidney tissue using antibodies against β-catenin. While tubule staining for β-catenin was not different between kidneys from Muc1 KO and control mice with no treatment, we did observe a notable increase of β-catenin staining in renal tubules from control mice at 72 h of recovery during IRI that was reduced in Muc1 KO mice (Fig. 4A). Cytoplasmic and nuclear staining of β-catenin was clearly evident in renal tubules from control mice at 72 h of recovery, whereas β-catenin was localized to the cytoplasm at the basolateral membrane of tubules from Muc1 KO mice after ischemia (Fig. 4A). Furthermore, immunoblot analysis of isolated nuclei and postnuclear supernatants from kidney homogenates clearly revealed β-catenin in the nuclei of control mice at 72 h of recovery (Fig. 4B). In contrast, we found no β-catenin in the nuclei from Muc1 KO mouse kidneys by immunblot analysis, even when using a more sensitive reagent to develop the blot (enhanced image in Fig. 4B). Altogether, these data support the hypothesis that Muc1 plays a key role in regulating the stability and nuclear localization of β-catenin during AKI.
Fig. 4.
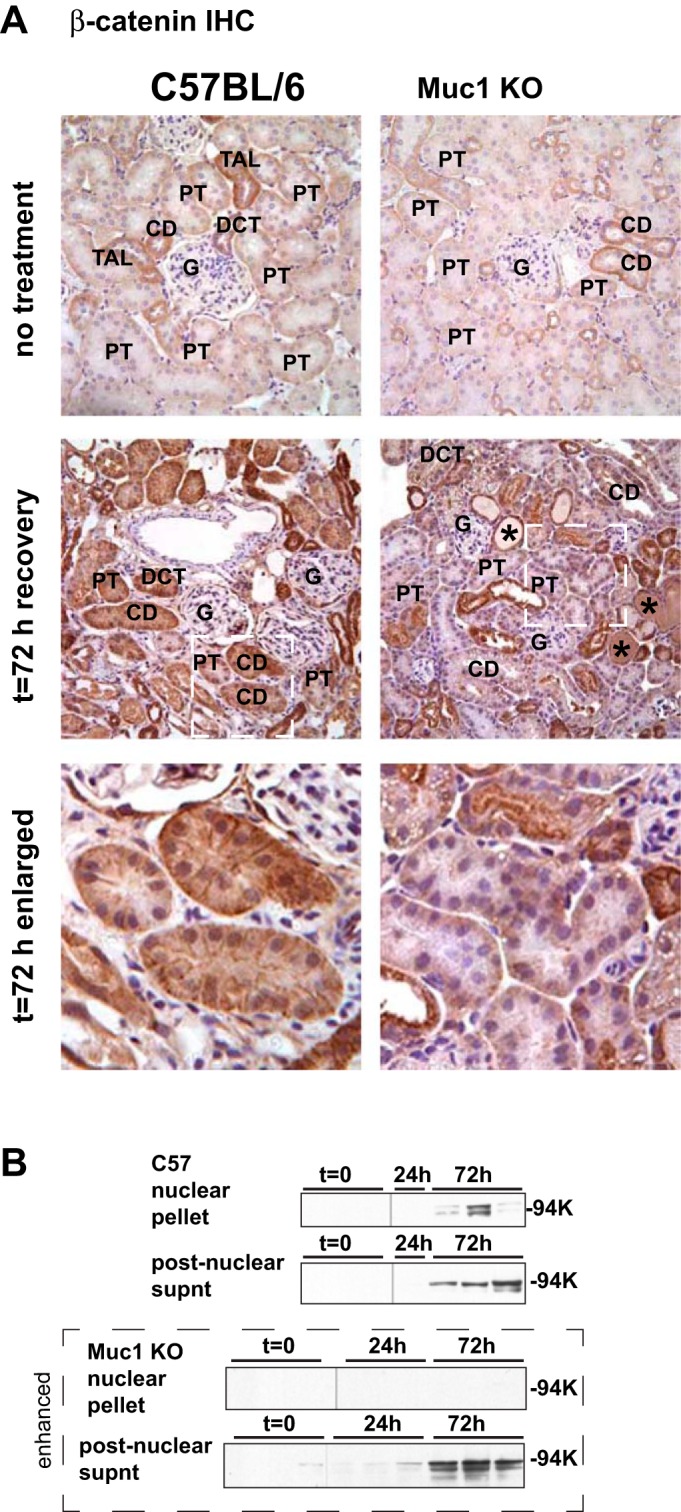
Nuclear targeting of β-catenin during IRI is Muc1 dependent. A: immunohistochemical (IHC) staining of kidneys at 72 h of recovery during IRI with anti-β-catenin antibodies revealed more overall staining and more nuclear staining in kidneys of control C57BL/6 mice than in Muc1 KO mice. Anti-β-catenin staining is brown; nuclear counterstain is blue. Images are shown for the kidney cortex from untreated (top) and 72-h recovered (middle) mice. The white dashed boxes in 72-h images are enlarged as indicated (bottom). Examples of casts (*), glomerulus (G), proximal tubules (PT), thick ascending limb (TAL), and collecting duct (CD) are indicated based on morphology. B: aliquots of nuclear extracts and postnuclear supernatants of some mouse kidneys were immunoblotted for β-catenin. Blots of aliquots from Muc1 KO mouse kidneys were enhanced by development with a more sensitive substrate than that used for developing the blots of aliquots from control mice (C57).
Muc1 regulates β-catenin activity during IRI.
We previously reported that Muc1 enhances the HIF-1 protective pathway in a mouse model of IRI through stabilization of HIF-1α and induction of expression from HIF-1 target genes (38). To understand if Muc1 affects transcription of the downstream targets of β-catenin in kidneys during IRI, we first assessed localization of the transcription factor TCF4 in Muc1 KO and control mouse kidneys during IRI. TCF4 is a member of the TCF/LEF factor family and the ultimate mediator of Wnt/β-catenin signaling (41). We found a significant increase of staining for TCF4 by immunohistochemistry in renal tubules from control mice at 24–72 h of recovery compared with Muc1 KO mice (Fig. 5). More significantly, we observed nuclear staining for TCF4 at 24 h of recovery in control mouse kidneys that was absent in Muc1 KO mouse kidneys.
Fig. 5.
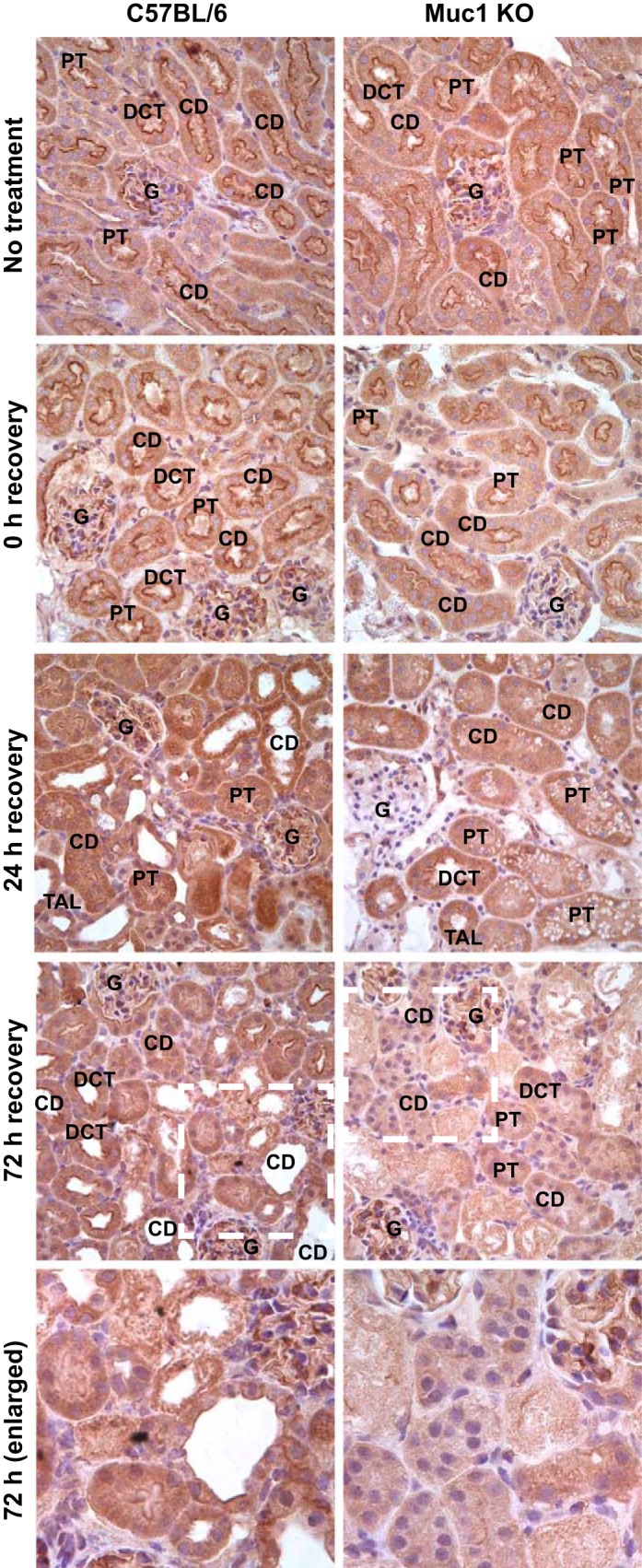
TCF4 levels and nuclear targeting during IRI are Muc1 dependent. Kidneys of Muc1 KO mice and control C57BL/6 mice were subjected to 19 min of ischemia and recovery for 0, 24, or 72 h or no treatment. Kidneys were fixed in paraformaldehyde and stained with anti-TCF4 antibodies using IHC. Anti-TCF4 staining is brown; nuclear counterstain is blue. Examples of the glomerulus (G), PT, TAL, DCT, and CD are indicated based on morphology. The white dashed boxes in 72-h images are enlarged as indicated.
To assess TCF4 activity in kidneys during IRI, tissue was subjected to immunohistochemistry using antibodies against cyclin D1, a downstream target of TCF4. We found that both cytoplasmic and nuclear levels of cyclin D1 were substantially higher in kidneys of control mice by 72 h of recovery during IRI compared with kidneys from Muc1 KO mice (Fig. 6A). This was confirmed by finding significantly higher levels of cyclin D1 in immunoblots of homogenates of control mouse kidneys during IRI compared with Muc1 KO kidneys (P < 0.001 by two-way ANOVA; Fig. 6B). Moreover, levels of cyclin D1 in kidney homogenates of Muc1 KO mice were unchanged and significantly lower (3-fold) than cyclin D1 levels in control kidneys at 72 h of recovery (P < 0.001; Fig. 6B). Altogether, these results indicate that Muc1 specifically stabilizes β-catenin, enhances its nuclear translocation during IRI, and augments its transcriptional activity, as indicated by the upregulation of TCF4 and its downstream target cyclin D1.
Fig. 6.
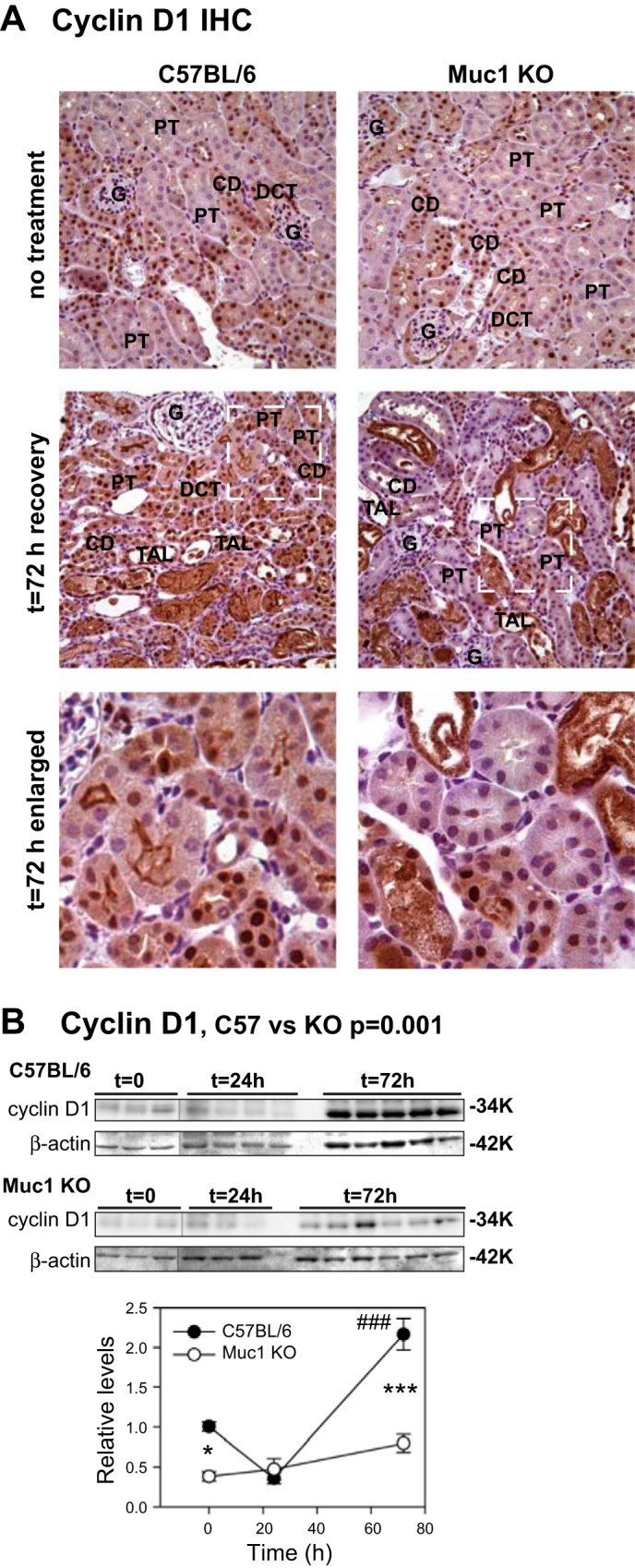
Induction of cyclin D1 during IRI is Muc1 dependent. IRI was carried out as described in Fig 2. A: IHC staining of kidneys at 72 h of recovery during IRI with anti-cyclin D1 antibodies revealed more overall staining and more nuclear staining in kidneys of control C57BL/6 mice than in Muc1 KO mice. Antibody staining is brown; nuclear counterstain is blue. Examples of the glomerulus (G), PT, TAL, DCT, and CD are indicated based on morphology. B: kidney extracts were immunblotted for cyclin D1 and β-actin as loading control. The level for control mice (C57BL/6) at t = 0 was set at 1, and data are presented as means ± SE. Levels of cyclin D1 were significantly increased in control mice by 72 h (###P < 0.001), whereas levels in KO mice were unchanged and different from control mice at t = 0 (*P < 0.05) and t = 72 h (***P < 0.001). Data analyzed by two-way ANOVA indicated a significantly reduced level of cyclin D1 in kidneys of Muc1 KO mice compared with kidneys of control mice (P < 0.001).
Muc1-dependent stabilization of β-catenin inhibits tubular cell apoptosis during IRI.
The cumulative published data have indicated that the protective action of β-catenin is attributed to the activation of prosurvival factors (phospho-Akt, survivin, and cyclin D1) and/or suppression of proapoptotic genes (p53, Bax, and caspase-3) (see Fig. 1) (56, 60). To fully elucidate how the β-catenin protective pathway is regulated in kidneys under metabolic stress conditions like IRI and to determine if efficient induction of β-catenin targets in our mouse model is protective during IRI, we immunoblotted kidney homogenates for the following β-catenin downstream targets: activated Akt, survivin, p53, active Bax, and cleaved caspase-3. Whereas levels of total Akt were significantly increased 2.2-fold in Muc1 KO mouse kidneys by 72 h (P < 0.05), levels of phosphorylated Akt were significantly increased 9.7-fold in control mouse kidneys at 24 h (P < 0.001), such that the levels of activated Akt (the ratio of active phosphorylated to total Akt) was significantly increased 10.4-fold by 24 h of recovery during IRI in control mice (P < 0.01; Fig. 7). The profile of activated Akt in Muc1 KO mice was clearly lower compared with the profile in control mice during IRI (P = 0.065 by two-way ANOVA; Fig. 7). Moreover, the level of activated Akt in Muc1 KO mice at 24 h of recovery was significantly lower compared with the level in control mice (P < 0.05; Fig. 7).
Fig. 7.
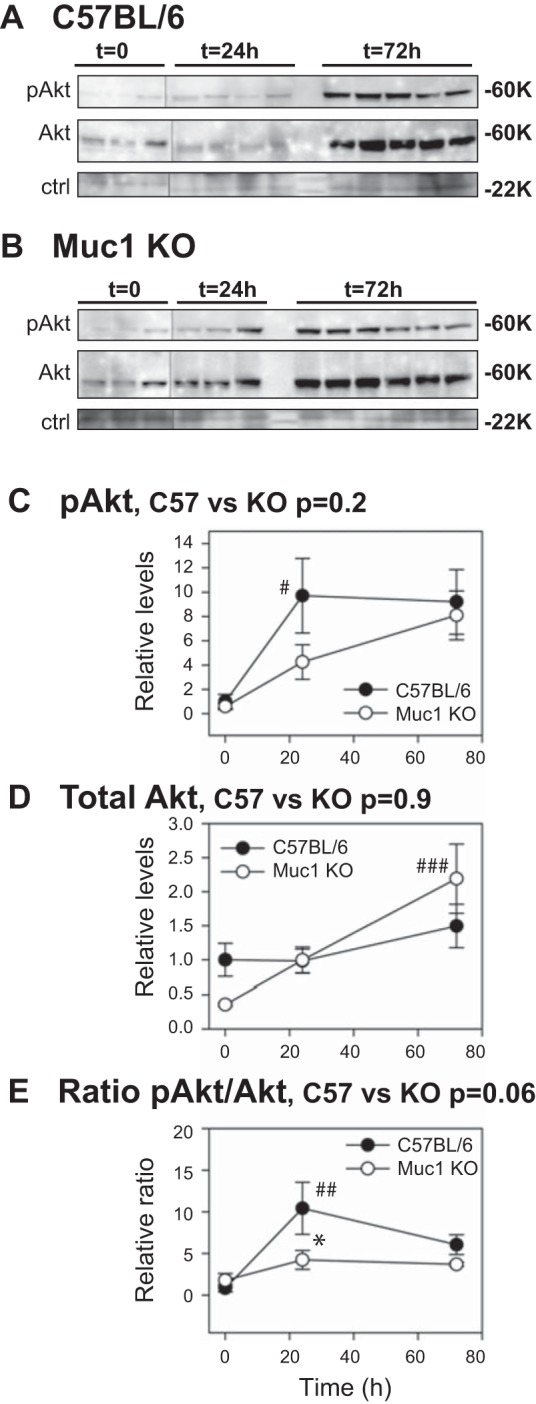
Activation of Akt is significantly reduced during IRI in the absence of Muc1. IRI was carried out as described in Fig 2. Kidney extracts from control C57BL/6 (A) and Muc1 KO mice (B) were immunoblotted for activated pAkt and then total Akt. A nonspecific band (ctrl, 22 kDa) was used as a loading control to normalize the pAkt data (C) and total Akt data (D). The normalized data at t = 0 for control mice (C57BL/6) was set at 1, and all data are presented as means ± SE. Data revealed a significant increase in total Akt in Muc1 KO kidneys at 72 h (###P < 0.001) and an increase in pAkt in control kidneys at 24 h (#P < 0.05), such that the ratio of activated to total Akt was significantly increased in control kidneys at 24 h (##P < 0.01) and significantly higher than the ratio in Muc1 KO kidneys (*P < 0.05). The overall profile for the ratio of activated to total Akt (P = 0.06) by two-way ANOVA is consistent with a blunted activation of Akt in Muc1 KO mouse kidneys during IRI.
We also found that the level of survivin, a member of the inhibitor of apoptosis protein family (10), in control mice at 72 h of recovery was higher compared with the level in Muc1 KO mice (P < 0.05; Fig. 8A). However, normalized data (survivin profiles) analyzed by two-way ANOVA indicated an approach to significance (P = 0.07). Levels of cleaved caspase-3, a downstream target of survivin, were significantly increased 8.5-fold in Muc1 KO mice by 24 h of recovery during IRI (P < 0.001; Fig. 8B). Whereas levels of cleaved caspase-3 in control kidneys were not significantly increased during IRI, levels were significantly lower from Muc1 KO kidneys only at 24 h of recovery (P < 0.001; Fig. 8B).
Fig. 8.
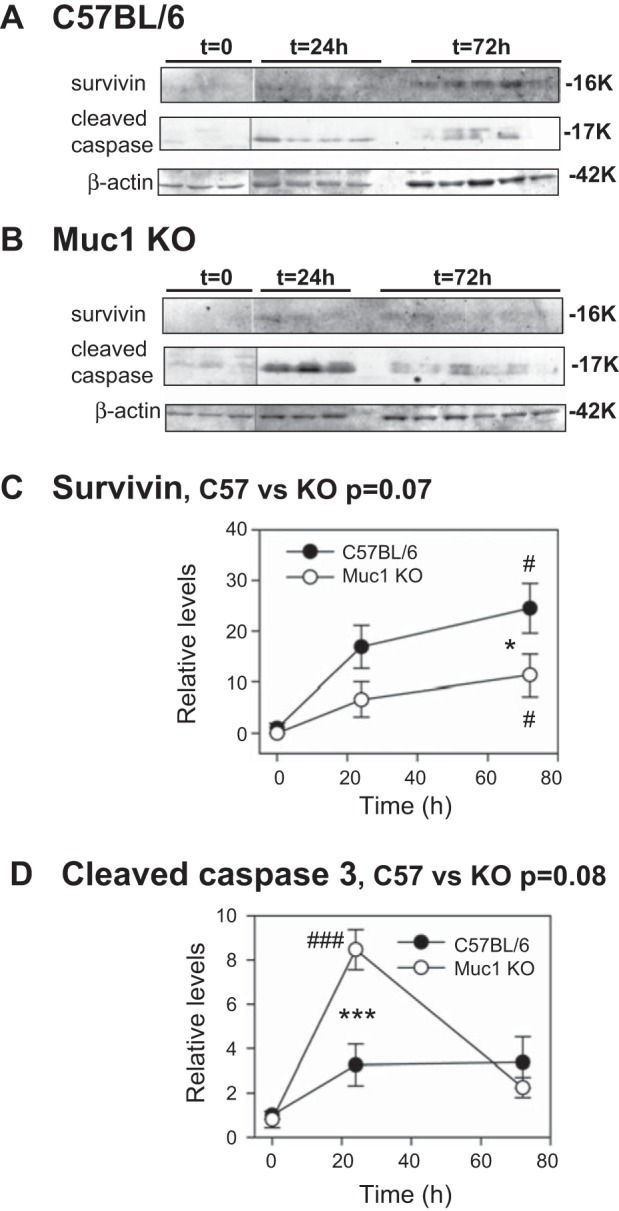
Levels of survivin and cleaved caspase-3 are significantly different during IRI in the absence of Muc1. IRI was carried out as described in Fig 2. Kidney extracts from control C57BL/6 mice (A) and Muc1 KO mice (B) were immunblotted for survivin, cleaved caspase-3, and β-actin as a loading control (same blot as Fig. 6). The level for control mice (C57BL/6) at t = 0 was set at 1 in each case, and data are presented as means ± SE. Levels of survivin were significantly increased by 72 h for both control and Muc1 KO mice (#P < 0.05) but were significantly less for Muc1 KO mice (*P < 0.05). Levels of cleaved caspase-3 were significantly increased by 24 h in Muc1 KO mice (###P < 0.001) and significantly higher than in control mice (***P < 0.001). The normalized data analyzed by two-way ANOVA indicated an approach to significance for a difference between profiles from control and Muc1 KO mice for levels of survivin (P = 0.07) and cleaved caspase-3 (P = 0.08).
We also observed a significant and sustained increase in p53 levels, an upstream regulator of Bax, in kidney homogenates of Muc1 KO mice compared with control kidneys (P < 0.01 by two-way ANOVA; Fig. 9A). In contrast, p53 levels in control kidneys were slightly but not significantly increased at 24 h of recovery and decreased at 72 h of recovery as well as significantly lower than levels in Muc1 KO mouse kidneys (P < 0.05; Fig. 9A). Moreover, we found that while levels of total BAX did not change during IRI, levels of active BAX (a proapoptotic form) were significantly higher in Muc1 KO mice compared with control mice (P < 0.05 by two-way ANOVA), such that the ratio of active to total BAX was also significantly different (P < 0.01 by two-way ANOVA; Fig. 9, B–F). The ratio of active to total BAX was also significantly different at time 0 and 72 h of recovery (P < 0.01 and P < 0.05).
Fig. 9.

Levels of p53 and active BAX are increased during IRI in the absence of Muc1. IRI was carried out as described in Fig 2. Kidney extracts were immunblotted for p53 (A) and β-actin as a loading control (same blot as Fig. 6) as well as active BAX, total BAX, and β-actin as loading control (same blot as Fig. 3; B–E). The level for control mice (C57BL/6) at t = 0 was set at 1 in each case, and data are presented as means ± SE. Levels of p53 were not changed during IRI in control or Muc1 KO mice but were significantly different at 72 h (*P < 0.05). Profiles of p53 for control and Muc1 KO mice were significantly different when analyzed by two-way ANOVA (P < 0.01). Levels of active BAX (D) were significantly different in control and Muc1 KO mice when analyzed by two-way ANOVA (P < 0.05), whereas levels of total BAX (E) were not different (P = 0.09). The ratio of active to total BAX was significantly different in control and Muc1 KO mice when analyzed by two-way ANOVA (P < 0.05), and the ratio was significantly different at t = 0 (**P < 0.01) and t = 72 h (*P < 0.05).
Our data are consistent with Muc1 protection of the kidney during IRI by enhancing the β-catenin protective pathway, as evidenced by Muc1-dependent activation of Akt, increased expression of survivin, TCF4, and its downstream target cyclin D1, and decreased levels of p53, active Bax, and cleaved caspase-3.
DISCUSSION
Several studies have shown that kidney survival and recovery during AKI is dependent on the β-catenin protective pathway (19, 56, 60). β-Catenin is a unique dual function protein that plays a key role in the coordination of cell-cell adhesion at the adherens junctions as well as modulation of canonical Wnt signaling through transactivation of gene transcription (3, 28). Wnt/β-catenin signaling plays a key role in cell survival, growth, and apoptosis by 1) modulating cascade pathways involving Bax and the serine/threonine kinase Akt, 2) modulating activity of transcription factors including p53 and TCF/LEF, and 3) inducing survivin, which blocks caspase-3 and caspase-9 activation (18, 30, 60).
Using a conditional KO mouse model, Zhou et al. (60) have shown that β-catenin plays a pivotal renoprotective role during AKI by balancing survival and apoptotic pathways. There is also evidence that pharmacological inhibition of GSK3β, a major upstream negative regulator of β-catenin, or RNA interference-mediated knockdown of GSK3β in different animal models (mouse and rats) and cell cultures of proximal tubule cells reduces tubular injury and promotes the recovery of renal epithelial cells (19, 56). Interestingly, in these studies (using both animals and cell cultures), the protective action of β-catenin was attributed to the activation of prosurvival factors (Akt, cyclin D1, and survivin) and/or suppression of proapoptotic genes (p53, caspase-3, and Bax; see Fig. 1) (56, 60).
Our group had previously reported that the HIF-1 protective pathway is enhanced by nuclear targeting of the cell surface glycoprotein Muc1 in a mouse kidney model of IRI. We found that levels of HIF-1α and expression of HIF-1 target genes associated with a shift in glucose metabolism to glycolysis were reduced in Muc1 KO mice during IRI. Consequently, Muc1 KO mice had reduced kidney function and more tubule damage with increased metabolic stress compared with control mice, consistent with a key role for Muc1 in protection against tubular cell injury during AKI (38). In the present study, we show, in the same model, that Muc1 specifically stabilizes β-catenin, enhances its nuclear translocation during IRI, and augments expression of its downstream targets. Our data are consistent with Muc1 protection of the kidney during IRI by enhancing the β-catenin protective pathway, as evidenced by the activation of prosurvival factors like activated Akt (phosphorylated) and the TCF/LEF target cyclin D1 and survivin as well as repression of proapoptotic factors such as p53, Bax, and cleaved caspase-3 (consistent with decreased apoptosis). In the future, it will be important to evaluate how Muc1/β-catenin modulates these prosurvival and proapoptotic factors and if it is at transcriptional or posttranscriptional levels. It would also be important to elucidate the specific role and mechanism of β-catenin signaling during kidney injury, recovery, and repair.
Taken together, the results presented in this study indicate that regulation of β-catenin during IRI is clearly Muc1 dependent. However, the reduction in the level of cleaved caspase-3 at 72 h of recovery in Muc1 KO mice likely reflects multiple pathways involved during IRI in regulating caspase-3 activation, which has been shown to be cellular context dependent (21). In addition, the reduction of cyclin D1 level even below the basal expression at 24 h of recovery in control mouse kidney epithelial cells might be explained by the cell entering into a cell cycle arrest phase due to the severe damage that occurred at this time point based on our published observations in the same model (38).
It is increasingly clear that apoptosis and not necrosis correlate with the severity of organ impairment and significantly contributes to an abrupt decline in kidney function (16). Our present data support the protective action of β-catenin and can be attributed to the activation of prosurvival factors (Akt, cyclin D1, and survivin) and suppression of proapoptotic genes (p53, Bax, and caspase-3) during IRI. Therefore, understanding the Muc1/β-catenin protective pathway will provide additional new targets for the development of therapies to treat patients with AKI.
In summary, we have established that Muc1 is protective during IRI through transactivation of the β-catenin protective pathway.
GRANTS
This work was supported by National Institutes of Health Grants DK-099345 (to T. A. Sutton), DK-084184 (to N. M. Pastor-Soler), CA-64389 (to S. J. Gendler), DK-62277, DK-100287, and DK-095498 (to S. P. Monga), DK-097889 (to M. M. Al-bataineh), DK107632 (to R. P. Hughey), DK-079307 (Pilot Project to R. P. Hughey), and P30-DK-079307 (to the Microscopy Core, N. M. Pastor-Soler, and S.I. Bastacky) as well as a Samuel and Emma Winters Foundation grant (to R. P. Hughey).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.M.A.-b., T.A.S., and R.P.H. conception and design of research; M.M.A.-b., C.L.K., P.A.P., T.A.S., H.E.M., S.I.B., S.J.G., C.S.M., S.S., and R.P.H. performed experiments; M.M.A.-b., C.L.K., P.A.P., N.M.P.-S., S.I.B., S.P.M., and R.P.H. analyzed data; M.M.A.-b., C.L.K., N.M.P.-S., S.P.M., and R.P.H. interpreted results of experiments; M.M.A.-b. and R.P.H. drafted manuscript; M.M.A.-b., N.M.P.-S., and R.P.H. edited and revised manuscript; M.M.A.-b., C.L.K., P.A.P., N.M.P.-S., T.A.S., H.E.M., S.I.B., S.J.G., C.S.M., S.S., S.P.M., and R.P.H. approved final version of manuscript; R.P.H. prepared figures.
REFERENCES
- 1.Ahmad R, Raina D, Joshi MD, Kawano T, Ren J, Kharbanda S, Kufe D. MUC1-C oncoprotein functions as a direct activator of the nuclear factor-kappaB p65 transcription factor. Cancer Res 69: 7013–7021, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-bataineh MM, Gong F, Marciszyn AL, Myerburg MM, Pastor-Soler NM. Regulation of proximal tubule vacuolar H+-ATPase by PKA and AMP-activated protein kinase. Am J Physiol Renal Physiol 306: F981–F995, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol 10: 468–477, 2009. [DOI] [PubMed] [Google Scholar]
- 4.Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet 380: 756–766, 2012. [DOI] [PubMed] [Google Scholar]
- 5.Brennan K, Gonzalez-Sancho JM, Castelo-Soccio LA, Howe LR, Brown AM. Truncated mutants of the putative Wnt receptor LRP6/Arrow can stabilize β-catenin independently of Frizzled proteins. Oncogene 23: 4873–4884, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brezis M, Rosen S. Hypoxia of the renal medulla–its implications for disease. N Engl J Med 332: 647–655, 1995. [DOI] [PubMed] [Google Scholar]
- 7.Chaika NV, Gebregiworgis T, Lewallen ME, Purohit V, Radhakrishnan P, Liu X, Zhang B, Mehla K, Brown RB, Caffrey T, Yu F, Johnson KR, Powers R, Hollingsworth MA, Singh PK. MUC1 mucin stabilizes and activates hypoxia-inducible factor 1α to regulate metabolism in pancreatic cancer. Proc Natl Acad Sci USA 109: 13787–13792, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chertow GM, Burdick E, Honour M, Bonventre JV, Bates DW. Acute kidney injury, mortality, length of stay, and costs in hospitalized patients. J Am Soc Nephrol 16: 3365–3370, 2005. [DOI] [PubMed] [Google Scholar]
- 9.Coca SG, Yusuf B, Shlipak MG, Garg AX, Parikh CR. Long-term risk of mortality and other adverse outcomes after acute kidney injury: a systematic review and meta-analysis. Am J Kidney Dis 53: 961–973, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deveraux QL, Reed JC. IAP family proteins–suppressors of apoptosis. Genes Dev 13: 239–252, 1999. [DOI] [PubMed] [Google Scholar]
- 11.Golestaneh L, Melamed ML, Hostetter TH. Uremic memory: the role of acute kidney injury in long-term outcomes. Kidney Int 76: 813–814, 2009. [DOI] [PubMed] [Google Scholar]
- 12.Gordon MD, Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem 281: 22429–22433, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Haase VH. Mechanisms of hypoxia responses in renal tissue. J Am Soc Nephrol 24: 537–541, 2013. [DOI] [PubMed] [Google Scholar]
- 14.Hansen MK, Gammelager H, Jacobsen CJ, Hjortdal VE, Layton JB, Rasmussen BS, Andreasen JJ, Johnsen SP, Christiansen CF. Acute kidney injury and long-term risk of cardiovascular events after cardiac surgery: a population-based cohort study. J Cardiothorac Vasc Anesth 29: 617–625, 2015. [DOI] [PubMed] [Google Scholar]
- 15.Hattrup CL, Gendler SJ. Structure and function of the cell surface (tethered) mucins. Annu Rev Physiol 70: 431–457, 2008. [DOI] [PubMed] [Google Scholar]
- 16.Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int 80: 29–40, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heyman SN, Evans RG, Rosen S, Rosenberger C. Cellular adaptive changes in AKI: mitigating renal hypoxic injury. Nephrol Dial Transplant 27: 1721–1728, 2012. [DOI] [PubMed] [Google Scholar]
- 18.Hoppler S, Kavanagh CL. Wnt signalling: variety at the core. J Cell Sci 120: 385–393, 2007. [DOI] [PubMed] [Google Scholar]
- 19.Howard C, Tao S, Yang HC, Fogo AB, Woodgett JR, Harris RC, Rao R. Specific deletion of glycogen synthase kinase-3β in the renal proximal tubule protects against acute nephrotoxic injury in mice. Kidney Int 82: 1000–1009, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang L, Chen D, Liu D, Yin L, Kharbanda S, Kufe D. MUC1 oncoprotein blocks glycogen synthase kinase 3β-mediated phosphorylation and degradation of β-catenin. Cancer Res 65: 10413–10422, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Kasten MM, Giordano A. pRb and the cdks in apoptosis and the cell cycle. Cell Death Different 5: 132–140, 1998. [DOI] [PubMed] [Google Scholar]
- 22.Kufe DW. Mucins in cancer: function, prognosis and therapy. Nat Rev Cancer 9: 874–885, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Levitin F, Stern O, Weiss M, Gil-Henn C, Ziv R, Prokocimer Z, Smorodinsky NI, Rubinstein DB, Wreschner DH. The MUC1 SEA module is a self-cleaving domain. J Biol Chem 280: 33374–33386, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Li Y, Ren J, Yu W, Li Q, Kuwahara H, Yin L, Carraway KL 3rd, Kufe D. The epidermal growth factor receptor regulates interaction of the human DF3/MUC1 carcinoma antigen with c-Src and β-catenin. J Biol Chem 276: 35239–35242, 2001. [DOI] [PubMed] [Google Scholar]
- 25.Liano F, Pascual J. Epidemiology of acute renal failure: a prospective, multicenter, community-based study. Madrid Acute Renal Failure Study Group. Kidney Int 50: 811–818, 1996. [DOI] [PubMed] [Google Scholar]
- 26.Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of β-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108: 837–847, 2002. [DOI] [PubMed] [Google Scholar]
- 27.Macao B, Johansson DG, Hansson GC, Hard T. Autoproteolysis coupled to protein folding in the SEA domain of the membrane-bound MUC1 mucin. Nat Struct Mol Biol 13: 71–76, 2006. [DOI] [PubMed] [Google Scholar]
- 28.MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell 17: 9–26, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandel LJ, Doctor RB, Bacallao R. ATP depletion: a novel method to study junctional properties in epithelial tissues. II. Internalization of Na+,K+-ATPase and E-cadherin. J Cell Sci 107: 3315–3324, 1994. [DOI] [PubMed] [Google Scholar]
- 30.Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404: 782–787, 2000. [DOI] [PubMed] [Google Scholar]
- 31.Mehta RL, Pascual MT, Soroko S, Savage BR, Himmelfarb J, Ikizler TA, Paganini EP, Chertow GM; Program to Improve Care in Acute Renal Disease. Spectrum of acute renal failure in the intensive care unit: the PICARD experience. Kidney Int 66: 1613–1621, 2004. [DOI] [PubMed] [Google Scholar]
- 32.Monga SP. β-Catenin signaling and roles in liver homeostasis, injury, and tumorigenesis. Gastroenterology 148: 1294–1310, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mukherjee P, Tinder TL, Basu GD, Gendler SJ. MUC1 (CD227) interacts with lck tyrosine kinase in Jurkat lymphoma cells and normal T cells. J Leukoc Biol 77: 90–99, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Munemitsu S, Albert I, Souza B, Rubinfeld B, Polakis P. Regulation of intracellular β-catenin levels by the adenomatous polyposis coli (APC) tumor-suppressor protein. Proc Natl Acad Sci USA 92: 3046–3050, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murugan R, Kellum JA. Acute kidney injury: what's the prognosis? Nat Rev Nephrol 7: 209–217, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nash K, Hafeez A, Hou S. Hospital-acquired renal insufficiency. Am J Kidney Dis 39: 930–936, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Orford K, Crockett C, Jensen JP, Weissman AM, Byers SW. Serine phosphorylation-regulated ubiquitination and degradation of β-catenin. J Biol Chem 272: 24735–24738, 1997. [DOI] [PubMed] [Google Scholar]
- 38.Pastor-Soler NM, Sutton TA, Mang HE, Kinlough CL, Gendler SJ, Madsen CS, Bastacky SI, Ho J, Al-Bataineh MM, Hallows KR, Singh S, Monga SP, Kobayashi H, Haase VH, Hughey RP. Muc1 is protective during kidney ischemia-reperfusion injury. Am J Physiol Renal Physiol 308: F1452–F1462, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Price VR, Reed CA, Lieberthal W, Schwartz JH. ATP depletion of tubular cells causes dissociation of the zonula adherens and nuclear translocation of β-catenin and LEF-1. J Am Soc Nephrol 13: 1152–1161, 2002. [DOI] [PubMed] [Google Scholar]
- 40.Quin RJ, McGuckin MA. Phosphorylation of the cytoplasmic domain of the MUC1 mucin correlates with changes in cell-cell adhesion. Int J Cancer 87: 499–506, 2000. [DOI] [PubMed] [Google Scholar]
- 41.Rajabi H, Ahmad R, Jin C, Kosugi M, Alam M, Joshi MD, Kufe D. MUC1-C oncoprotein induces TCF7L2 transcription factor activation and promotes cyclin D1 expression in human breast cancer cells. J Biol Chem 287: 10703–10713, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science 272: 1023–1026, 1996. [DOI] [PubMed] [Google Scholar]
- 43.Ryan MJ, Johnson G, Kirk J, Fuerstenberg SM, Zager RA, Torok-Storb B. HK-2: an immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int 45: 48–57, 1994. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt-Ott KM, Barasch J. WNT/β-catenin signaling in nephron progenitors and their epithelial progeny. Kidney Int 74: 1004–1008, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schroeder JA, Adriance MC, Thompson MC, Camenisch TD, Gendler SJ. MUC1 alters β-catenin-dependent tumor formation and promotes cellular invasion. Oncogene 22: 1324–1332, 2003. [DOI] [PubMed] [Google Scholar]
- 46.Schroeder JA, Thompson MC, Gardner MM, Gendler SJ. Transgenic MUC1 interacts with epidermal growth factor receptor and correlates with mitogen-activated protein kinase activation in the mouse mammary gland. J Biol Chem 276: 13057–13064, 2001. [DOI] [PubMed] [Google Scholar]
- 47.Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell 148: 399–408, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Siew ED, Davenport A. The growth of acute kidney injury: a rising tide or just closer attention to detail? Kidney Int 87: 46–61, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singh PK, Hollingsworth MA. Cell surface-associated mucins in signal transduction. Trends Cell Biol 16: 467–476, 2006. [DOI] [PubMed] [Google Scholar]
- 50.Spicer AP, Duhig T, Chilton BS, Gendler SJ. Analysis of mammalian MUC1 genes reveals potential functionally important domains. Mamm Genome 6: 885–888, 1995. [DOI] [PubMed] [Google Scholar]
- 51.Sutton TA, Hato T, Mai E, Yoshimoto M, Kuehl S, Anderson M, Mang H, Plotkin Z, Chan RJ, Dagher PC. p53 is renoprotective after ischemic kidney injury by reducing inflammation. J Am Soc Nephrol 24: 113–124, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thadhani R, Pascual M, Bonventre JV. Acute renal failure. N Engl J Med 334: 1448–1460, 1996. [DOI] [PubMed] [Google Scholar]
- 53.Thompson MD, Monga SP. WNT/β-catenin signaling in liver health and disease. Hepatology 45: 1298–1305, 2007. [DOI] [PubMed] [Google Scholar]
- 54.Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, Tolwani A, Ronco C; Beginning and Ending Supportive Therapy for the Kidney (BEST Kidney) Investigators. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA 294: 813–818, 2005. [DOI] [PubMed] [Google Scholar]
- 55.Wang H, Lillehoj EP, Kim KC. Identification of four sites of stimulated tyrosine phosphorylation in the MUC1 cytoplasmic tail. Biochem Biophys Res Commun 310: 341–346, 2003. [DOI] [PubMed] [Google Scholar]
- 56.Wang Z, Havasi A, Gall J, Bonegio R, Li Z, Mao H, Schwartz JH, Borkan SC. GSK3β promotes apoptosis after renal ischemic injury. J Am Soc Nephrol 21: 284–294, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wen Y, Caffrey TC, Wheelock MJ, Johnson KR, Hollingsworth MA. Nuclear association of the cytoplasmic tail of MUC1 and β-catenin. J Biol Chem 278: 38029–38039, 2003. [DOI] [PubMed] [Google Scholar]
- 58.Xiao L, Zhou D, Tan RJ, Fu H, Zhou L, Hou FF, Liu Y. Sustained activation of Wnt/β-catenin signaling drives AKI to CKD progression. J Am Soc Nephrol; doi: 10.1681/ASN.2015040449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamamoto M, Bharti A, Li Y, Kufe D. Interaction of the DF3/MUC1 breast carcinoma-associated antigen and beta-catenin in cell adhesion. J Biol Chem 272: 12492–12494, 1997. [DOI] [PubMed] [Google Scholar]
- 60.Zhou D, Li Y, Lin L, Zhou L, Igarashi P, Liu Y. Tubule-specific ablation of endogenous β-catenin aggravates acute kidney injury in mice. Kidney Int 82: 537–547, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zrihan-Licht S, Baruch A, Elroy-Stein O, Keydar I, Wreschner DH. Tyrosine phosphorylation of the MUC1 breast cancer membrane proteins. Cytokine receptor-like molecules. FEBS Lett 356: 130–136, 1994. [DOI] [PubMed] [Google Scholar]