

Abstract
Cisplatin, a chemotherapeutic used for the treatment of solid cancers, has nephrotoxic side effects leading to acute kidney injury (AKI). Cisplatin cannot be given to patients that have comorbidities that predispose them to an increased risk for AKI. Even without these comorbidities, 30% of patients administered cisplatin will develop kidney injury, requiring the oncologist to withhold or reduce the next dose, leading to a less effective therapeutic regimen. Although recovery can occur after one episode of cisplatin-induced AKI, longitudinal studies have indicated that multiple episodes of AKI lead to the development of chronic kidney disease, an irreversible disease with no current treatment. The standard mouse model of cisplatin-induced AKI consists of one high dose of cisplatin (>20 mg/kg) that is lethal to the animal 3 days later. This model does not accurately reflect the dosing regimen patients receive nor does it allow for the long-term study of kidney function and biology. We have developed a repeated dosing model whereby cisplatin is given once a week for 4 wk. Comparison of the repeated dosing model with the standard dosing model demonstrated that inflammatory cytokines and chemokines were induced in the repeated dosing model, but levels of cell death were lower in the repeated dosing model. The repeated dosing model had increased levels of fibrotic markers (fibronectin, transforming growth factor-β, and α-smooth muscle actin) and interstitial fibrosis. These data indicate that the repeated dosing model can be used to study the AKI to chronic kidney disease progression as well as the mechanisms of this progression.
Keywords: acute kidney injury, chronic kidney disease, cisplatin, fibrosis, nephrotoxicity
acute kidney injury (AKI), the rapid loss of kidney function, has many medical complications, a high mortality rate, and limited therapeutic interventions beyond palliative care (45). One of the most prominent causes of AKI is pharmaceutical-induced nephrotoxicity, which accounts for 19% of all cases of AKI (44). Cisplatin is a potent chemotherapeutic used for the treatment of many cancers but induces AKI in 30% of patients even in the absence of comorbidities such as advanced age or preexisting kidney diseases (35). Clinically, cisplatin is given at low doses in multiple rounds to try to avoid nephrotoxic side effects, but even with this precaution in place, AKI still occurs (3). Blood urea nitrogen (BUN) and serum creatinine (SCr) are clinical tests used to monitor kidney function but are highly insensitive (9, 10). Elevated BUN or SCr levels in patients during the course of cisplatin treatment requires that the dose of cisplatin be lowered or delayed or, alternatively, that the patient be switched to a potentially less effective chemotherapeutic that lacks nephrotoxic side effects (3). None of these options are favorable and can result in a less efficacious cancer treatment.
Until recently, it was assumed that patients that survive AKI and don't require dialysis are able to achieve full recovery of kidney function (23). However, recent large-scale longitudinal studies that assessed the impact of AKI on long-term renal function have indicated that patients that had AKI are more likely to develop CKD and end-stage renal disease (ESRD) than patients with no history of AKI. Furthermore, the occurrence of CKD/ESRD was directly proportional to the level and frequency of AKI experienced by these patients (1, 11, 12, 16, 28–30, 42). Additionally, the incidence of AKI and CKD/ESRD has significantly increased in the past decade (16, 27, 47), as the overall age of our population is also increasing (18). This is of importance because kidney function declines with normal aging even in the absence of obvious kidney disease (2). The majority of individuals receiving nephrotoxic chemotherapeutics are middle aged or older and already have increased exposure to renal stressors, enhanced susceptibility to injury, and decreased ability to repair after injury. With improved diagnosis and treatment of cancers, there is also increased long-term patient survival. Hence, the percentage of cancer survivors expected to develop CKD/ESRD will increase, placing a major burden on patient quality of life and our health care system.
Gaining an understanding of the cellular processes involved in the development of CKD after cisplatin-induced AKI would be useful for developing renoprotective agents. Unfortunately, the standard dosing mouse model of AKI does not allow for long-term studies. In the standard dosing model, a single high dose of cisplatin (>20 mg/kg) is administered once to 8- to 10-wk-old mice. This dose is lethal to the mouse beyond 72 h, and, as a result, examining the AKI to CKD transition is not possible. Furthermore, this model does not accurately represent the repeated dosing regimen used in the clinic. Thus, there is a need for a more clinically relevant mouse model that enables the study of the cellular processes involved in the progression of cisplatin-induced AKI to CKD.
We have developed a model of cisplatin-induced kidney injury that reflects the repeated low dosing of cisplatin used in the clinic and allows for detailed analysis of repair, recovery, and long-term kidney function. Here, mice were treated with 7 mg/kg cisplatin once a week for 4 wk and euthanized 3 days after the last injection. We compared this dosing regimen with mice that were treated according to the standard dosing model of AKI (one dose of 25 mg/kg cisplatin and euthanized 3 days later). We analyzed markers of kidney function and injury, inflammatory cytokines and chemokines, indicators of endoplasmic reticulum (ER) stress and cell death, and profibrotic indicators. While the standard dosing model of AKI and the repeated dosing model have similarities in their effects on kidney function, kidney damage is less severe in the repeated dosing model, enabling mice to survive beyond the 24-day course of treatment. Data also indicated that interstitial fibrosis occurred in the repeated dosing model but not in the standard dosing model. Data suggested that the increased incidence of CKD after cisplatin-induced AKI may be a result of repeated injury leading to fibrosis.
MATERIALS AND METHODS
Reagents and antibodies.
The following antibodies were purchased from Cell Signaling (Beverly, MA) unless otherwise noted: cleaved caspase-3 (no. 9664), C/EEBP homologous protein (CHOP; no. 2895), JNK (no. 9258), phosphorylated (p-)JNK (no. 4668), transforming growth factor (TGF)-β (no. 3712S), fibronectin (F3648, Sigma-Aldrich, St. Louis, MO), p-SMAD3 (no. 12747), α-tubulin (SC-23948, Santa Cruz Biotechnology, Dallas, TX), and β-actin (SC-47778, Santa Cruz Biotechnology). Cisplatin (P4394, Sigma-Aldrich) was used for experiments comparing the effects of a high single dose (euthanization 3 days later) with the new repeated dosing model. Pharmaceutical grade cisplatin (purchased directly from the University of Louisville hospital pharmacy) was used for experiments comparing the effects of single versus repeated injury from cisplatin. Similar effects were observed for both sources of cisplatin.
Animals.
FVB/n mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were maintained on a 12:12-h light-dark cycle and provided food and water ad libitum. Animals were maintained under standard laboratory conditions. All animal procedures were approved by the Institutional Animal Care and Use Committee of the University of Louisville and followed the guidelines of the American Veterinary Medical Association. Cisplatin at 25 mg/kg in PBS (200 μl/animal) was administered by an intraperitoneal injection. Seventy-two hours after cisplatin injection, these mice were euthanized. Another cohort of mice received either 7 or 9 mg/kg cisplatin administered by an intraperitoneal injection once a week for 4 wk. For these survival experiments, mice were monitored for weight loss and signs of discomfort/distress on a daily basis. Mice exhibiting a weight loss of 20% or more total body weight or high levels of discomfort and stress were euthanized. Mice that survived the course of treatment were euthanized 72 h after the fourth injection of cisplatin. Serum was prepared and stored at −80°C. The kidneys were flash frozen in liquid nitrogen or fixed in 10% neutral buffered formalin.
BUN and SCr determination.
BUN (DIUR-500) and SCr (C7548-120) were determined using kits from Bioassay Systems (Hayward, CA) and Point Scientific (Canton, MI), respectively, following the manufacturers' instructions. For SCr, this specific assay kit uses a two-reagent enzymatic assay system to eliminate interference by endogenous creatine and ascorbic acid.
Protein quantification, Western blot analysis, and ELISAs.
Homogenates were made from the kidney cortex by homogenization in cell extraction buffer [10 mM Tris·HCl (pH 8.0), 150 mM NaCl,1 mM imidazole, 1 mM magnesium acetate, 20 mM EGTA, and 10 mM β-mercaptoethanol] containing a Complete Protease Inhibitor Cocktail Tablet and Phosphatase Inhibitor Cocktail Tablets (Roche, Indianapolis, IN). Homogenates were centrifuged at 15,000 g for 10 min at 4°C. Supernatants were removed, aliquoted, and stored at −80°C until use. Protein concentrations were determined using Bradford Reagent (Bio-Rad, Hercules, CA). Kidney homogenate (40 μg) was separated on 4–12% gradient Tris-glycine-SDS polyacrylamide gels and transferred to polyvinylidene difluoride membranes, which were blocked with 5% (wt/vol) dried milk in Tris-buffered saline containing 0.1% Tween 20 (TBST) for 1 h. Membranes were incubated with 1:5,000 dilutions of primary antibody overnight at 4°C. The next morning, membranes were washed three times for 5 min each with TBST containing 5% (wt/vol) dried milk. After incubation for 1 h at room temperature with secondary antibodies conjugated with horseradish peroxidase [1:50,000 in TBST containing 1.25% (wt/vol) dried milk], membrane proteins were detected by chemiluminiscence substrate. ELISAs for kidney injury molecule (KIM)-1 (DY1817, R&D Systems, Minneapolis, MN) and neutrophil gelatinase-associated lipocalin (NGAL; DY1857, R&D Systems) were performed on the urine as directed by the manufacturer.
Gene expression.
Total RNA was isolated using RNA-STAT 60 (TEL-TEST, Friendswood, TX) combined with mini-bead-beater glass beads and a Mini Bead Beater machine (Cole-Palmer, Vernon Hills, IL) following the manufacturer's protocol. cDNA was made from 1 μg total RNA using High-Capacity Reverse Transcriptase (Life Technologies, Grand Island, NY) following the manufacturer's instructions. Gene-specific cDNAs were quantitated using real-time PCR and predesigned TaqMan assays. TNF-α (Mm00443258_m1), IL-6 (Mm00446190_m1), IL-1β (Mm0043228_m1), chemokine (C-X-C motif) ligand (CXCL)1 (Mm04207460_m1), monocyte chemotactic protein (MCP)-1 (Mm00441242_m1), plasminogen activator inhibitor (PAI)-1 (Mm00435860_m1), α-smooth muscle actin (α-SMA; Mm1546133_m1), bone morphogenetic protein (BMP)-7 (Mm00432102_m1), collagen type IV α1-chain (Col4α1; Mm01210125_m1), cyclin-dependent kinase inhibitor (CDK)N2A (Mm00491449-m1), connective tissue growth factor (Mm01192932_g1), collagen type I-α1 (Col1α1; Mm00801666_g1), and normalization genes [β2-microglubilin (Mm00437762_m1) and β-actin (Mm01205647-g1)] were purchased from Life Technologies and used in combination with 2× Gene Expression Master Mix (Life Technologies).
Histology.
Kidney sections (5 μm thick) from cisplatin-treated and untreated animals were stained with hematoxylin and eosin as well as periodic acid-Schiff, and the degree of morphological changes was determined by light microscopy in a blinded fashion by a renal pathologist. The following measures were chosen as an indication of morphological damage to the kidney after treatment with vehicle or cisplatin: proximal tubular necrosis, loss of the brush border, proximal tubule degradation, tubular casts, presence of inflammatory cells, and interstitial fibrosis. These measures were evaluated on a scale from 0 to 4, which ranged from not present (0) to mild (1), moderate (2), severe (3), and very severe (4).
Immunohistochemistry.
Kidney sections (5 μm thick) were rehydrated in Histoclear followed by an ethanol gradient. Antigen retrieval was performed in citric acid buffer (pH 6.0) at 95°C in a steamer for 30 min. Endogenous peroxidases were inhibited with 3% hydrogen peroxide and dual endogenous enzyme blocker (Dako) for 10 min followed by two 5-min PBS washes. Slides were then blocked with avidin for 10 min followed by a PBS wash and then biotin for 10 min followed by a wash in PBS (Dako). Slides were further blocked with 5% normal goat serum in 0.1% TBST for 1 h at room temperature. α-SMA primary rabbit antibody (Abcam) was added to slides at a concentration of 0.5 μg/ml and allowed to incubate at 4°C overnight. Slides were rinsed with PBS for 5 min, three times. Biotinylated goat anti-rabbit IgG antibody (1: 25,000, BA-1000, Vector Laboratories) was added to each section and incubated for 30 min at room temperature. Slides were rinsed twice with PBS (5 min each). Vector ABC reagent (PK-7100, Vector Laboratories) was added to each section and incubated for 30 min at room temperature. Slides were rinsed two times with PBS followed by the addition of 100 µl of DAB substrate for 5-7 min to detect horseradish peroxidase (SK-4800, Vector Laboratories). Slides were rinsed in distilled water for 5 min, counterstained with modified Mayer's hematoxylin (no. 72804, Thermo Scientific), and then dehydrated in an ethanol gradient to Histoclear followed by mounting with Permount (SP15, Fisher Scientific). Positive staining for α-SMA was quantified using Metamorph Image Analysis software, and the percentage of positive pixels was calculated as follows: [threshold area/(total area − acellular area)].
Sirius red/fast green staining.
Kidney sections (5 μm thick) were rehydrated in Histoclear followed by an ethanol gradient. Slides were then dipped into a Coplin jar containing PBS with 0.1% Tween 20 and incubated for 5 min. Slides were washed with distilled water twice for 5 min each and then incubated in saturated picric acid containing 0.1% Sirius red and 0.1% fast green. Sirius red/direct red 80 (catalog no. 365548) and fast green FCF (catalog no. F7258) were from Sigma, whereas picric acid [saturated ∼1.2% (wt/vol)] was from Ricca Chemicals (catalog no. 5860-32). Slides were washed with 5% glacial acetic water until the water ran clear. Tissue samples were then dehydrated and fixed using Permount (F-SP15-100, Fisher Scientific). Positive staining for Sirius red was quantified using Metamorph Image Analysis software, and the percentage of positive pixels was calculated as follows: [threshold area/(total area − acellular area)].
Statistical analysis.
Data are expressed as means ± SE for all experiments. Multiple comparisons of normally distributed data were analyzed by one-way ANOVA, as appropriate, and group means were compared using Tukey posttests. Single comparisons were analyzed by Student's t-test where appropriate. For statistical analysis of the survival curve, a log-rank (Mantel-Cox) test was used. The criterion for statistical differences was P < 0.05, P < 0.01, P < 0.001, and P < 0.0001.
RESULTS
Effects of dosing regimens on mouse survival.
The current model used to study cisplatin-induced AKI does not allow for the analysis of long-term effects on kidney function, nor does it recapitulate the repeated nature of the dosing regimen of cisplatin in the clinic. We hypothesized that administration of a low dose of cisplatin once a week for several weeks would be more clinically relevant and allow for the analysis of long-term effects on kidney function. To test this, we compared survival of mice given a single high dose of cisplatin (standard dosing model, 25 mg/kg) with mice given a dose of cisplatin (7 or 9 mg/kg) once a week for 4 wk. All mice subjected to the standard dosing regimen were euthanized 3 days after cisplatin injection due to moribund status. Mice treated with the 7 mg/kg repeated dosing regimen survived the course of treatment and were euthanized 3 days after the fourth treatment (day 24). Ninety percent of mice treated with the 9 mg/kg repeated dosing regimen survived until day 24 (Fig. 1). Statistical analyses comparing survival curves of 7- and 9 mg/kg-treated mice revealed no statistical significance, but there was statistical significance between repeated dosing and standard dosing survival curves (Fig. 1). These data indicate that the repeated dosing model with 7 mg/kg enables the survival of all treated mice for long-term studies of kidney function.
Fig. 1.

Survival curve of animals treated with the standard dosing model and repeated dosing model. Eight-week-old male FVB mice were injected (intraperitoneally) with saline vehicle or cisplatin (7 or 9 mg/kg) once a week for 4 wk (repeated dosing model) or with 25 mg/kg cisplatin given once (standard dosing model). Mice were monitored daily for weight loss and changes in overall well-being and were euthanized when moribund in accordance to Institutional Animal Care and Use Committee guidelines. At day 24, surviving mice were euthanized and analyzed.
Effects of dosing regimens on kidney injury and function.
To assess the impact of repeated dosing on the kidney, we measured markers of kidney function and injury in the serum and urine of mice, respectively. BUN levels of mice treated with the standard dosing model were significantly increased at 72 h posttreatment (Fig. 2A). In the repeated dosing model, BUN also increased, but not significantly (Fig. 2A). SCr levels were significantly increased for both the repeated and standard dosing models, but SCr levels were higher in the standard dosing model (Fig. 2A). Urinary KIM-1 and NGAL levels were examined, as they are more sensitive biomarkers of AKI than BUN and SCr (5, 6). Urinary KIM-1 and NGAL levels were significantly increased in the standard dosing model, but only NGAL levels were significantly increased in the repeated dosing model, albeit to a lesser extent than in the standard model (Fig. 2B). These data indicate that the repeated dosing model of cisplatin induced less kidney injury and less loss of kidney function compared with the standard dosing model.
Fig. 2.

Comparison of kidney function and injury between the standard dosing model and repeated dosing model. Eight-week-old male FVB mice were injected (intraperitoneally) with saline vehicle or 25 mg/kg cisplatin given once (standard dosing model) or with cisplatin (7 mg/kg) once a week for 4 wk (repeated dosing model). Animals were euthanized 72 h after the last injection. A: levels of blood urea nitrogen (BUN) and serum creatinine (SCr) measured in the serum. B: kidney injury molecule (KIM)-1 and neutrophil gelatinase-associated lipocalin (NGAL) measured in the urine. Data are expressed as means ± SE; n = 10. Statistical significance was determined by Student's t-test. **P < 0.01; ***P < 0.001.
Effects of dosing regimen on inflammatory cytokine and chemokine levels.
One component of the standard dosing model of AKI is a large inflammatory response (37, 41). We compared the levels of inflammatory cytokines and chemokines between the standard and repeated dosing models. TNF-α is a potent cytokine that mediates inflammatory tissue damage in the kidney and activates downstream cytokines and chemokines, particularly IL1-β, MCP-1, and IL-6 (21, 41). CXCL1 plays a role in neutrophil recruitment to sites of tissue inflammation (15). mRNA levels of TNF-α, IL-1β, MCP-1, and CXCL1 were increased significantly and approximately to the same extent in both the standard and repeated dosing models (Fig. 3, A and C–E). IL-6 mRNA was significantly increased in the standard dosing model but only increased 5.51 ± 2.01-fold in the repeated dosing model (Fig. 3B). These data suggest similar effects of both dosing regimens on inflammatory cytokines and chemokines.
Fig. 3.

Comparison of inflammatory markers between the standard dosing model and repeated dosing model. Eight-week-old male FVB mice were injected (intraperitoneally) with saline vehicle or 25 mg/kg cisplatin given once (standard model) or with cisplatin (7 mg/kg) once a week for 4 wk (repeated dosing model). Animals were euthanized 72 h after the last injection. mRNA levels of TNF-α (A), IL-6 (B), IL-1β (C), monocyte chemotactic protein (MCP)-1 (D), and chemokine (C-X-C motif) ligand (CXCL)1 (E) were assessed in the kidney cortex via real-time quantitative RT-PCR and were normalized to their vehicle control. Data are expressed as means ± SE; n = 10. Statistical significance was determined by Student's t-test. *P < 0.05; **P < 0.01; ***P < 0.001.
Effect of dosing regimen on activation of ER stress and cell death proteins.
Cell death and ER stress are characteristic of cisplatin-induced AKI. It is known that inhibition or deletion of key players in pathways of apoptosis or ER stress protects the kidney from cisplatin-induced injury in the standard dosing model (22, 25, 39). Therefore, we assessed cellular markers of ER stress and cell death proteins in both models. JNK phosphorylation and activation are associated with ER stress-induced apoptosis (37, 49). We found that p-JNK was elevated in both models (Fig. 4). CHOP is also associated with ER stress and was also activated in both models (Fig. 4) (14). However, cleaved caspase-3, a marker of apoptosis, was not increased in the repeated dosing model (Fig. 4). These data suggest that while both models show similar trends in activation of ER stress proteins, there may be less cell death activation in the repeated dosing model.
Fig. 4.
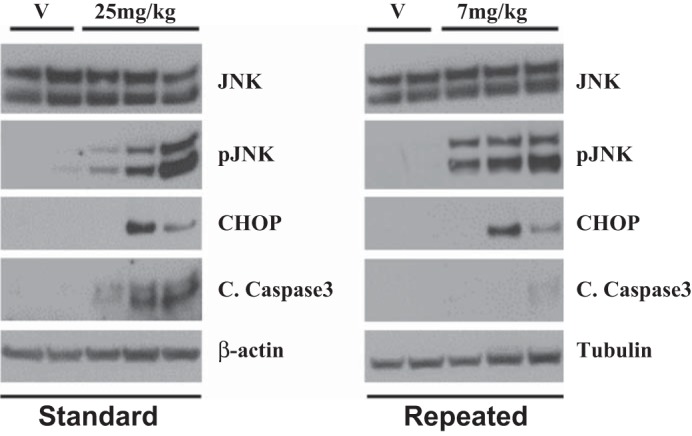
The repeated dosing model shows similar increases in markers of endoplasmic reticulum stress but not cell death markers compared with the standard dosing model. Eight-week-old male FVB mice were injected (intraperitoneally) with saline vehicle (V) or 25 mg/kg cisplatin given once (standard model) or with cisplatin (7 mg/kg) once a week for 4 wk (repeated dosing model). Animals were euthanized 72 h after the last injection. Markers of endoplasmic reticulum stress and cell death were assessed in kidney cortex homogenates via Western blot analysis. p-JNK, phosphorylated JNK; CHOP, C/EBP homologous protein; C caspase 3, cleaved caspase-3.
Effect of dosing regimen evident in tissue pathology.
The standard dosing model of cisplatin-induced AKI is associated with changes in kidney pathology. We compared kidney pathology of the standard and repeated dosing models by examining tubular necrosis, loss of brush borders, tubule dilation, cast formation, presence of inflammatory cells, and interstitial fibrosis, all of which are indicative of kidney injury and damage. Blinded analysis by a certified pathologist indicated that tubular necrosis was significantly higher in the standard dosing model compared with the repeated dosing model (Fig. 5A). In contrast, there was a significant loss of brush borders (Fig. 5B), an increase in tubular dilation (Fig. 5C), and an increase in cast formation (Fig. 5D) in both models. Interestingly, only the repeated dosing model displayed a significant increase in the presence of inflammatory cells and interstitial fibrosis (Fig. 5, E and F). These data demonstrate that there are key differences in kidney pathology between the standard and repeated dosing models.
Fig. 5.

Qualitative analysis of kidney pathology indexes. Renal histological changes were assessed on hematoxylin and eosin- and periodic acid-Schiff-stained kidney sections (5 μm thick). Eight-week-old male FVB mice were injected (intraperitoneally) with saline vehicle or cisplatin (7 mg/kg) once a week for 4 wk (repeated dosing model) or with 25 mg/kg cisplatin given once (standard dosing model). Animals were euthanized 72 h after the last injection. A: tubular necrosis. B: loss of proximal tubule brush borders. C: proximal tubule dilation. D: proximal tubule cast formation. E: presence of inflammatory cells. F: interstitial fibrosis. In A–F, scoring of the sections was performed in a blinded manner by a renal pathologist (J. Megyesi) using a scale of 0–4 (where 0 = not present, 1 = mild, 2 = moderate, 3 = severe, and 4 = very severe renal histological changes in proximal tubules). Data are means ± SE; n = 5–10. Statistical significance was determined by one-way ANOVA followed by Tukey's multiple-comparison test. ***P < 0.001.
Fibrotic markers and fibrosis in the repeated dosing model.
Since pathology of kidney sections revealed tubulointerstitial fibrosis in the repeated dosing model but not in the standard model, we examined known markers of fibrosis in this model. After kidney injury, TGF-β is released from immune cells (32, 37). TGF-β can then signal through its receptor, leading to the phosphorylation of SMAD3, thereby activating pathways that increase extracellular matrix protein deposition, particularly fibronectin (8, 40). BMP-7 is also a member of the TGF-β superfamily and works to counteract the profibrotic activity of TGF-β (33). TGF-β, p-SMAD3, and fibronectin were all increased at the protein level in the repeated dosing model (Fig. 6A). Conversely, BMP-7 mRNA expression was significantly decreased (Fig. 6B). Col1α1, which encodes for collagen type I protein and is a transcript marker of fibrosis, also significantly increased at the message level in the repeated dosing model (Fig. 6B) (4). PAI-1 is produced by resident and infiltrating inflammatory cells and inhibits fibrinolysis, leading to the accumulation of scar tissue in the kidney (20). mRNA expression of PAI-1 was increased in the repeated dosing model (Fig. 6B). CDKN2A encodes for p16, and increased expression of CDKN2A is associated with cell cycle arrest and cellular senescence (49). mRNA expression of CDKN2A was significantly increased after repeated dosing (Fig. 6B). Furthermore, collagen deposition as the result of extracellular matrix production can be quantified with Sirius red and fast green staining, which stains collagen red. We performed Sirius red and fast green staining and found that collagen levels increased in the kidneys after repeated dosing of cisplatin (Fig. 6C). α-SMA is a marker of myofibroblasts, which are known to deposit collagen (31, 34). α-SMA immunohistochemistry indicated increased myofibroblasts after repeated dosing of cisplatin (Fig. 6D). Taken together, these data indicate that there are alterations in key mediators of kidney fibrosis in the repeated dosing model.
Fig. 6.

Assessment of fibrosis and fibrotic markers. Eight-week-old male FVB mice were injected (intraperitoneally) with saline vehicle or cisplatin (7 mg/kg) once a week for 4 wk (repeated dosing model). Animals were euthanized 72 h after the last injection. A: markers of fibrosis in the kidney cortex assessed via Western blot analysis. TGF-β, transforming growth factor-β; p-Smad, phosphorylated Smad. B: measurement of mRNA levels of fibrotic markers in the kidney cortex as assessed via quantitative RT-PCR. PAI-1, plasminogen activator inhibitor-1; COL1A1, collagen type I-α1; CDKN2A, cyclin-dependent kinase inhibitor 2A (CDK)N2A; BMP-7, bone morphogenetic protein-7. C and D: Sirius red and fast green (SR/FG) staining of kidney sections and quantitation of staining (C) as well as α-SMA immunohistochemical staining in the kidney cortex and quantitation of staining (D). Data are expressed as means ± SE; n = 5–10. Statistical significance was determined by Student's t-test. *P < 0.05; **P < 0.01.
Comparison of a single low dose of cisplatin (7VVV) with the repeated dosing model.
To determine if fibrosis is a result of a single low dose of cisplatin or rather repeated injury from several low doses, mice were administered a low dose of cisplatin (7 mg/kg) followed by three weekly vehicle injections (7VVV) and compared with mice that received four weekly cisplatin injections (7777). BUN and SCr both increased significantly in the repeated dosing model but not after a single low dose of cisplatin (Fig. 7A). Likewise, mRNA expression levels of TNF-α and IL-6 increased with repeated dosing of cisplatin (7777) but not after a single dose (7VVV; Fig. 7C). Western blot analysis of TGF-β and fibronectin in the kidney indicated that a single dose was not sufficient to cause an increase in these fibrotic markers (Fig. 7B). mRNA expression of PAI-1, CDKN2A, and Col1α1 did not increase, and mRNA levels of BMP-7 did not decrease in the single low-dose regimen (Fig. 7C). Quantification of Sirius red and fast green staining for collagen deposition and immunohistochemical staining for α-SMA for myofibroblasts also indicated that a single low dose of cisplatin was insufficient to elicit changes (Fig. 7, D and E). These data suggest that fibrosis is a result of repeated injury to the kidney by several low doses of cisplatin.
Fig. 7.

Comparison of single low dose and repeated dosing. Eight-week-old male FVB mice were injected (intraperitoneally) with saline vehicle or cisplatin (7 mg/kg) once (VVVV and 7VVV, respectively) or with cisplatin (7 mg/kg; 7777) once a week for 4 wk (repeated dosing model). Animals were euthanized 72 h after the last injection. A: levels of BUN and SCr assessed in the serum. B: markers of kidney fibrosis assessed via Western blot analysis. C: mRNA levels of inflammatory cytokines and fibrotic markers in the kidney cortex as measured by qRT-PCR. D: SR/FG staining of kidney sections and quantification of staining. E: α-SMA immunohistochemical staining in the kidney and quantitation of staining. Statistical significance was determined by one-way ANOVA followed by Tukey's multiple-comparison test. *P < 0.05; **P < 0.01; ***P < 0.001.
DISCUSSION
Treatment of human cancers with cisplatin often leads to nephrotoxic side effects that are cumulative and dose dependent. AKI occurs in some individuals even after one low dose of cisplatin, and multiple episodes of AKI can cause CKD (13). Whereas Kobayashi et al. (31) performed repeated cisplatin dosing in mice to determine circadian changes related to drug administration, in the present study, we developed a model to study the effects of multiple “hits” of cisplatin-induced AKI and the subsequent development of CKD. The standard dosing model of cisplatin-induced AKI has limitations that cannot be overcome for studying the AKI to CKD progression. For one, a single high-dose regimen is not clinically relevant. Patients are administered multiple low doses of cisplatin over an extended period of time. Second, the standard dosing model cannot be aged out to study long-term effects associated with repeated AKI, namely, fibrosis, the underlying pathology of CKD (7). In the present study, we report a model for studying the nephrotoxic effects of cisplatin that mimics the repeated administration of cisplatin given clinically and allows for analysis of long-term effects on kidney function. Data obtained from this model indicated that repeated cisplatin injury induces interstitial fibrosis and suggest that targeting fibrotic mediators may prevent both short- and long-term renal side effects of cisplatin.
The standard dosing model of cisplatin induces high levels of kidney injury and cell death through apoptosis and necrosis. This, in turn, results in a rapid loss of kidney function. With the repeated dosing model, cleaved caspase-3 as a measure of apoptosis is low, and pathology reveals a low level of tubular necrosis. This translates to lower injury levels and a smaller decline in overall kidney function. These lower levels of injury and the maintenance of kidney function with the repeated dosing model may be key to explaining how mice treated with multiple low doses of cisplatin are able to survive for 24 days and perhaps even beyond that.
In the standard dosing model of cisplatin-induced AKI there is a strong inflammatory response, which involves TNF-α elevation and elevation of its downstream targets. In the repeated dosing model, a similar inflammatory response is observed. However, IL-6 expression is not as elevated in the repeated dosing model. IL-6 plays a role in mounting an effective immune response and has been indicated in AKI. Particularly, IL-6 expression is correlated with the onset and severity of AKI and has been indicated as a potential urinary and plasma biomarker of AKI (19, 36, 50). The low levels of IL-6 mRNA measured in the repeated dosing model may be indicative of a less severe form of AKI and help explain why less injury is occurring in this model.
Pathology indicates that there is a significant increase in infiltrating immune cells in the repeated dosing model, despite less injury. It has been shown that rapid increase in the macrophage population results in the development of fibrosis (48). Macrophages are known to play a major role in mounting an effective repair response postinjury and have also been indicated in maladaptive repair (24). Whereas M2 macrophages play a role in normal repair, an increase in M1 macrophages has been associated with maladaptive repair (24). Future studies of this model will focus on identifying the type of infiltrating immune cells, whether the M1 to M2 transition is inhibited in macrophages, and whether this inhibition of phenotype change is responsible for the onset of fibrosis.
The repeated administration of low-dose cisplatin induces fibrosis, and this is a physiologically relevant process that could be targeted therapeutically. Grgic et al. (26) have shown that kidney function can recover after a single round of injury induced by diphtheria toxin to transgenic mice expressing the diphtheria toxin receptor in proximal tubule cells. However, repeated injury in this model culminated in fibrosis, as determined by increased levels of TGF-β1, fibronectin, and Col1α1 (26). In our model of repeated cisplatin dosing, we also found increased protein levels of TGF-β and fibronectin and increased mRNA expression of Col1α1.
G2/M cell cycle arrest, cellular senescence, and fibrosis have been indicated in the ischemia-reperfusion and unilateral ureteral obstruction mouse models (49). We found increased mRNA expression of CDKN2A, which is suggestive of cellular senescence. Furthermore, p-JNK has been indicated as a downstream target of G2/M cell cycle arrest and is believed to also play a role in promoting renal fibrosis (49). While further studies are needed to determine whether or not G2/M arrest is indeed occurring after repeated cisplatin dosing, we did observe an increase in p-JNK without evidence of apoptosis. Thus, the increase in p-JNK may be indicative of G2/M arrest occurring and, as a result, potentially maladaptive repair. Taken together, these data suggest that potential mechanisms such as senescence and G2/M arrest warrant future indepth investigation in this model to identify the mechanisms by which fibrosis is induced.
Fibrosis plays a major role in our repeated dosing model and is indicative that this model can be used to perform extensive mechanistic studies of the AKI to CKD progression. For example, studies should be completed to determine the type and role for infiltrating immune cells in fibrosis in the kidney. Furthermore, it would also be worthwhile to examine kidney function, injury, fibrosis, and inflammatory markers throughout the course of cisplatin treatment rather than just at the end of the repeated dosing regimen. This would provide further insights into when fibrosis occurs temporally. Along with determination of the temporal timeline by which fibrosis occurs, structural studies looking at remodeling of the extracellular matrix during this process would provide insights into the morphological processes that occur within kidney tissue. Fortunately, Torres et al. (43), through the application of clearing multiphoton microscopy, have gained new, detailed morphological insights into the pathophysiology involved in the AKI to CKD transition. While Torres et al. (43) used a dosing regimen consisting of only two relatively high doses of cisplatin, they found that significant remodeling of the extracellular matrix was occurring, although they did not document major increases in collagen levels.
Fibrosis not only plays a role in CKD but also has been indicated in cancer metastasis (17). Our repeated cisplatin dosing model could be adapted to a cancer model to also look at the fibrosis both in the kidneys and cancer. Pabla et al. (38) have shown that repeated administration of 10 mg/kg cisplatin for 4 wk does alter the tumor size in a cancer xenograft model and also leads to an increase in BUN and SCr levels, indicating loss of kidney function. While they did not look at fibrosis in this model, it would be interesting to determine whether cancer-associated fibrosis occurs with our repeated dosing model. In the present study, we used mice on the FVB background rather than the C57BL/6 mouse strain as mice on the C57BL/6 background are extremely resistant to developing interstitial fibrosis (46). In addition, numerous transgenic mouse models of cancer are available on the FVB background for studying the effects of repeated low-dose cisplatin in mice with cancers that develop in the proper tumor microenvironment. Thus, the data presented in this study serve as the foundation for future studies aiming to determine the impact of repeated cisplatin dosing on the tumor and the kidney. Perhaps by further elucidating the mechanism by which fibrosis occurs, we can find new, desirable targets for development of both renoprotective and cancer therapeutics.
GRANTS
Support for this work was provided by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-093462 (to L.J. Siskind).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: C.N.S., M.A.D., T.V.D., L.J.B., and L.J.S. conception and design of research; C.N.S., M.A.D., T.V.D., P.P.S., M.S., D.S., J.M., L.J.B., and L.J.S. performed experiments; C.N.S., M.A.D., T.V.D., P.P.S., G.E.A., J.M., L.J.B., and L.J.S. analyzed data; C.N.S., M.A.D., T.V.D., P.P.S., L.J.B., and L.J.S. interpreted results of experiments; C.N.S., M.A.D., L.J.B., and L.J.S. prepared figures; C.N.S., M.A.D., L.J.B., and L.J.S. drafted manuscript; C.N.S., M.A.D., P.P.S., L.J.B., and L.J.S. edited and revised manuscript; C.N.S., M.A.D., T.V.D., P.P.S., M.S., D.S., G.E.A., J.M., L.J.B., and L.J.S. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank all members of the laboratories of L. J. Siskind and L. J. Beverly for the constructive criticism and support.
REFERENCES
- 1.Amdur RL, Chawla LS, Amodeo S, Kimmel PL, Palant CE. Outcomes following diagnosis of acute renal failure in U.S. veterans: focus on acute tubular necrosis. Kidney Int 76: 1089–1097, 2009. [DOI] [PubMed] [Google Scholar]
- 2.Anderson S, Eldadah B, Halter JB, Hazzard WR, Himmelfarb J, Horne FM, Kimmel PL, Molitoris BA, Murthy M, O'Hare AM, Schmader KE, High KP. Acute kidney injury in older adults. J Am Soc Nephrol 22: 28–38, 2011. [DOI] [PubMed] [Google Scholar]
- 3.Bennis Y, Savry A, Rocca M, Gauthier-Villano L, Pisano P, Pourroy B. Cisplatin dose adjustment in patients with renal impairment, which recommendations should we follow? Int J Clin Pharm 36: 420–429, 2014. [DOI] [PubMed] [Google Scholar]
- 4.Bielesz B, Sirin Y, Si H, Niranjan T, Gruenwald A, Ahn S, Kato H, Pullman J, Gessler M, Haase VH, Susztak K. Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest 120: 4040–4054, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonventre JV. Kidney injury molecule-1 (KIM-1): a specific and sensitive biomarker of kidney injury. Scand J Clin Lab Invest Suppl 241: 78–83, 2008. [DOI] [PubMed] [Google Scholar]
- 6.Bonventre JV. Kidney injury molecule-1 (KIM-1): a urinary biomarker and much more. Nephrol Dial Transplant 24: 3265–3268, 2009. [DOI] [PubMed] [Google Scholar]
- 7.Boor P, Floege J. Chronic kidney disease growth factors in renal fibrosis. Clin Exp Pharmacol Physiol 38: 441–450, 2011. [DOI] [PubMed] [Google Scholar]
- 8.Branton MH, Kopp JB. TGF-β and fibrosis. Microbes Infect 1: 1349–1365, 1999. [DOI] [PubMed] [Google Scholar]
- 9.Breyer MD, Qi Z. Better nephrology for mice–and man. Kidney Int 77: 487–489, 2010. [DOI] [PubMed] [Google Scholar]
- 10.Briggs JD, Kennedy AC, Young LN, Luke RG, Gray M. Renal function after acute tubular necrosis. Br Med J 3: 513–516, 1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bydash JR, Ishani A. Acute kidney injury and chronic kidney disease: a work in progress. Clin J Am Soc Nephrol 6: 2555–2557, 2011. [DOI] [PubMed] [Google Scholar]
- 12.Chawla LS, Amdur RL, Amodeo S, Kimmel PL, Palant CE. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int 79: 1361–1369, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med 371: 58–66, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Brandizzi F. IRE1: ER stress sensor and cell fate executor. Trends Cell Biol 23: 547–555, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chung AC, Lan HY. Chemokines in renal injury. J Am Soc Nephrol 22: 802–809, 2011. [DOI] [PubMed] [Google Scholar]
- 16.Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int 81: 442–448, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech 4: 165–178, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Cabo R, Carmona-Gutierrez D, Bernier M, Hall MN, Madeo F. The search for antiaging interventions: from elixirs to fasting regimens. Cell 157: 1515–1526, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dennen P, Altmann C, Kaufman J, Klein CL, Andres-Hernando A, Ahuja NH, Edelstein CL, Cadnapaphornchai MA, Keniston A, Faubel S. Urine interleukin-6 is an early biomarker of acute kidney injury in children undergoing cardiac surgery. Crit Care 14: R181, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eddy AA, Fogo AB. Plasminogen activator inhibitor-1 in chronic kidney disease: evidence and mechanisms of action. J Am Soc Nephrol 17: 2999–3012, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Faubel S, Lewis EC, Reznikov L, Ljubanovic D, Hoke TS, Somerset H, Oh DJ, Lu L, Klein CL, Dinarello CA, Edelstein CL. Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1β, IL-18, IL-6, and neutrophil infiltration in the kidney. J Pharmacol Exp Ther 322: 8–15, 2007. [DOI] [PubMed] [Google Scholar]
- 22.Faubel S, Ljubanovic D, Reznikov L, Somerset H, Dinarello CA, Edelstein CL. Caspase-1-deficient mice are protected against cisplatin-induced apoptosis and acute tubular necrosis. Kidney Int 66: 2202–2213, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol 11: 264–276, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat Rev Nephrol 11: 264–276, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Francescato HD, Costa RS, Junior FB, Coimbra TM. Effect of JNK inhibition on cisplatin-induced renal damage. Nephrol Dial Transplant 22: 2138–2148, 2007. [DOI] [PubMed] [Google Scholar]
- 26.Grgic I, Campanholle G, Bijol V, Wang C, Sabbisetti VS, Ichimura T, Humphreys BD, Bonventre JV. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int 82: 172–183, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsu CY, McCulloch CE, Fan D, Ordonez JD, Chertow GM, Go AS. Community-based incidence of acute renal failure. Kidney Int 72: 208–212, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishani A, Nelson D, Clothier B, Schult T, Nugent S, Greer N, Slinin Y, Ensrud KE. The magnitude of acute serum creatinine increase after cardiac surgery and the risk of chronic kidney disease, progression of kidney disease, and death. Arch Intern Med 171: 226–233, 2011. [DOI] [PubMed] [Google Scholar]
- 29.Ishani A, Xue JL, Himmelfarb J, Eggers PW, Kimmel PL, Molitoris BA, Collins AJ. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol 20: 223–228, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.James MT, Hemmelgarn BR, Wiebe N, Pannu N, Manns BJ, Klarenbach SW, Tonelli M;. Alberta Kidney Disease Network. Glomerular filtration rate, proteinuria, and the incidence and consequences of acute kidney injury: a cohort study. Lancet 376: 2096–2103, 2010. [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi M, To H, Yuzawa M, Hakamata Y, Higuchi S, Tokue A, Fujimura A, Kobayashi E. Effects of dosing time and schedule on cisplatin-induced nephrotoxicity in rats. J Pharm Pharmacol 52: 1233–1237, 2000. [DOI] [PubMed] [Google Scholar]
- 32.Kondo S, Kagami S, Urushihara M, Kitamura A, Shimizu M, Strutz F, Muller GA, Kuroda Y. Transforming growth factor-beta1 stimulates collagen matrix remodeling through increased adhesive and contractive potential by human renal fibroblasts. Biochim Biophys Acta 1693: 91–100, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Meng XM, Chung AC, Lan HY. Role of the TGF-β/BMP-7/Smad pathways in renal diseases. Clin Sci 124: 243–254, 2013. [DOI] [PubMed] [Google Scholar]
- 34.Meran S, Steadman R. Fibroblasts and myofibroblasts in renal fibrosis. Int J Exp Pathol 92: 158–167, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller RP, Tadagavadi RK, Ramesh G, Reeves WB. Mechanisms of cisplatin nephrotoxicity. Toxins 2: 2490–2518, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nechemia-Arbely Y, Barkan D, Pizov G, Shriki A, Rose-John S, Galun E, Axelrod JH. IL-6/IL-6R axis plays a critical role in acute kidney injury. J Am Soc Nephrol 19: 1106–1115, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ozkok A, Edelstein CL. Pathophysiology of cisplatin-induced acute kidney injury. Biomed Res Int 2014: 967826, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pabla N, Dong G, Jiang M, Huang S, Kumar MV, Messing RO, Dong Z. Inhibition of PKCδ reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J Clin Invest 121: 2709–2722, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 73: 994–1007, 2008. [DOI] [PubMed] [Google Scholar]
- 40.Qu X, Li X, Zheng Y, Ren Y, Puelles VG, Caruana G, Nikolic-Paterson DJ, Li J. Regulation of renal fibrosis by Smad3 Thr388 phosphorylation. Am J Pathol 184: 944–952, 2014. [DOI] [PubMed] [Google Scholar]
- 41.Ramesh G, Reeves WB. Inflammatory cytokines in acute renal failure. Kidney Int Suppl 2004: S56–S61, 2004. [DOI] [PubMed] [Google Scholar]
- 42.Salahudeen AK, Doshi SM, Pawar T, Nowshad G, Lahoti A, Shah P. Incidence rate, clinical correlates, and outcomes of AKI in patients admitted to a comprehensive cancer center. Clin J Am Soc Nephrol 8: 347–354, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Torres R, Velazquez H, Chang JJ, Levene MJ, Moeckel G, Desir GV, Safirstein R. Three-dimensional morphology by multiphoton microscopy with clearing in a model of cisplatin-induced CKD. J Am Soc Nephrol; doi: 10.1681/ASN.2015010079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uchino S, Kellum JA, Bellomo R, Doig GS, Morimatsu H, Morgera S, Schetz M, Tan I, Bouman C, Macedo E, Gibney N, Tolwani A, Ronco C. Beginning and ending supportive therapy for the kidney. I. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA 294: 813–818, 2005. [DOI] [PubMed] [Google Scholar]
- 45.Waikar SS, Liu KD, Chertow GM. Diagnosis, epidemiology and outcomes of acute kidney injury. Clin J Am Soc Nephrol 3: 844–861, 2008. [DOI] [PubMed] [Google Scholar]
- 46.Walkin L, Herrick SE, Summers A, Brenchley PE, Hoff CM, Korstanje R, Margetts PJ. The role of mouse strain differences in the susceptibility to fibrosis: a systematic review. Fibrogenesis Tissue Repair 6: 18, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xue JL, Daniels F, Star RA, Kimmel PL, Eggers PW, Molitoris BA, Himmelfarb J, Collins AJ. Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001. J Am Soc Nephrol 17: 1135–1142, 2006. [DOI] [PubMed] [Google Scholar]
- 48.Yamate J, Ishida A, Tsujino K, Tatsumi M, Nakatsuji S, Kuwamura M, Kotani T, Sakuma S. Immunohistochemical study of rat renal interstitial fibrosis induced by repeated injection of cisplatin, with special reference to the kinetics of macrophages and myofibroblasts. Toxicol Pathol 24: 199–206, 1996. [DOI] [PubMed] [Google Scholar]
- 49.Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 1 p following 143, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang WR, Garg AX, Coca SG, Devereaux PJ, Eikelboom J, Kavsak P, McArthur E, Thiessen-Philbrook H, Shortt C, Shlipak M, Whitlock R, Parikh CR; TRIBE-AKI Consortium. Plasma IL-6 and IL-10 concentrations predict AKI and long-term mortality in adults after cardiac surgery. J Am Soc Nephrol 26: 3123–3132, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]