

liver fibrosis is the common response to chronic liver injury and is due to the abnormal accumulation of extracellular matrix (ECM). In response to injury, activated hepatic stellate cells (HSCs) proliferate and produce excess amounts of ECM in response to inflammatory cytokines and growth factors (1, 2, 7). Among these mediators, transforming growth factor-β (TGF-β) is the major profibrogenic cytokine, upregulating the expression of α-smooth muscle actin (α-SMA) and type 1 collagen-α (COL1A) synthesis via Smad signaling pathway, while platelet-derived growth factor (PDGF) is a potent mitogen for HSCs via activation of extracellular signal-regulated protein kinase/mitogen-activated protein kinase and phosphoinositide-3-kinase pathways (2). Currently, inhibition of TGF-β and/or PDGF signaling pathways is considered as a promising antifibrotic strategy.
The PDGF ligand family consists of four different types, named PDGF-A, -B, -C, and -D, respectively. During liver injury, PDGF ligands produced by activated HSCs, infiltrating macrophages, Kupffer cells, and biliary epithelial cells stimulate HSCs in an autocrine and paracrine manner (1, 2). Produced homo- or heterodimerized PDGF ligands (PDGF-AA, -AB, -BB, -CC, and -DD) elicit their biological effects following binding to PDGFR-α and PDGFR-β tyrosine kinase receptors, which dimerize into either PDGFR-αα, -αβ, or -ββ, depending on the type of PDGF dimers. Each PDGF-AA and -DD ligand specifically binds to PDGFR-αα and PDGFR-ββ, respectively, whereas the PDGF-CC chain interacts with isomers of PDGFR-αα and PDGFR-αβ. PDGF-BB can bind to all three receptors, which likely induces different effects of PDGF isomers on liver fibrosis (2). HSCs express both PDGFR-α and -β, but only PDGFR-β expression is upregulated during HSC activation in vivo and in vitro (9), and accordingly, activation of PDGFR-β plays a major role in liver fibrosis. Thus, PDGFR-β binding to PDGF ligands, such as PDGF-B and -D chains, have been speculated as important factors in liver fibrosis (2). However, PDGF-A or PDGF-C transgenic mice develop spontaneous liver fibrosis by upregulating TGF-β, whereas specific transgenic PDGF-B expression and PDGF-D-treated HSCs accelerate activation of HSCs without increased levels of TGF-β, indicating a TGF-β-independent manner of activation (2, 3). In addition, TGF-β1 is reported to increase the mitotic effects of PDGF-BB by upregulating PDGFR-β expression in HSCs (1). These findings are suggested to involve TGF-β/Smad signaling in the induction of PDGFR-β expression at the transcriptional level. However, the reversal effect of PDGFR-mediated signaling that may regulate the TGF-β/Smad pathway remains unclear.
In this issue of American Journal of Physiology-Cell Physiology, Lee et al. (5) provide convincing evidence supporting the significant role of PDGF-C in the progression of liver fibrosis. By using PDGF-C transgenic mice crossed with Smad3-deficient mice, the authors report that these mice show reduced liver fibrosis compared with PDGF-C transgenic mice. Decreased fibrosis is associated with decreased expression of α-SMA, COL1A, and connective tissue growth factor (CTGF) without modification of PDGFRs and TGF-β1 in HSCs. These findings suggest that Smad3 signaling is associated in part with PDGF-C-mediated liver fibrosis and activation of HSCs. In addition, the authors demonstrate that Smad3-deficient HSC showed decreased proliferation in response to PDGF-CC treatment compared with HSCs in wild-type mice. Together, these findings postulate that PDGF-C stimulates proliferation and expression of α-SMA, COL1A, and CTGF in HSCs through Smad3 (Fig. 1).
Fig. 1.
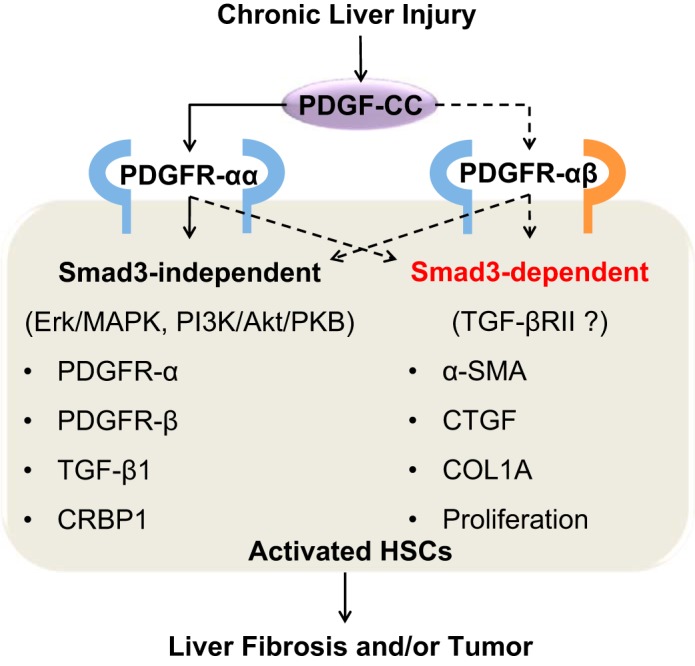
Postulated mechanism of PDGF-C-mediated liver fibrosis and tumor in Smad3-dependent and -independent signaling pathways. In response to liver injury, PDGF-C is produced by various types of cells including hepatic stellate cells (HSCs). PDGF-C binds to PDGF receptors, named PDGFR-αα and -αβ, in HSCs. PDGFR-αα signals increase expression of PDGFR-α, PDGFR-β, transforming growth factor (TGF)-β1, and cellular retinol binding protein 1 (CRBP1) in Smad3-independent pathway. In contrast, heterodimerized receptor PDGFR-αβ may activate Smad3 signaling with TGF-β receptor type II (TGF-βRII), leading to increased expression of α-smooth muscle actin (α-SMA), connective tissue growth factor (CTGF), type I collagen-α (COL1A), and proliferation of HSCs. Altogether, Smad3-dependent and -independent pathways in HSCs induce liver fibrosis or drive progression of liver fibrosis into hepatocellular carcinoma. Solid lines are clarified but dot lines remain to be addressed.
The present study undoubtedly provides several insightful findings regarding the crucial roles of PDGF-C in Smad3 signaling of activated HSCs. However, several unanswered questions remain to be addressed. In particular, the authors did not reveal whether activation of the Smad3 signaling pathway was directly or indirectly activated by PDGF-CC. An interesting paper recently addressed a plausible mechanism, indicating that PDGFR-α regulates Smad3 signaling by direct binding with TGF-β receptor II (6). This likely reveals the implication of PDGF-C in Smad3 activation due to the activity of PDGF-C binding with PDGFR-α. Conversely, PDGFR-β expression significantly increased in proportion to activation of HSCs in vitro (6), suggesting less importance of PDGF-C ligand and its receptor PDGFR-α for the therapeutic targets in patient with liver fibrosis.
Another important question that could not be addressed in the present study, due to premature mortality of PDGF-C transgenic mice, is the possible impact of PDGF-C in hepatocellular carcinoma (HCC). In this regard, PDGF-C, but not PDGFR-β in HSCs, has been considered an important factor in the development of HCC (3, 10). Various anti-PDGF signaling strategies have been developed to inhibit liver fibrosis, including neutralizing antibodies, recombinant soluble PDGFR extracellular domain or aptamers, and tyrosine kinase inhibitors (2). The present study provides evidence that induction of Smad7 to inhibit PDGF-C-mediated Smad3 activation could also be an interesting strategy.
As neutralization of TGF-β is not applicable due to adverse effects, induction of interferon-γ by direct treatment or activation of hepatic natural killer cells may be useful to inhibit Smad3 signaling due to the well-known effects of interferon-γ as not only a suppressor against activated HSCs but also an inducer of Smad7 through the STAT1 pathway (4, 8). Therefore, further investigation on the exact role of interferon-γ against the PDGF-C-mediated Smad3 signaling pathway is needed at the onset of HCC development from fibrotic liver. In summary, the present study reveals the existence of a novel pathway where inhibition of Smad3 signaling may become a dual therapeutic target for not only PDGF-C-mediated fibrosis but also HCC.
GRANTS
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (NFR-2015R1A2A1A10055551), and the Korea Mouse Phenotyping Project (NRF-2014 M3A9D5A01073556) of the Ministry of Science, ICT and Future Planning through the National Research Foundation, Republic of Korea.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
W.S. and W.I.J. prepared figure; drafted manuscript; edited and approved final version of manuscript.
REFERENCES
- 1.Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev 15: 255–273, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Borkham-Kamphorst E, Weiskirchen R. The PDGF system and its antagonists in liver fibrosis. Cytokine Growth Factor Rev 2015. doi: 10.1016/j.cytogfr.2015.10.002 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 3.Campbell JS, Hughes SD, Gilbertson DG, Palmer TE, Holdren MS, Haran AC, Odell MM, Bauer RL, Ren HP, Haugen HS, Yeh MM, Fausto N. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc Natl Acad Sci USA 102: 3389–3394, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeong WI, Park O, Radaeva S, Gao B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology 44: 1441–1451, 2006. [DOI] [PubMed] [Google Scholar]
- 5.Lee JI, Wright JH, Johnson MM, Bauer RL, Sorg K, Yuen S, Hayes BJ, Nguyen L, Riehle KJ, Campbell JS. Role of Smad3 in platelet-derived growth factor-C-induced liver fibrosis. Am J Physiol Cell Physiol. doi: 10.1152/ajpcell.00423.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu C, Li J, Xiang X, Guo L, Tu K, Liu Q, Shah VH, Kang N. PDGF receptor-α promotes TGF-β signaling in hepatic stellate cells via transcriptional and posttranscriptional regulation of TGF-β receptors. Am J Physiol Gastrointest Liver Physiol 307: G749–G759, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mallat A, Lotersztajn S. Cellular Mechanisms of Tissue Fibrosis. 5. Novel insights into liver fibrosis. Am J Physiol Cell Physiol 305: C789–C799, 2013. [DOI] [PubMed] [Google Scholar]
- 8.Ulloa L, Doody J, Massague J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature 397: 710–713, 1999. [DOI] [PubMed] [Google Scholar]
- 9.Wong L, Yamasaki G, Johnson RJ, Friedman SL. Induction of beta-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. J Clin Invest 94: 1563–1569, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wright JH, Johnson MM, Shimizu-Albergine M, Bauer RL, Hayes BJ, Surapisitchat J, Hudkins KL, Riehle KJ, Johnson SC, Yeh MM, Bammler TK, Beyer RP, Gilbertson DG, Alpers CE, Fausto N, Campbell JS. Paracrine activation of hepatic stellate cells in platelet-derived growth factor C transgenic mice: evidence for stromal induction of hepatocellular carcinoma. Int J Cancer 134: 778–788, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]