

Peroxisome proliferator-activated receptor (PPAR)-γ is anti-inflammatory, and we show here that treatment of the aorta with IL-1β activates NF-κB and NF-κB-dependent transcription. We show that the protective actions of PPAR-γ activation, induced by either rosiglitazone or overexpression of PPAR-γ in the endothelium, occurs independently of NF-κB activity, by reducing oxidative stress and preserving the phospho-endothelial nitric oxide synthase-to-endothelial nitric oxide synthase ratio.
Keywords: peroxisome proliferator-activated receptor-γ, vascular dysfunction, endothelium, inflammation, interleukin-1β
Abstract
Loss of peroxisome proliferator-activated receptor (PPAR)-γ function in the vascular endothelium enhances atherosclerosis and NF-κB target gene expression in high-fat diet-fed apolipoprotein E-deficient mice. The mechanisms by which endothelial PPAR-γ regulates inflammatory responses and protects against atherosclerosis remain unclear. To assess functional interactions between PPAR-γ and inflammation, we used a model of IL-1β-induced aortic dysfunction in transgenic mice with endothelium-specific overexpression of either wild-type (E-WT) or dominant negative PPAR-γ (E-V290M). IL-1β dose dependently decreased IκB-α, increased phospho-p65, and increased luciferase activity in the aorta of NF-κB-LUC transgenic mice. IL-1β also dose dependently reduced endothelial-dependent relaxation by ACh. The loss of ACh responsiveness was partially improved by pretreatment of the vessels with the PPAR-γ agonist rosiglitazone or in E-WT. Conversely, IL-1β-induced endothelial dysfunction was worsened in the aorta from E-V290M mice. Although IL-1β increased the expression of NF-κB target genes, NF-κB p65 inhibitor did not alleviate endothelial dysfunction induced by IL-1β. Tempol, a SOD mimetic, partially restored ACh responsiveness in the IL-1β-treated aorta. Notably, tempol only modestly improved protection in the E-WT aorta but had an increased protective effect in the E-V290M aorta compared with the aorta from nontransgenic mice, suggesting that PPAR-γ-mediated protection involves antioxidant effects. IL-1β increased ROS and decreased the phospho-endothelial nitric oxide synthase (Ser1177)-to-endothelial nitric oxide synthase ratio in the nontransgenic aorta. These effects were completely abolished in the aorta with endothelial overexpression of WT PPAR-γ but were worsened in the aorta with E-V290M even in the absence of IL-1β. We conclude that PPAR-γ protects against IL-1β-mediated endothelial dysfunction through a reduction of oxidative stress responses but not by blunting IL-1β-mediated NF-κB activity.
NEW & NOTEWORTHY
Peroxisome proliferator-activated receptor (PPAR)-γ is anti-inflammatory, and we show here that treatment of the aorta with IL-1β activates NF-κB and NF-κB-dependent transcription. We show that the protective actions of PPAR-γ activation, induced by either rosiglitazone or overexpression of PPAR-γ in the endothelium, occurs independently of NF-κB activity, by reducing oxidative stress and preserving the phospho-endothelial nitric oxide synthase-to-endothelial nitric oxide synthase ratio.
thiazolidinediones (TZDs), a class of synthetic ligands of peroxisome proliferator-activated receptor (PPAR-γ), were previously considered highly effective oral medications for type 2 diabetes due to their robust insulin-sensitizing activities (23). PPAR-γ is a ligand-activated transcription factor of the nuclear hormone receptor superfamily that is expressed in many tissues, including adipocyte, macrophage, vascular endothelial cell (EC), and smooth muscle cell (SMC) lineages (1, 21). Classically, PPAR-γ is known to induce adipocyte differentiation and to regulate lipid metabolism (43). In macrophages, PPAR-γ exerts strong anti-inflammatory effects (45).
Accumulating evidence from human and animal studies has shown that TZDs also attenuate vascular diseases including atherosclerosis. TZD treatment inhibits the formation of atherosclerotic lesions in the aorta and aortic root of apolipoprotein E (ApoE)-deficient and low-density lipoprotein receptor-deficient mouse models of hypercholesterolemia (10, 26). The PROactive trial, the largest clinical trial to measure cardiovascular end points in pioglitazone-treated populations, reported significantly lower blood pressure and reduced rates of all-cause mortality, myocardial infarction, and stroke (9). In contrast, patients with dominant negative (DN) mutations in PPAR-γ (V290M or P467L) (4) exhibit severe early onset hypertension and insulin resistance. Other mutations in human PPAR-γ (R165T or L339X) cause hypertension and lipodystrophies (3). Taken together, these observations indicate a significant requirement for functional PPAR-γ in the regulation of cardiovascular homeostasis.
To explore the mechanistic importance of PPAR-γ in the vasculature, we generated several mouse models expressing DN PPAR-γ (human PPAR-γ P467L or V290M) specifically in SMCs or ECs. Our data clearly support the concept that PPAR-γ plays a critical role in both SMCs and ECs but that it has a distinct mechanism of action in each cell type. Interference with PPAR-γ in SMCs caused a significant increase in arterial blood pressure and severe vascular dysfunction at baseline (15). In the aorta, loss of PPAR-γ function in SMCs decreased RhoBTB1 expression and cullin-3 activity, which increased RhoA/Rho kinase activity (36). Loss of PPAR-γ in the mesenteric circulation decreased the expression of regulator of G protein signaling-5 mRNA, which enhanced myogenic tone and ANG II-mediated contraction (20). In contrast, EC-specific interference with PPAR-γ (E-V290M) led to cerebral vascular dysfunction in response to a high-fat diet (5) through decreased nitric oxide (NO) bioavailability caused by an increase in superoxide. Loss of PPAR-γ function in ECs caused an upregulation of NADPH oxidase (NOX) subunits and a decrease in catalase and SOD expression (5, 21). Atherosclerosis was exacerbated and inflammatory markers were significantly increased in the aorta when both mouse models were crossed with ApoE-deficient mice and treated with a high-cholesterol diet (35). The precise mechanism by which loss of PPAR-γ function exacerbates inflammatory signals and augments atherosclerosis remains unclear.
Inflammation is a risk factor for cardiovascular diseases, and NF-κB is known as a central regulator of inflammation. NF-κB can be detected in the cytoplasm of many cells in association with IκB factors, which inhibit their DNA-binding activity (31). Cellular activation by cytokines (33) or virus (6) can induce the phosphorylation of IκB, leading to its dissociation and release of active NF-κB transcription factors. Activation of NF-κB increases proinflammatory mediators such as ICAM-1, VCAM, and monocyte chemoattractant protein (MCP)-1 in vascular cells (14, 37). NF-κB activity is also augmented in ECs, SMCs, monocytes/macrophages, and T lymphocytes in atherosclerotic plaques (7). PPAR-γ has been reported to regulate NF-κB activity in macrophages by a transrepression mechanism involving an interaction between PPAR-γ and NF-κB that does not require binding of the PPAR-γ/retinoid X receptor heterodimer to a PPAR response element (34). Moreover, PPAR-γ has recently been reported to act as an E3 ubiquitin ligase that regulates the stability of the p65 subunit of NF-κB (18). Taken together, we hypothesized that vascular PPAR-γ protects against inflammation by decreasing NF-κB activity. In the present study, we assessed the interactions between PPAR-γ and NF-κB in a model of IL-1β-induced vascular dysfunction. IL-1β is a NF-κB activator in ECs (37) and contributes to the development of atherosclerosis (29). This study was facilitated using transgenic mouse models that specifically expressed either wild-type (WT) PPAR-γ overexpressed in the endothelium (E-WT) or E-V290M in ECs, which caused a significant decrease in PPAR-γ target gene expression in aortic ECs (5). Our data show that PPAR-γ in ECs protects against IL-1β-induced endothelial dysfunction. However, we conclude that the mechanism mediating this protection does not depend on interference with NF-κB activity but rather functions through EC PPAR-γ-dependent regulation of ROS.
MATERIALS AND METHODS
Animals.
The mice used in this study included male C57BL/6J (12–20 wk of age), male and female NF-κB-LUC mice that expressed luciferase under the control of the NF-κB responsive promoter (a gift from Dr. Timothy Blackwell, Vanderbilt University) (40), and male and female transgenic mice (3–7 mo of age) carrying E-WT or the E-V290M form of human PPAR-γ under the control of the endothelium-specific vascular cadherin promoter as previously described (5). Age-matched nontransgenic (NT) littermates were used as controls. Care of these mice met standards set forth by the National Institutes of Health guidelines for the care and use of experimental animals. All procedures were approved by Animal Care and Use Committee of the University of Iowa.
Western blot analysis.
The frozen aorta (excluding perivascular fat) was homogenized in lysis buffer containing 50 mmol/l Tris·Cl buffer, 0.1 mmol/l EDTA (pH 7.5), 1% (wt/vol) Na deoxycholic acid, 1% (vol/vol) Nonidet P-40, and 0.1% (vol/vol) SDS with protease inhibitor (Roche) and phosphatase inhibitors (Roche). Supernatants were collected after sonication for 10 s and centrifuged (20,000 g) for 10 min at 4°C. The protein concentration in the lysis buffer was determined by a Lowry assay (Bio-Rad). Equal amounts of proteins (15–35 μg) were separated by SDS-PAGE (8–12%) and transferred to a nitrocellulose membrane (GE Healthcare). After being blocked with 5% skim milk, membranes were incubated with primary antibodies at 4°C overnight and then visualized using horseradish peroxidase-conjugated secondary antibodies (1:10,000 dilution, 1 h). Anti-phospho-p65 (no. 3033, Cell Signaling), anti-p65 (no. 3034, Cell Signaling), anti-IκB-α (no. 9242, Cell Signaling), endothelial NO synthase (eNOS; no. 9572, Cell Signaling), and phospho-eNOS (no. 9571, Cell Signaling) were used for these experiments. β-Actin was used as a loading control (ab16039, Abcam).
Bioluminescence imaging.
A luciferase assay was performed using NF-κB-LUC mice. After treatment of the isolated aorta with IL-1β (0–500 pg/ml) for 24 h, samples were washed using ice-cold Dulbecco's PBS (dPBS) and incubated with dPBS including 1.5 mg/ml d-luciferin (Gold Biotechnology) (42). Bioluminescence imaging was performed on a Xenogen IVIS-200 System. Luminescence was quantitated where the peak of the luminescent signal occurred.
Vascular function.
Aortic function was assessed using a wire myograph preparation. The thoracic aorta was dissected free of perivascular fat and cut into four segments. After three washes with sterile PBS, aorta rings were placed in DMEM-F-12 supplemented with 1% penicillin-streptomycin in the absence (control) or presence of IL-1β (0.1–500 pg/ml, 30 min–24 h). Aortic rings were pretreated with the indicated agents (rosiglitazone, NF-κB p65 inhibitor, tempol, or vehicle) for 1 h at 37°C in an atmosphere of 95% air-5% CO2 and then incubated in the absence (control) or presence of IL-1β (0.1–500 pg/ml, 24 h). Aortic rings were then equilibrated for 45 min under a resting tension of 0.5 g, and vasoconstriction was recorded in response to KCl (10–100 mM). Concentration-dependent response curves to ACh (1 nM–30 μM) or sodium nitroprusside (SNP; 0.1 nM–30 μM) were performed after an initial submaximal precontraction (40–60%) with PGF2α (3–10 μM).
Real-time RT-PCR.
Total RNA was extracted from the thoracic aorta using RNeasy spin columns (RNeasy Mini Kit, Qiagen). cDNA was synthesized from 500–800 ng of total RNA by RT-PCR using Superscript III (Invitrogen), RNaseOUT (Invitrogen), and oligo(dT) primers. Quantitative PCRs were performed in duplicate using Taqman Fast Advanced Master Mix (Applied Biosystems), TaqMan Gene Expression Assays (Applied Biosystems), and 10 ng of cDNA in a total volume of 10 μl. For Taqman assays, the Applied Biosystems StepOnePlus System was used (4352932-0905028 for GAPDH). In some experiments, quantitative PCRs were performed using Fast SYBR Green Master Mix (Applied Biosystems), target gene primers, and 10 ng of cDNA in a total volume of 10 μl. The primers used were as follows: GAPDH, forward 5′-GCTACAGCTTCACCACCACA-3′ and reverse 5′-AAGGAAGGCTGGAAAAGAGC-3′; murine MCP-1, forward 5′-CCCAATGAGTAGGCTGGAGA-3′ and reverse 5′-TCTGGACCCATTCCTTCTTG-3′; murine ICAM, forward 5′-TTCACACTGAATGCCAGCTC-3′ and reverse 5′-GTCTGCTGAGACCCCTCTTG-3′; and inducible NO synthase, forward 5′-CACCTTGGAGTTCACCCAGT-3′ and reverse 5′-CGCCTTGGAGTTCACCCAGT-3′. ΔΔCT values (where CT is threshold cycle) were calculated using GAPDH or β-actin as a reference gene to determine relative mRNA expression levels.
Fluorometric measurement of ROS.
ROS accumulation was assessed using dihydroethidium (DHE; Invitrogen). Aortic rings were pretreated with the indicated agents (apocynin or vehicle) for 1 h at 37°C and then incubated in the absence (control) or presence of IL-1β (20–500 pg/ml, 24 h) before treatment with DHE. ImageJ software was used for quantitative analysis.
Chemicals.
IL-1β was purchased from R&D Systems (Minneapolis, MN). ACh, SNP, KCl, and tempol were from Sigma (St. Louis, MO). PGF2α were acquired from Pfizer (New York, NY). We used the NF-κB p65 inhibitor NBP2-29321 from Novus Biologicals (Littleton, CO). Rosiglitazone was from Cayman (Ann Arbor, MI) and dissolved in DMSO according to the manufacturer's instruction.
Statistical analysis.
Experiments were performed in similar numbers between male and female mice. We observed that there was no difference between the sex of mice; therefore, data were pooled from both. Results are expressed as means ± SE. Statistical evaluation of the data was performed using GraphPad Prism. Where appropriate, a paired or unpaired Student's t-test was used to compare between two groups. In other experiments, ANOVA followed by Tukey's test for comparisons was performed. Differences were considered significant when P values were <0.05.
RESULTS
IL-1β induces NF-κB activity and vascular dysfunction.
To examine whether IL-1β activates NF-κB, isolated aortas from C57BL/6J mice were treated with IL-1β (0.1–100 pg/ml) in vitro, and NF-κB activity was examined. IL-1β, starting at 5 pg/ml, increased p65 phosphorylation without affecting total expression of p65 and reduced the levels of IκB-α (Fig. 1A). This indicates that IL-1β activated NF-κB signaling in the whole aorta. IL-1β treatment of aortas isolated from NF-κB-LUC mice consistently caused a dose-dependent increase in NF-κB transcriptional activity (Fig. 1, B and C).
Fig. 1.

Effect of IL-1β on NF-κB activity in the aorta. A: Western blot detecting phosphorylated and total p65, total IκB-α, and β-actin in IL-1β (0.1–100 pg/ml, 2 h)-treated aortas from C57BL/6J mice. B and C: NF-κB activity determined by luciferase assay in IL-1β (5–500 pg/ml, 24 h)-treated aortas isolated from NF-κB-LUC reporter mice and nontransgenic (NT) littermate controls (n = 6). All data are means ± SE. #P < 0.05 and *P < 0.01 vs. no treatment.
Aortas isolated from C57BL/6J mice were incubated with IL-1β in vitro to determine if IL-1β can induce endothelial dysfunction. IL-1β dose dependently impaired vasodilation to ACh (Fig. 2). IL-1β treatment caused a much smaller decrease in endothelium-independent vasodilation induced by SNP, suggesting possible additional impairment of smooth muscle function (Fig. 2). KCl and PGF2α-induced contraction were not altered by IL-1β at any concentrations (1–500 pg/ml, n = 5–6, data not shown). These data show that IL-1β treatment of the aorta ex vivo causes endothelial dysfunction.
Fig. 2.

Effect of IL-1β on vascular relaxation. Isometric tension experiments were performed using thoracic aortic rings from C57BL/6J mice treated ex vivo with or without low (1–20 pg/ml; A) or high (50–500 pg/ml; B) doses of IL-1β (1–500 pg/ml) for 24 h. Concentration-dependent relaxation to ACh (1 nM–30 μM) or sodium nitroprusside (SNP; 0.1 nM–30 μM) was recorded after precontraction with PGF2α. All data are means ± SE; n = 5–6. #P < 0.05 and *P < 0.01 vs. control.
PPAR-γ in ECs protects against IL-1β-induced vascular dysfunction.
Next, we investigated whether PPAR-γ activation can protect against IL-1β-induced EC dysfunction. Rosiglitazone, a potent PPAR-γ agonist, did not affect relaxation to ACh or SNP in vehicle-treated aortas from C57BL/6J mice (Fig. 3A). However, pretreatment with rosiglitazone before IL-1β modestly but significantly improved ACh-induced vasodilation. To eliminate the possibility of an offtarget effect of rosiglitazone, we used a transgenic mouse model that expressed WT PPAR-γ specifically in ECs. At baseline, there was no difference in ACh- or SNP-induced vasodilation between E-WT and NT littermate controls (Fig. 3, A and B). KCl-induced contraction was similar between genotypes (NT + vehicle: 0.84 ± 0.08 g, E-WT + vehicle: 0.95 ± 0.11 g, NT + IL-1β: 0.82 ± 0.09 g, and E-WT + IL-1β: 0.81 ± 0.06 g, n = 6–8, P > 0.05). Consistent with the results from the rosiglitazone experiment, overexpression of WT PPAR-γ in the endothelium significantly improved ACh-induced relaxation in IL-1β (500 pg/ml)-treated aortas (Fig. 3B). A higher dose of IL-1β was used in the E-WT experiments because overexpression of WT PPAR-γ provides protection from IL-1β-induced endothelial dysfunction. There was no further improvement in vasodilation by rosiglitazone treatment in aortas from E-WT mice (Fig. 3C).
Fig. 3.

Vasomotor function with peroxisome proliferator-activated receptor (PPAR)-γ activation. A: ACh (1 nM–30 μM) or SNP (0.1 nM–30 μM) responses in aortas from NT mice. Aortic rings were treated with or without rosiglitazone (Rosi; 1 μM) for 1 h before IL-1β (100 pg/ml, 24 h) or control treatment (n = 6–8). B: concentration-dependent relaxation to ACh or SNP in aortas from NT or transgenic mice overexpressing wild-type (WT) PPAR-γ specifically in the endothelium (E-WT). Aortic rings were treated with or without IL-1β (500 pg/ml) for 24 h (n = 6). C: ACh or SNP response in aortas from E-WT mice. Aortic rings were treated with or without Rosi (1 μM) for 1 h before IL-1β (500 pg/ml, 24 h) or control treatments (n = 6). All data are means ± SE. *P < 0.01, IL-1β vs. Rosi + IL-1β (A) or NT IL-1β vs. E-WT IL-1β (B).
Conversely, overexpression of a DN mutant form of PPAR-γ specifically in the endothelium exacerbated endothelial dysfunction caused by low-dose IL-1β (20 pg/ml; Fig. 4). Low-dose IL-1β was used in experiments using aortas from E-V290M mice because interference with PPAR-γ caused increased susceptibility to IL-1β-induced endothelial dysfunction. Indeed, even very low doses of IL-1β (5 pg/ml) caused a modest impairment in ACh-induced vasodilation in aortas from E-V290M compared with NT mice (maximum relaxation by ACh: 68.21 ± 4.41% in NT mice and 53.30 ± 4.01% in E-V290M mice, n = 6, P < 0.05). In contrast, high-dose IL-1β (500 pg/ml) caused an almost complete ablation of ACh-induced vasodilation in NT aortas, and this maximal level of dysfunction could not be worsened in aortas from E-V290M mice (maximum relaxation by ACh: −5.17 ± 9.96% in NT mice and 3.90 ± 6.25% in E-V290M mice, n = 3). There were no differences in SNP-induced relaxation (Fig. 4) and KCl-induced contraction (data not shown) between NT and E-V290M mice. These data support the hypothesis that endothelial PPAR-γ functions to protect against IL-1β-induced endothelial dysfunction in the aorta.
Fig. 4.
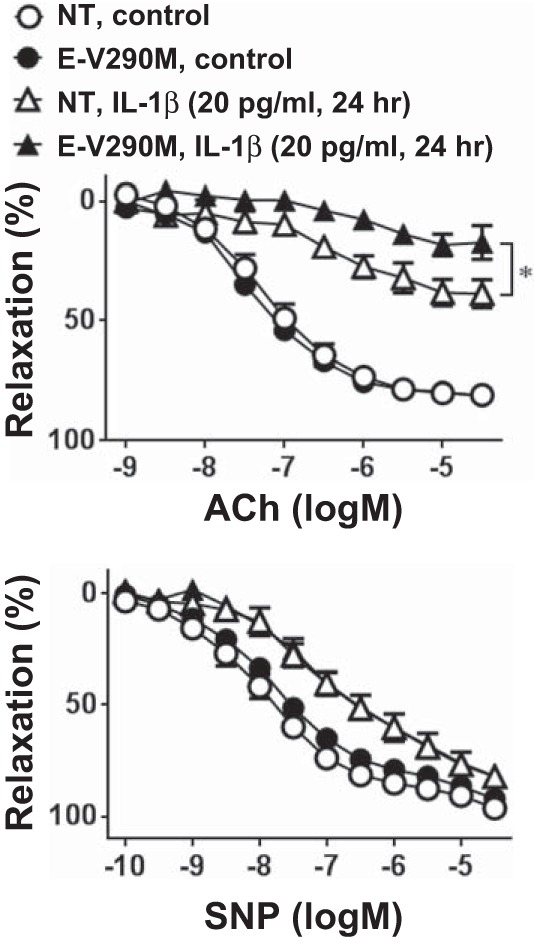
Vasomotor function with PPAR-γ interference. Concentration-dependent relaxation to ACh (1 nM–30 μM) or SNP (0.1 nM–30 μM) in aortas from NT or transgenic mice expressing dominant negative PPAR-γ specifically in the endothelium (E-V290M) is shown. Aortic rings were treated with IL-1β (20 pg/ml) or control for 24 h (n = 6). All data are means ± SE. *P < 0.01, NT + IL-1β vs. E-V290M + IL-1β.
NF-κB activity does not affect EC dysfunction by IL-1β.
We showed that IL-1β induces NF-κB activity in the aorta (Fig. 1). To determine if IL-1β-induced endothelial dysfunction requires activation of NF-κB, we used a NF-κB p65 inhibitory peptide at a concentration that blunted IL-1β (20 pg/ml)-induced luciferase activity in aortas from NF-κB-LUC mice (Fig. 5A). However, the NF-κB inhibitor did not affect endothelial dysfunction induced by any dose of IL-1β in aortas from NT mice (Fig. 5, B and C). The inhibitor modestly improved endothelium-independent relaxation in aortas treated with low-dose IL-1β (Fig. 5B), suggesting that NF-κB activity may contribute to IL-1β-induced dysfunction in SMCs. Importantly, IL-1β-induced impairment of vasodilation to ACh was also not altered by the NF-κB inhibitor in aortas from E-WT mice (n = 6, data not shown), suggesting that the protective actions of endothelial PPAR-γ are not dependent on altering NF-κB activity. Consistent with this, endothelium-specific overexpression of WT PPAR-γ in E-WT mice did not blunt the induction of the NF-κB target genes iNOS, MCP-1, or ICAM-1 in IL-1β-treated aortas (Fig. 6). Similarly, there was no effect on IL-1β-induced iNOS protein levels in aortas from E-WT mice (NT + vehicle: 1.00 ± 0.90, NT + IL-1β: 4.02 ± 0.67, E-WT + vehicle: 1.08 ± 0.69, and E-WT + IL-1β: 3.57 ± 0.33, n = 3).
Fig. 5.

Effect of NF-κB inhibition on vascular dysfunction induced by IL-1β. Aortas were pretreated with vehicle (control) or the NF-κB p65 inhibitor (50 μM) for 1 h before exposure to IL-1β or control treatments. A: NF-κB activity was determined by luciferase assay in aortas from NF-κB-LUC mice treated with IL-1β (20 pg/ml, 24 h) in the presence or absence of the p65 inhibitor (n = 3). B and C: aortas from NT mice were treated with low (B; 20 pg/ml, 24 h) or high (C; 500 pg/ml, 24 h) doses of IL-1β in the presence or absence of the p65 inhibitor. Isometric tension experiments were then performed with ACh (1 nM–30 μM) or SNP (0.1 nM–30 μM). All data are means ± SE; n = 3–6. *P < 0.05, IL-1β vs. p65 inhibitor + IL-1β.
Fig. 6.

Analysis of NF-κB target genes. Relative mRNA expression of mouse monocyte chemoattractant protein (MCP)-1 (A), ICAM-1 (B), and inducible nitric oxide synthase (iNOS; C) was determined by quantitative real-time RT-PCR in aortas from NT or E-WT mice treated ex vivo with or without IL-1β (500 pg/ml) for 24 h (n = 3). Data were normalized to the NT value, which was set to 1.0. All data are means ± SE. *P < 0.01, control vs. IL-1β. Differences between IL-1β-treated NT and E-WT were not significant (NS).
IL-1β induces vascular dysfunction via increasing ROS.
Oxidative stress has been implicated in endothelial dysfunction and cardiovascular diseases. We next used a ROS scavenger to test if increased ROS plays a role in IL-1β-mediated endothelial dysfunction. Tempol significantly improved ACh-mediated dilation of IL-1β-treated aortas from NT mice (Fig. 7A) but had a blunted effect on aortas from E-WT mice (Fig. 7B) when both were treated with the same dose of IL-1β (500 ng/ml). Although the maximal relaxation in response to tempol was similar in both groups (maximum relaxation by ACh: 47.98 ± 4.57% for NT tempol + IL-1β and 51.59 ± 5.55% for E-WT tempol + IL-1β), the effect size of tempol was greater on NT aortas compared with E-WT aortas (difference in maximum relaxation by ACh between tempol + IL-1β and IL-1β alone: 54.39 ± 4.03% in NT aortas and 19.86 ± 7.75% in E-WT aortas, P < 0.05). Analysis of the area under the curve also showed that the effect of tempol was greater on NT aortas (IL-1β: 1,083 ± 24 vs. tempol + IL-1β: 810 ± 75) compared with E-WT aortas (IL-1β: 904 ± 34 vs. tempol + IL-1β: 806 ± 29). This suggests that there is at least some redundancy in the protection mediated by tempol and endothelial PPAR-γ. Consistent with this, tempol also improved IL-1β-induced impairment in E-V290M aortas (Fig. 7D) but with a larger effect compared with NT aortas (Fig. 7C) when tested at the same low dose of IL-1β (20 pg/ml, difference in maximum relaxation by ACh between tempol + IL-1β and IL-1β alone: 12.79 ± 5.43% in NT aortas and 25.65 ± 12.42% in E-V290M aortas). Similarly, analysis of the area under the curve showed that tempol caused a greater improvement in the IL-1β-induced impairment in E-V290M aortas (IL-1β: 999 ± 42 vs. tempol + IL-1β: 812 ± 33) compared with NT aortas (IL-1β: 835 ± 35 vs. tempol + IL-1β: 765 ± 38). Tempol had no effect on the response to SNP in any group (Fig. 7).
Fig. 7.

Effect of ROS scavenger on vascular dysfunction induced by IL-1β. Aortas from NT, E-WT, and E-V290M mice were pretreated with or without tempol (1 mM) for 1 h before high (500 pg/ml; A and B) or low (20 pg/ml; C and D) doses of IL-1β or control treatments for 24 h. Isometric tension experiments were performed with ACh (1 nM–30 μM) or SNP (0.1 nM–30 μM). All data are means ± SE; n = 5–6. *P < 0.01, IL-1β vs. tempol + IL-1β.
To determine if IL-1β can induce ROS accumulation in the aorta, DHE staining was performed in aortas from NT mice. ROS was increased by IL-1β (Fig. 8A), and its accumulation was significantly blunted in the presence of the NOX inhibitor apocynin. This suggests that this effect depends on NOX activity. We next investigated whether PPAR-γ in the endothelium decreases IL-1β-induced ROS generation. Consistent with the vascular function data, ROS accumulation in response to IL-1β was reduced in E-WT compared with NT aortas (Fig. 8B). In contrast, DN PPAR-γ in ECs significantly increased ROS in aortas even without IL-1β treatment (Fig. 9). Taken together, these data suggest that IL-1β causes oxidative stress and that the oxidative stress component of IL-1β-induced endothelial dysfunction is blunted by PPAR-γ.
Fig. 8.

Effect of IL-1β on ROS generation. Dihydroethidium (DHE) staining was used to detect ROS generation in the aorta. A: aortas from NT mice were pretreated with or without apocynin (0.3 mM) for 1 h before IL-1β (500 pg/ml, 24 h) or control treatments (n = 6). B: aortas from NT or E-WT were treated with IL-1β (500 pg/ml, 24 h) alone (n = 7). Samples labeled 1 and 2 refer to two independent replicates showing the range of DHE staining in the experiments. All data are means ± SE. *P < 0.05, IL-1β vs. no treatment or apocynin + IL-1β.
Fig. 9.
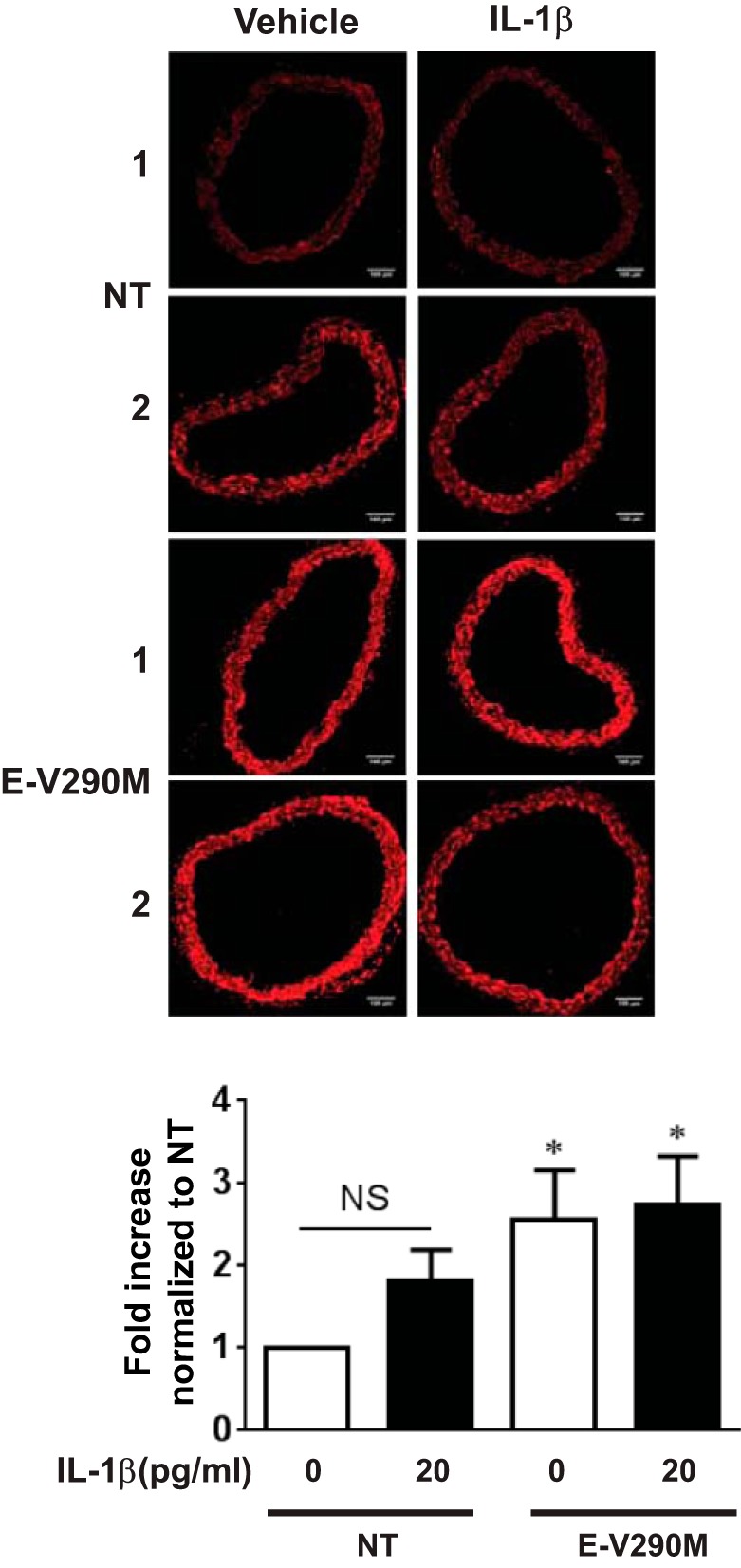
ROS generation with PPAR-γ interference. ROS generation in aortas from NT or E-V290M mice was detected by DHE staining. Aortic rings were treated with IL-1β (20 pg/ml) or control for 24 h (n = 7). Samples labeled 1 and 2 refer to two independent replicates showing the range of DHE staining in the experiments. All data are means ± SE. *P < 0.05 vs. NT no treatment.
NOX is one of the major sources of ROS in vascular tissues (24), and antioxidant enzymes such as catalase and SOD are known as critical determinants of the redox status in the vasculature (8). IL-1β decreased expression of catalase and NOX4 and increased expression of SOD2 without changing expression of NOX1, NOX2, SOD1, and SOD3 mRNA (n = 3, data not shown). Although IL-1β-induced ROS accumulation was dependent on NOX activity (Fig. 8A), there was no difference in the IL-1β-induced changes in oxidant or antioxidant gene expression between NT and E-WT mice. These data suggest that the reduced ROS accumulation mediated by PPAR-γ activity is not dependent on changes in the expression of these genes.
Overexpression of endothelial PPAR-γ preserved activity of eNOS.
In the aorta, ACh-induced relaxation mainly depends on eNOS activity, which is partly determined by the prevalence of phospho-Ser1177 within the total eNOS population (11). In aortas from NT mice, IL-1β (500 pg/ml) caused an increase in eNOS expression but with a concomitant decrease in the phospho-eNOS-to-eNOS ratio, consistent with marked EC dysfunction (Fig. 10). Interestingly, the ratio of phospho-eNOS to eNOS was preserved in IL-1β-treated aortas from E-WT mice. Conversely, loss of PPAR-γ function in the endothelium (E-V290M) resulted in a sharp reduction in the ratio of phospho-eNOS to eNOS even in the absence of IL-1β. These data suggest that endothelial PPAR-γ preserves eNOS activity, which is reduced in aortas of NT mice in response to IL-1β.
Fig. 10.

Analysis of endothelial nitric oxide synthase (eNOS) activity. A: Western blot detecting phosphorylated (p-eNOS) and total eNOS as well as β-actin in IL-1β (500 pg/ml, 24 h)-treated aortas from NT, E-WT, or E-V290M mice. B: total eNOS expression, its phosphorylated form, and the phospho-eNOS-to-total eNOS ratio were determined from Western blots of aortas treated with or without IL-1β (500 pg/ml, 24 h). All data are means ± SE. #P < 0.05 and *P < 0.01, IL-1β vs. untreated control or indicated comparisons. M, marker.
DISCUSSION
TZDs are pharmacological activators of PPAR-γ used to treat patients with type 2 diabetes. As reported in the PROactive clinical trial, TZDs not only exhibited unparalleled glycemic control but also decreased macrovascular events and lowered blood pressure and cardiovascular risk (9). In contrast, we have previously shown that interference with PPAR-γ function in SMCs or ECs exacerbated aortic atherosclerosis with increased NF-κB target gene expression in ApoE-deficient mice (35). Because IL-1β is an NF-κB activator and contributes to the development of atherosclerosis (29), the present study examined the effect of EC PPAR-γ on vascular dysfunction induced by IL-1β using transgenic mice with overexpression of WT or DN PPAR-γ in ECs. Treatment with IL-1β impaired vascular function, which was improved by increased endothelial PPAR-γ activity. Our results suggest that IL-1β-induced EC dysfunction was dependent on ROS formation but not on NF-κB activity and that PPAR-γ-mediated protection involves suppressing effects of IL-1β-induced ROS.
NF-κB is a transcription factor known as a central regulator of inflammation, and several lines of evidence suggest that PPAR-γ might exert anti-inflammatory effects by interfering with NF-κB activity in vascular cells. Indeed, expression of constitutively active PPAR-γ in cultured ECs was reported to reduce NF-κB activity (46), although the mechanism of this effect was not determined. However, direct effects of PPAR-γ on NF-κB have been documented in other cell types. In macrophages, inflammatory gene expression is inhibited through a “transrepression” mechanism in which PPAR-γ interacts with nuclear receptor corepressor-histone deacetylase-3 complexes on NF-κB or activator protein-1 (34). It has also been reported that PPAR-γ has a RING domain similar to E3 ubiquitin ligases and can directly bind to the p65 subunit of NF-κB, causing its ubiquitination and degradation (18). However, we did not find evidence to support these mechanisms as mediators of PPAR-γ protection from IL-1β-induced EC dysfunction. Specifically, neither expression of p65 protein nor its activating phosphorylation (Ser536) was altered by overexpression of WT PPAR-γ in IL-1β-treated aortas (n = 3, data not shown). We also did not detect any changes in NF-κB-mediated gene expression associated with PPAR-γ activity in the aorta. Thus, we were not able to demonstrate that reported mechanisms of PPAR-γ-mediated interference with NF-κB are operative in vascular ECs. We also did not find evidence that NF-κB activity contributed to EC dysfunction induced by inflammatory cytokines, since this dysfunction was not reduced by a NF-κB p65 inhibitor.
Similarly, suppression of vascular endothelium-specific NF-κB did not decrease blood pressure in a complex model of hypertension involving ANG II infusion, high salt, and treatment with N-nitro-l-arginine methyl ester (16). Conversely, activation of NF-κB was reported to protect against endothelial apoptosis (25) through its target gene, A1 (41), or the immediate early response gene X-1 (39). These evidences indicate that NF-κB activity in vascular ECs might not impair endothelium-dependent vasodilation directly and that other pathways may be responsible for IL-1β-mediated vascular dysfunction.
The pathway mediating ACh-induced relaxation in the aorta mainly depends on eNOS and its activity, which is regulated by protein-protein interactions and multisite phosphorylation (22). Activating (Ser1177) and inhibitory (Thr495) phosphorylations are the most thoroughly studied sites (11), and Ser1177 phosphorylation has been shown to be decreased in ECs in models of atherosclerosis (30) and hypertension (2, 22). Moreover, cardiovascular risk factors, including atherosclerosis (19), diabetes (17) and hypertension (44), are associated with an increase in eNOS expression rather than a decrease (27). Expression of eNOS is also increased in the presence of superoxide, through H2O2-mediated transcriptional and posttranscriptional mechanisms (13). Consistent with these reports, our present study showed that an inflammatory cytokine robustly decreased the phospho-eNOS (Ser1177)-to-eNOS ratio. These findings are consistent with marked endothelial dysfunction caused by an IL-1β-induced increased in ROS. Importantly, the noninflammatory level of eNOS activity was preserved in aortas from E-WT mice.
We have provided several lines of evidences to indicate that endothelial PPAR-γ diminished the effects of oxidative stress induced by IL-1β. First, a ROS scavenger, tempol, significantly lessened the impairment of ACh relaxation induced by IL-1β in aortas from NT mice. Second, the magnitude of this tempol-mediated protection was markedly blunted in aortas from E-WT mice compared with aortas from NT mice. Third, endothelial PPAR-γ prevented the NOX-dependent increase in ROS induced by IL-1β. This suggests that overexpression of PPAR-γ in the endothelium may have contributed to the protection from oxidative stress-induced endothelial dysfunction. However, this protection was not associated with altered expression of NOX or antioxidant genes in IL-1β-treated aortas, suggesting that PPAR-γ reduces ROS production through some other mechanism. The ability of EC PPAR-γ to preserve a noninflammatory profile of eNOS activity suggests possible protection against the uncoupling of eNOS activity associated with IL-1β-induced ROS. However, we also showed that ROS accumulation and decreased eNOS activity in aortas from E-V290M mice did not cause endothelial dysfunction in the absence of IL-1β. These data indicate that increased ROS and reduced eNOS activity by themselves do not cause vascular dysfunction but increase susceptibility to IL-1β-induced dysfunction. Moreover, the fact that neither tempol nor expression of WT PPAR-γ did not fully correct endothelial dysfunction mediated by IL-1β suggests there may be an oxidant stress-independent component to the endothelial dysfunction caused by IL-1β.
In addition to causing vascular dysfunction, it is known that IL-1β contributes to the development of atherosclerosis, resulting from alterations of lipid metabolism and plaque development (29). Several rodent and human studies have demonstrated that circulating IL-1β levels are also correlated with cardiovascular risk factors such as obesity, diabetes, and hypertension. Notably, inflammatory cytokines, such as IL-1β and IL-6, are secreted from expanding adipose tissue and are elevated in obesity (47). Functionally, IL-1β has been reported to contribute to the development of type II diabetes mellitus and insulin resistance (38). An effect of inflammatory cytokines on vascular function has also been suggested by a human study (12) where serum IL-1β levels were correlated with essential hypertension, although other possible risk factors of atherosclerosis could not be excluded. Concentrations of 500 pg/ml IL-1β are also commonly seen in a systemic inflammatory response syndrome, which is associated with endothelial dysfunction in the rat aorta (28). Serum levels of 30 pg/ml IL-1β have also been observed in young db/db mice compared with control mice, a threefold increase compared with control mice (32). Thus, the concentrations of IL-1β (5–500 pg/ml) used in the present study have pathobiological relevance. Given the protective effects of PPAR-γ observed in the present study against IL-1β-mediated EC dysfunction, future studies closely examining the interaction between IL-1β and PPAR-γ on cardiovascular disease are warranted.
EC dysfunction is a marker of cardiovascular disease and is closely associated with inflammation. Loss of PPAR-γ function in ECs enhances atherosclerosis in ApoE-deficient mice fed a high-fat diet (35). This study shows that PPAR-γ also protects against inflammatory cytokine-mediated vascular dysfunction through a reduction of ROS-mediated effects rather than direct antagonism of NF-κB activities. This contributes to an increasing understand for the potential roles of EC PPAR-γ in the pathogenesis of vascular diseases.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute grants HL-084207, HL-048058, HL-062984, and HL-125603 (to C.D. Sigmund). The authors gratefully acknowledge the generous research support of the Roy J. Carver Trust.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.M., P.K., F.W.Q., and C.D.S. conception and design of research; M.M., M.S., P.K., and C.H. performed experiments; M.M., M.S., P.K., C.H., and C.D.S. analyzed data; M.M., P.K., C.H., and C.D.S. interpreted results of experiments; M.M. and C.H. prepared figures; M.M. and C.D.S. drafted manuscript; M.M., M.S., P.K., C.H., F.W.Q., and C.D.S. edited and revised manuscript; M.M., M.S., P.K., C.H., F.W.Q., and C.D.S. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Bill Paradee, Norma Sinclair, JoAnne Schwarting, and Patricia Yarolem for genotyping mice. Transgenic mice were generated at the University of Iowa Genome Editing Facility, which was supported in part by grants from the National Institutes of Health and from the Roy J. and Lucille A. Carver College of Medicine. The authors thank Deborah Davis and Dr. Frank M. Faraci, Dr. Justin L. Grobe, Dr. Jianqiang Shao, and Dr. Henry L. Keen at the University of Iowa for the suggestions. The authors also acknowledge the use of equipment and assistance from the Central Microscopy Facility at the University of Iowa.
REFERENCES
- 1.Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, Evans RM. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med 19: 557–566, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atochin DN, Wang A, Liu VW, Critchlow JD, Dantas AP, Looft-Wilson R, Murata T, Salomone S, Shin HK, Ayata C, Moskowitz MA, Michel T, Sessa WC, Huang PL. The phosphorylation state of eNOS modulates vascular reactivity and outcome of cerebral ischemia in vivo. J Clin Invest 117: 1961–1967, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Auclair M, Vigouroux C, Boccara F, Capel E, Vigeral C, Guerci B, Lascols O, Capeau J, Caron-Debarle M. Peroxisome proliferator-activated receptor-γ mutations responsible for lipodystrophy with severe hypertension activate the cellular renin-angiotensin system. Arterioscler Thromb Vasc Biol 33: 829–838, 2013. [DOI] [PubMed] [Google Scholar]
- 4.Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, Chatterjee VK, O'Rahilly S. Dominant negative mutations in human PPARγ associated with severe insulin resistance, diabetes mellitus and hypertension. Nature 402: 880–883, 1999. [DOI] [PubMed] [Google Scholar]
- 5.Beyer AM, de Lange WJ, Halabi CM, Modrick ML, Keen HL, Faraci FM, Sigmund CD. Endothelium-specific interference with peroxisome proliferator activated receptor γ causes cerebral vascular dysfunction in response to a high-fat diet. Circ Res 103: 654–661, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bohnlein E, Lowenthal JW, Siekevitz M, Ballard DW, Franza BR, Greene WC. The same inducible nuclear proteins regulates mitogen activation of both the interleukin-2 receptor-α gene and type 1 HIV. Cell 53: 827–836, 1988. [DOI] [PubMed] [Google Scholar]
- 7.Brand K, Page S, Rogler G, Bartsch A, Brandl R, Knuechel R, Page M, Kaltschmidt C, Baeuerle PA, Neumeier D. Activated transcription factor nuclear factor-κB is present in the atherosclerotic lesion. J Clin Invest 97: 1715–1722, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res 116: 531–549, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Charbonnel B, Dormandy J, Erdmann E, Massi-Benedetti M, Skene A, Group PRS. The prospective pioglitazone clinical trial in macrovascular events (PROactive): can pioglitazone reduce cardiovascular events in diabetes? Study design and baseline characteristics of 5238 patients. Diabetes Care 27: 1647–1653, 2004. [DOI] [PubMed] [Google Scholar]
- 10.Chen Z, Ishibashi S, Perrey S, Osuga J, Gotoda T, Kitamine T, Tamura Y, Okazaki H, Yahagi N, Iizuka Y, Shionoiri F, Ohashi K, Harada K, Shimano H, Nagai R, Yamada N. Troglitazone inhibits atherosclerosis in apolipoprotein E-knockout mice: pleiotropic effects on CD36 expression and HDL. Arterioscler Thromb Vasc Biol 21: 372–377, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Chen ZP, Mitchelhill KI, Michell BJ, Stapleton D, Rodriguez-Crespo I, Witters LA, Power DA, Ortiz de Montellano PR, Kemp BE. AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett 443: 285–289, 1999. [DOI] [PubMed] [Google Scholar]
- 12.Dalekos GN, Elisaf M, Bairaktari E, Tsolas O, Siamopoulos KC. Increased serum levels of interleukin-1β in the systemic circulation of patients with essential hypertension: additional risk factor for atherogenesis in hypertensive patients? J Lab Clin Med 129: 300–308, 1997. [DOI] [PubMed] [Google Scholar]
- 13.Drummond GR, Cai H, Davis ME, Ramasamy S, Harrison DG. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ Res 86: 347–354, 2000. [DOI] [PubMed] [Google Scholar]
- 14.Gareus R, Kotsaki E, Xanthoulea S, van der Made I, Gijbels MJ, Kardakaris R, Polykratis A, Kollias G, de Winther MP, Pasparakis M. Endothelial cell-specific NF-κB inhibition protects mice from atherosclerosis. Cell Metab 8: 372–383, 2008. [DOI] [PubMed] [Google Scholar]
- 15.Halabi CM, Beyer AM, de Lange WJ, Keen HL, Baumbach GL, Faraci FM, Sigmund CD. Interference with PPAR γ function in smooth muscle causes vascular dysfunction and hypertension. Cell Metab 7: 215–226, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Henke N, Schmidt-Ullrich R, Dechend R, Park JK, Qadri F, Wellner M, Obst M, Gross V, Dietz R, Luft FC, Scheidereit C, Muller DN. Vascular endothelial cell-specific NF-κB suppression attenuates hypertension-induced renal damage. Circ Res 101: 268–276, 2007. [DOI] [PubMed] [Google Scholar]
- 17.Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, Skatchkov M, Thaiss F, Stahl RA, Warnholtz A, Meinertz T, Griendling K, Harrison DG, Forstermann U, Munzel T. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res 88: E14–22, 2001. [DOI] [PubMed] [Google Scholar]
- 18.Hou Y, Moreau F, Chadee K. PPARγ is an E3 ligase that induces the degradation of NFκB/p65. Nat Commun 3: 1300, 2012. [DOI] [PubMed] [Google Scholar]
- 19.Kano H, Hayashi T, Sumi D, Esaki T, Asai Y, Thakur NK, Jayachandran M, Iguchi A. A HMG-CoA reductase inhibitor improved regression of atherosclerosis in the rabbit aorta without affecting serum lipid levels: possible relevance of up-regulation of endothelial NO synthase mRNA. Biochem Biophys Res Commun 259: 414–419, 1999. [DOI] [PubMed] [Google Scholar]
- 20.Ketsawatsomkron P, Lorca RA, Keen HL, Weatherford ET, Liu X, Pelham CJ, Grobe JL, Faraci FM, England SK, Sigmund CD. PPARγ regulates resistance vessel tone through a mechanism involving RGS5-mediated control of protein kinase C and BKCa channel activity. Circ Res 111: 1446–1458, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ketsawatsomkron P, Sigmund CD. Molecular mechanisms regulating vascular tone by peroxisome proliferator activated receptor γ. Curr Opin Nephrol Hypertens 24: 123–130, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kolluru GK, Siamwala JH, Chatterjee S. eNOS phosphorylation in health and disease. Biochimie 92: 1186–1198, 2010. [DOI] [PubMed] [Google Scholar]
- 23.Kung J, Henry RR. Thiazolidinedione safety. Expert Opin Drug Saf 11: 565–579, 2012. [DOI] [PubMed] [Google Scholar]
- 24.Lassegue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 30: 653–661, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee R, Collins T. Nuclear factor-κB and cell survival: IAPs call for support. Circ Res 88: 262–264, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. Peroxisome proliferator-activated receptor γ ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest 106: 523–531, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, Wallerath T, Munzel T, Forstermann U. Regulation of endothelial-type NO synthase expression in pathophysiology and in response to drugs. Nitric Oxide 7: 149–164, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Loughrey JP, Laffey JG, Moore BJ, Lynch F, Boylan JF, McLoughlin P. Interleukin-1β rapidly inhibits aortic endothelium-dependent relaxation by a DNA transcription-dependent mechanism. Crit Care Med 31: 910–915, 2003. [DOI] [PubMed] [Google Scholar]
- 29.McCarty S, Frishman W. Interleukin 1β: a proinflammatory target for preventing atherosclerotic heart disease. Cardiol Rev 22: 176–181, 2014. [DOI] [PubMed] [Google Scholar]
- 30.Napp A, Brixius K, Pott C, Ziskoven C, Boelck B, Mehlhorn U, Schwinger RH, Bloch W. Effects of the β3-adrenergic agonist BRL 37344 on endothelial nitric oxide synthase phosphorylation and force of contraction in human failing myocardium. J Card Fail 15: 57–67, 2009. [DOI] [PubMed] [Google Scholar]
- 31.Nolan GP, Ghosh S, Liou HC, Tempst P, Baltimore D. DNA binding and IκB inhibition of the cloned p65 subunit of NF-κB, a rel-related polypeptide. Cell 64: 961–969, 1991. [DOI] [PubMed] [Google Scholar]
- 32.O'Neill CM, Lu C, Corbin KL, Sharma PR, Dula SB, Carter JD, Ramadan JW, Xin W, Lee JK, Nunemaker CS. Circulating levels of IL-1B+IL-6 cause ER stress and dysfunction in islets from prediabetic male mice. Endocrinology 154: 3077–3088, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor α and interleukin 1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor κ B. Proc Natl Acad Sci USA 86: 2336–2340, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 437: 759–763, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pelham CJ, Keen HL, Lentz SR, Sigmund CD. Dominant negative PPARγ promotes atherosclerosis, vascular dysfunction, and hypertension through distinct effects in endothelium and vascular muscle. Am J Physiol Regul Integr Comp Physiol 304: R690–R701, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pelham CJ, Ketsawatsomkron P, Groh S, Grobe JL, de Lange WJ, Ibeawuchi SR, Keen HL, Weatherford ET, Faraci FM, Sigmund CD. Cullin-3 regulates vascular smooth muscle function and arterial blood pressure via PPARγ and RhoA/Rho-kinase. Cell Metab 16: 462–472, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 7: 803–815, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Rocha VZ, Libby P. The multiple facets of the fat tissue. Thyroid 18: 175–183, 2008. [DOI] [PubMed] [Google Scholar]
- 39.Schafer H, Diebel J, Arlt A, Trauzold A, Schmidt WE. The promoter of human p22/PACAP response gene 1 (PRG1) contains functional binding sites for the p53 tumor suppressor and for NFκB. FEBS Lett 436: 139–143, 1998. [DOI] [PubMed] [Google Scholar]
- 40.Singh MV, Kapoun A, Higgins L, Kutschke W, Thurman JM, Zhang R, Singh M, Yang J, Guan X, Lowe JS, Weiss RM, Zimmermann K, Yull FE, Blackwell TS, Mohler PJ, Anderson ME. Ca2+/calmodulin-dependent kinase II triggers cell membrane injury by inducing complement factor B gene expression in the mouse heart. J Clin Invest 119: 986–996, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stroka DM, Badrichani AZ, Bach FH, Ferran C. Overexpression of A1, an NF-κB-inducible anti-apoptotic bcl gene, inhibits endothelial cell activation. Blood 93: 3803–3810, 1999. [PubMed] [Google Scholar]
- 42.Sun X, He S, Wara AK, Icli B, Shvartz E, Tesmenitsky Y, Belkin N, Li D, Blackwell TS, Sukhova GK, Croce K, Feinberg MW. Systemic delivery of microRNA-181b inhibits nuclear factor-kappaB activation, vascular inflammation, and atherosclerosis in apolipoprotein E-deficient mice. Circ Res 114: 32–40, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARγ. Annu Rev Biochem 77: 289–312, 2008. [DOI] [PubMed] [Google Scholar]
- 44.Vaziri ND, Ni Z, Oveisi F. Upregulation of renal and vascular nitric oxide synthase in young spontaneously hypertensive rats. Hypertension 31: 1248–1254, 1998. [DOI] [PubMed] [Google Scholar]
- 45.Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab 23: 351–363, 2012. [DOI] [PubMed] [Google Scholar]
- 46.Wang N, Verna L, Chen NG, Chen J, Li H, Forman BM, Stemerman MB. Constitutive activation of peroxisome proliferator-activated receptor-γ suppresses pro-inflammatory adhesion molecules in human vascular endothelial cells. J Biol Chem 277: 34176–34181, 2002. [DOI] [PubMed] [Google Scholar]
- 47.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112: 1796–1808, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]