

Abstract
The olefination of aldehydes constitutes a most valuable and widely adopted strategy for constructing carbon-carbon double bonds in organic chemistry. While various synthetic methods have been made available for this purpose, no biocatalysts are known to mediate this transformation. Here, we report that engineered myoglobin variants can catalyze the olefination of aldehydes in the presence of α-diazoesters with high catalytic efficiency (up to 4,900 turnovers) and excellent E-diastereoselectivity (92–99.9% de). This transformation could be applied to the olefination of a variety of substituted benzaldehydes, heteroaromatic and alkyl aldehydes, also in combination with different alkyl α-diazoacetate reagents. This work provides a first example of biocatalytic aldehyde olefination and extends the spectrum of synthetically valuable chemical transformations accessible using metalloprotein-based catalysts.
Keywords: Biocatalysis, Wittig reaction, myoglobin, carbene transfer, protein engineering, aldehyde olfination
Graphical Abstract
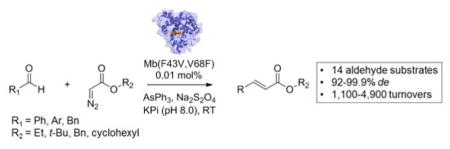
Biocatalytic Wittig olefination: Engineered variants of sperm whale myoglobin can serve as biocatalysts for the conversion of aldehydes and α-diazo esters to the corresponding α,β-unsaturated esters. This transformation proceeds with high catalytic efficiency and high E-diastereoselectivity and could be applied to a variety of different aldehyde substrates and α-diazo-esters.
The Wittig reaction[1] represents one of the most valuable and broadly adopted route for the construction of olefinic bonds during the synthesis of organic molecules.[2] Classically, this method involves the reaction between carbonyl compounds and phosphonium ylides, which are prepared by deprotonation of the corresponding phosphonium salts[3]. Because of the basic conditions required for the latter process, there has been a significant interest toward developing alternative methods to enable the olefination of aldehydes under milder, neutral conditions. In this regard, the transition metal catalyzed transformation of carbonyls in the presence of diazo compounds and tertiary phosphines has provided a particularly attractive strategy due to the readily accessibility of these reagents.[4] Over the recent years, a number of organometallic catalysts including Mo[4a], Re[4b–d], Rh[4e], Ir[4f], Ru[4g, 4h], Cu[4i] and Fe[4j–o] complexes have proven useful in this transformation, yielding E-olefins with modest to good catalytic activity (typically, 50–300 turnovers) and moderate to high E-selectivity (typically, 70–98% de). In contrast to the important progress made in the development of synthetic catalysts for aldehyde olefination, no natural enzyme or artificial biocatalysts[5] has been reported to promote this valuable transformation. An aldehyde olefination biocatayst would thus represent a valuable addition to the toolbox of currently available enzymes for asymmetric synthesis[6].
We and others have recently reported the ability of heme-dependent metalloproteins such as cytochrome P450s[7] and myoglobin[8] to engage diazo-containing reagents in carbene transfer reactions. In particular, we recently discovered that engineered variants of myoglobin can provide particularly efficient catalysts for olefin cyclopropanation[8a], carbene N–H insertion[8b], and carbene S–H insertion reactions[8c] in the presence of α-diazo ester reagents. Our mechanistic studies supported the intermediacy of an electrophilic heme-carbene complex[8a] which reacts with a nucleophilic olefin, amine or mercaptan to yield the carbene insertion adduct. These studies also showed the possibility to generate a transient sulfonium ylide intermediate upon attack of a thiol substrate to the myoglobin-bound carbenoid species.[8c] Building upon these findings and inspired by pioneering studies conducted by Woo and coworkers with metalloporphyrins[4j, 4l], we hypothesized that an analogous process could be exploited in the presence of tertiary phosphine nucleophiles to yield a myoglobin-bound phosphonium ylide. We further envisioned the latter could react with an aldehyde to yield an olefin via a Wittig reaction, with the active site of the protein potentially furnishing an asymmetric environment to influence the stereoselectivity of the reaction. Here, we report that engineered variants of myoglobin can mediate aldehyde olefination reactions across a range of aldehydes and α-diazoacetates with high catalytic activity and E-selectivity. This transformation proceeds in buffer and at room temperature, thus providing an extremely mild biocatalytic route for the olefination of aryl and alkyl aldehydes.
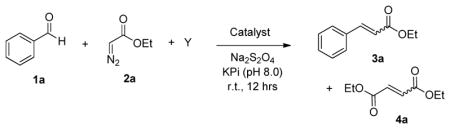
Guided by the hypothesis outlined above, we began our studies by testing the ability of wild-type sperm whale myoglobin to promote the conversion of benzaldehyde 1a and ethyl α-diazo acetate (EDA, 2a) to ethyl cinnamate 3a in the presence of triphenylphosphine (PPh3). To our delight, we observed formation of the desired product 3a with good diastereoselectivity (76% de for E-isomer), albeit with only modest activity (31 turnovers or TON) (Table 1, Entry 3). Both reducing (Na2S2O4) and oxygen-free conditions were found to be required for the observed Mb-dependent aldehyde olefination activity, consistent with the idea that the ferrous form of the hemoprotein is involved in the activation of the diazo compound. Additional experiments showed that hemin can also promote this transformation, but with reduced catalytic efficiency (22 TON) and lower diastereoselectivity (65% de) as compared to Mb (Table 1, Entry 1). In addition, the hemin reaction is much less chemoselective, yielding larger amounts of the carbene dimerization byproducts, diethyl fumarate and diethyl maleate (TON(3a)/TON(4a): 0.4 vs. 2.8 with Mb, Table 1). In an effort to improve the efficiency and selectivity of the Mb-mediated olefination reaction, a variety of trialkyl phosphines (e.g., PEt3, P(t-Bu)3, P(n-Bu)3) as well as heavier congeners of PPh3 (i.e., AsPh3, AsPh3, and BiPh3) were tested as a substitute for triphenylphosphine (Table 1). Interestingly, whereas neither BiPh3 nor any of the trialkyl phosphines led to the desired olefin product, the reaction in the presence of AsPh3 exhibited excellent diastereoselectivity, leading to the formation of trans-3a as the only detectable isomer (>99.9% de).
Table 1.
Catalytic activity of hemin and wild-type sperm whale myoglobin (Mb) in the olefination of benzaldehyde with ethyl α-diazoacetate (EDA).[a]
| |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Y | TON[b] (3a) | de (E)[c] | TON(3a)/TON(4a) |
| 1 | Hemin | PPh3 | 22 | 64% | 0.4 |
| 2 | Hemin[d] | AsPh3 | 4 | 91% | 0.1 |
| 3 | WT Mb | PPh3 | 31 | 76% | 2.8 |
| 4 | WT Mb | AsPh3 | 27 | 99.9% | 0.7 |
| 5 | WT Mb | SbPh3 | 5 | 99.9% | 0.4 |
| 6 | WT Mb | BiPh3 | 0 | - | - |
Reactions were carried out under anaerobic conditions with 10 mM 1a, 10 mM 2a, 20 μM catalyst, 10 mM Na2S2O4, and 10 mM Y for 12 hours at room temperature.
= nmol olefin / nmol catalyst. Errors in reported values are within ± 10%.
As determined by chiral gas chromatography.
With hemin at 60 μM.
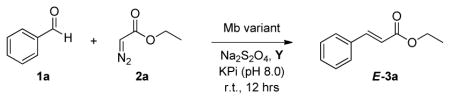
Encouraged by these results, we extended our investigations to a panel of Mb variants containing one or two mutations at the level of the protein active site. In sperm whale Mb, five amino acid residues (Leu29, Phe43, His64, Val68, Ile107) define the cavity located above the distal face of the heme cofactor (Figure S1).[9] Previously, we found that mutagenesis of these residues had a profound impact on the selectivity and activity of Mb-catalyzed carbene transfer reactions.[8] Accordingly, Mb active site variants were tested for their relative activity and selectivity in the olefination of benzaldehyde with EDA in the presence of either PPh3 or AsPh3. As summarized in Table 2, the active site mutations were found to have noticeable effects on the catalytic efficiency (TON), diastereoselectivity, and chemoselectivity of the reaction. Among the Mb variants tested, the double mutant Mb(F43V,V68F), used in combination with AsPh3, emerged as the most promising catalyst for this reaction, exhibiting 3-fold higher TON compared to wild-type Mb, excellent diasteroselectivity (>99.9% de), and high chemoselectivity toward aldehyde olefination over carbene dimerization. At a catalyst loading of 0.01 mol%, Mb(F43V,V68F) was determined to support over 1,100 catalytic turnovers for the conversion of 1 to E-3a, featuring an initial rate of 320 and 40 turnovers min−1 over the first minute and first 15 minutes, respectively (Figure S2). Importantly, nearly absolute E-selectivity as well as high chemoselectivity (TON(olefin):TON(dimer) > 4) are maintained under these conditions, the latter being achieved without the need for slow addition of the diazo compound as typically required for metalloporphyrin catalysts[4h, 4j, 4n] in order to minimize carbene dimerization.
Table 2.
Catalytic activity and selectivity of myoglobin variants in benzaldehyde olefination with EDA.[a]
| |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Y | TON | de (E) | TON(3a)/TON(4a) |
| 1 | WT Mb | PPh3 | 31 | 76% | 2.8 |
| AsPh3 | 27 | 99.9% | 0.7 | ||
| 2 | Mb(L29A) | PPh3 | 16 | 79% | 0.8 |
| AsPh3 | 34 | 99.9% | 0.5 | ||
| 3 | Mb(F43V) | PPh3 | 20 | 69% | 1.2 |
| AsPh3 | 35 | 99.9% | 1.1 | ||
| 4 | Mb(F43W) | PPh3 | 30 | 68% | 6.0 |
| AsPh3 | 56 | 99.9% | 2.7 | ||
| 5 | Mb(H64V) | PPh3 | 37 | 69% | 1.7 |
| AsPh3 | 35 | 99.9% | 0.5 | ||
| 6 | Mb(V68A) | PPh3 | 13 | 69% | 0.5 |
| AsPh3 | 36 | 99.9% | 1.1 | ||
| 7 | Mb(V68F)[b] | PPh3 | 28 | 64 | 0.7 |
| AsPh3 | 82 | 94 | 1.9 | ||
| 8 | Mb(L29A,H64V) | PPh3 | 57 | 70% | 1.4 |
| AsPh3 | 50 | 80% | 0.5 | ||
| 9 | Mb(H64V,V68A) | PPh3 | 7 | 73% | 0.8 |
| AsPh3 | 16 | 57% | 0.2 | ||
| 10 | Mb(F43V,V68F) | PPh3 | 38 | 64% | 2.9 |
| AsPh3 | 92 | 99.9% | 3.3 | ||
| 11 | Mb(F43V,V68F)[c] | AsPh3 | 1,170 | 99.9% | 4.2 |
Reaction conditions are the same as in Table 1.
With 5 μM catalyst (0.05 mol%).
With 1 μM catalyst (0.01 mol%).
Across nearly all Mb variants, the AsPh3-supported reactions consistently furnished higher degrees of diastereoselectivity as compared to those performed in the presence of PPh3 (Table 2). The only exception was Mb(H64V,V68A), for which a reversal of this trend was observed (70% vs. 57% de for reaction with PPh3 vs. AsPh3). Intriguing is also the differential effect of the active site mutations in the context of this reaction as compared to the carbene-mediated transformations previously investigated by our group.[8] For example, while the double mutation in Mb(H64V,V68A) greatly enhanced the reactivity and selectivity of Mb toward olefin cyclopropanation,[8a] the same mutations led to a reduction in TON, diastereo- and chemoselectivity for the aldehyde olefination reaction (Entry 9 vs. 1, Table 2). These differences highlight the peculiar active site requirements for favouring high reactivity and selectivity in the context of these related yet mechanistically distinct reactions.
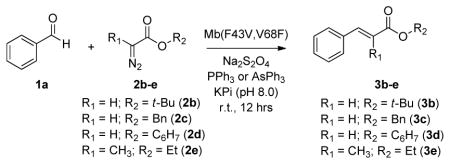
To investigate the scope of Mb(F43V,V68F) as aldehyde olefination catalyst, the reaction with benzaldehyde was carried out in the presence of other α-diazo esters, including tert-butyl (2b), benzyl (2c), and cyclohexyl (2d) α-diazo acetate as well as ethyl α-diazo-propanoate (2e). Notably, despite their variable alkyl chain, all of the α-diazo-acetates (2b–2d) could be readily processed by the biocatalyst to yield the corresponding trans β-aryl-α,β-unsaturated ester products, 3b–3d, with good (79–83% de) to excellent (98–99.9% de) selectivity in the presence of PPh3 and AsPh3, respectively (Table 3). In combination with 3c, Mb(F43V,V68F) gave the highest TON value (4,920) and conversion ratio (49%), whereas the use of the Mb(F43V,V68F)/3d/AsPh3 system provided an optimal combination of high catalytic activity (4,200 TON) with excellent stereocontrol (99.9% de). As such, the latter system was maintained for further studies on the scope of this hemoprotein across different aldehydes (vide infra). Under these optimized conditions, the TON values supported by Mb(F43V,V68F) in water and at room temperature are one to two orders of magnitudes higher than those previously reported for similar transformations catalyzed by organometallic complexes in organic solvent and at elevated temperature (50–300 TON[4a–k, 4m, 4n]). The only exception is the Fe(TPP)Cl-catalyzed olefination of benzaldehyde with EDA and PPh3 in toluene at 80°C reported by Zhang and coworkers, for which even higher TON (8,900) but also lower diastereoselectivity (84% de) were measured.[4h] In contrast to 2b–2d, no olefination product was observed in the presence of 2e, indicating that α-substitutions on the carbene moiety are not tolerated by the Mb catalyst in the context of this reaction.
Table 3.
Catalytic activity and selectivity of Mb(F43V,V68F) variants in benzaldehyde olefination with different α-diazo esters.[a]
| |||||
|---|---|---|---|---|---|
| Entry | Product | Y | TON | % de | % conv.[b] |
| 1 |
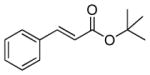 3b |
PPh3 | 160 | 79 | 32 |
| 2 | AsPh3 | 40 | 99.9 | 8 | |
| 3[c] | AsPh3 | 175 | 99.9 | 1.7 | |
| 3 |
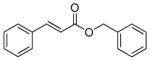 3c |
PPh3 | 185 | 83 | 37 |
| 4 | AsPh3 | 155 | 98 | 31 | |
| 5[c] | AsPh3 | 4,920 | 94 | 49 | |
| 5 |
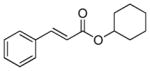 3d |
PPh3 | 155 | 79 | 31 |
| 6 | AsPh3 | 205 | 99.9 | 41 | |
| 7[c] | AsPh3 | 4,230 | 99.9 | 42 | |
Reaction conditions as described in Table 1 using 20 μM catalyst (0.2 mol%).
GC conversion.
Using 1 μM catalyst (0.01 mol%).
Next, the scope of Mb(F43V,V68F)-catalyzed olefination across different aldehyde substrates was investigated. As summarized in Scheme 1, a variety of monosubstituted benzyaldehyde derivatives (5a–13a) could be readily converted to the corresponding cyclohexyl trans-cinnamate esters 5b–13b with very good to excellent diastereoselectivity (99–99.9% de), with the Mb catalyst supporting from 1,110 (13b) to 3,400 turnovers (5b and 7b). Insights into the impact of electronic factors on the efficiency of the reaction could be gained from side-by-side comparison of the TON values for benzaldehyde derivatives carrying substituents of similar size but with different electronic properties. In particular, electron-deficient benzaldehydes were found to be consistently less reactive than their isosteric, electron-richer counterparts, as indicated by the lower TON measured for 8b vs. 3b, for 11b vs. 6b, and 9b vs. 7b. This trend contrasts with the higher reactivity of electron-poor aldehydes in transition metal-catalyzed olefination reactions in organic solvents[4h, 4j] and it can be rationalized on the basis of the higher level of hydration expected for benzaldehydes carrying electronwithdrawing groups in water.[10] A higher degree of hydration is expected to reduce the effective concentration of aldehyde susceptible to nucleophilic attack by the phoshonium ylide (vide infra), thus reducing the overall efficiency of the reaction.
Scheme 1.
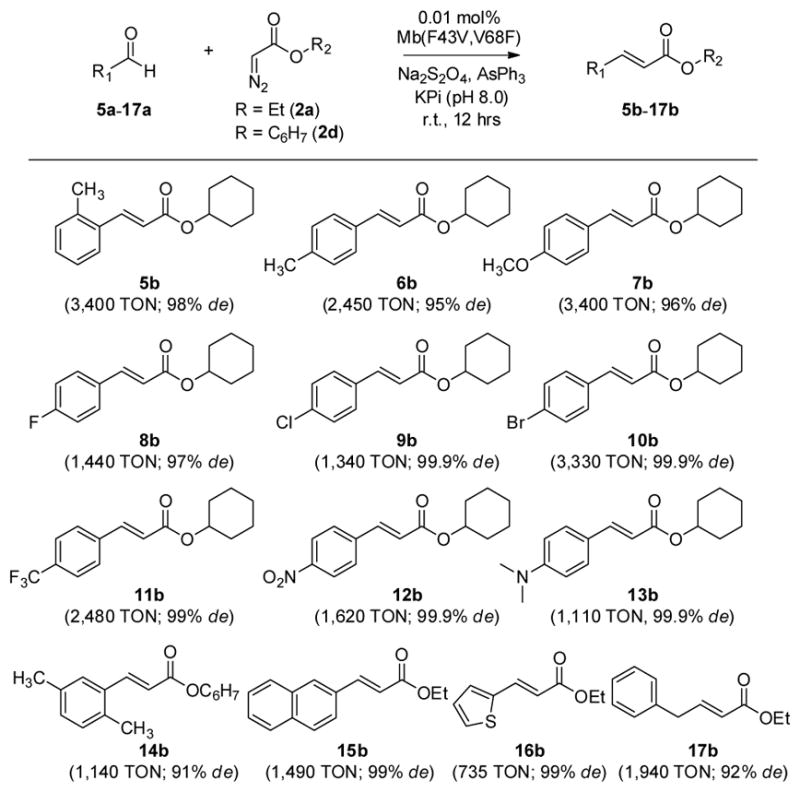
Substrate scope for Mb(H64V,V68A)-catalyzed aldehyde olefination. Reaction conditions: 10 mM aryl aldehyde, 1 μM Mb(F43V,V68F), 10 mM cyclohexyl α-diazo-acetate (2d), 10 mM AsPh3, 10 mM Na2S2O4.
The sucessful conversion of 14a to 14b showed that disubstituted benzyaldehydes could be also processed by the Mb(F43V,V68F) catalyst, albeit with lower efficiency (1,140 vs. 3,400 TON) and selectivity (91% vs. 98% de) compared to the monosubstituted counterpart, 5b. Substrates such as 2-naphthaldehyde (15a) and thiophene-2-carbaldehyde (16a) could also be converted to the corresponding trans olefin products 15b and 16b, respectively, with excellent selectivity (99% de), further supporting the broad substrate scope of Mb(F43V,V68F) across structurally different aryl aldehydes. Finally, the successful olefination of phenylacetaldehyde (17a) to give 17b (1,940 TON; 92% de) demonstrated the reactivity of the catalyst also toward alkyl aldehydes.
A proposed mechanism for the Mb-catalyzed aldehyde olefination reaction is presented in Scheme 2. Starting from the catalytically active ferrous form of the hemoprotein, the first step is envisioned to involve the formation of a heme-bound carbenoid intermediate (II in Scheme 2) upon reaction with the diazo reagent. This intermediate can be formally described as an iron(IV)-carbene complex or as a FeII←{:CHCO2R} complex, the latter being predicted to be a more stable resonance form at least in the context of synthetic iron-porphyrin systems.[11] Regardless of its exact nature, our previous studies showed that this species has electrophilic character[8a] and can react with thiol nucleophiles to generate a transient sulfonium ylide.[8c] Accordingly, attack of the nucleophilic PPh3 (or AsPh3) to the heme-carbene intermediate is envisioned to ensue giving rise to a phosphonium ylide (species III, Scheme 2). The latter would then react with the aryl aldehyde to generate a oxaphosphetane intermediate[12] (species IV, Scheme 2), whose rearrengement yields the olefin product and phosphine oxide as the byproduct.
Scheme 2.
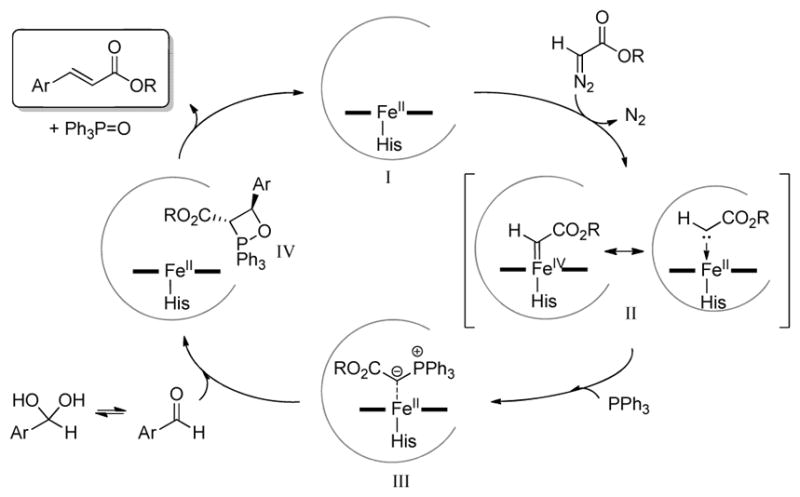
Proposed mechanism and catalytic steps for the myoglobin-catalyzed olefination of aryl aldehydes.
In view of the mechanistic model of Scheme 2, a number of considerations can be made in regard of the results described earier. A first one concerns the role of the biocatalyst on influencing the stereoselectivity of the reaction. Importantly, the Mb reaction with EDA and PPh3 in the absence of aldehyde was found to accumulate the phosphorane intermediate (Ph3P=CHCO2Et) in solution, thus supporting the occurrence of the steps I → II → III proposed in Scheme 2. Insightfully, a reaction of premade phosphorane with benzaldehyde yielded E-3b in 80% de both in the presence and in the absence of the Mb catalyst. Such diastereoselectivity differs from that observed in the olefination reactions with nearly all of the Mb variants starting from benzaldehyde and EDA (64–76% de, Table 2). This result together with the higher diastereoselectivity obtained with Mb vs. hemin (Table 1) and the effect of active site mutations on the E : Z ratio of the olefin product (Table 2) support the involvement of the protein environment in affecting the stereochemical outcome of the reaction. Since the stereoselectivity of the Wittig reaction is largely dictated by the relative orientation of the ylide and aldehyde during formation of the oxaphosphetane intermediate,[2b, 2c, 12b, 15] the asymmetric induction imposed by the hemoprotein scaffold most likely occurs during the conversion of III to IV. Possibly, this effect is mediated by coordination of the ylide to the heme iron and/or by interaction of the ylide with the distal cavity of the protein. While Mb has naturally evolved to bind small ligands (i.e. O2), the steric feasibility of the putative intermediate III is suggested by our previous finding that the distal heme pocket in Mb can accomodate rather bulky ligands and substrates.[8, 13] This process can be further facilitated by the ability of the distal histidine, His64 (Figure S2), to swing open upon ligand binding[14], thereby creating a larger cavity above the heme.
Another interesting point concerns the structure-reactivity data obtained with the different diazo compounds (Table 3). These studies showed that while relatively large and bulky groups within the ester group of the carbenoid moiety are well tolerated by the Mb(F43V,V68F) catalyst, α-substitutions are not. Since we previosly established that ethyl α-diazo-propanoate (2e) is a viable carbene donor in Mb-catalyzed olefin cyclopropanation,[8a] it can be derived that α-substitutions negatively affect catalytic steps downstream of the formation of the heme-carbene complex during aldehyde olefination. Reasonably, the increased steric hindrance provided by the α-methyl group may disfavour attack of the PPh3 to the heme-carbene (II→III, Scheme 2), thus preventing formation of the key phosphonium ylide intermediate.
In spite of the high TON supported by Mb(F43V,V68F), the aldehyde-to-olefin conversion in this reaction was surprisingly found to not exceed 50%. Increasing the α-diazo ester : aldehyde ratio did not improve the yield and resulted in a larger amount of the carbene dimerization product. Through control experiments, catalyst inhibition by action of the aldehyde, phosphine, or olefin product could be ruled out as a possible cause for this phenomenon. A reduction in TON was observed however upon addition of increasing amounts of phosphine oxide to the Mb(F43V,V68F)-catalyzed reaction (Figure S3). Overcoming the inhibitory effect exerted by the phosphine/arsine oxide could thus provide a way to further enhance the efficiency of this Mb-mediated transformation in the future.
In summary, our results show that engineered myoglobins can provide efficient and selective biocatalysts for the olefination of aldehydes under mild and neutral conditions. To our knowledge, this report represents the first example of a biocatalytic strategy for aldehyde olefination. Using the most promising Mb-based catalyst identified in this work, Mb(F43V,V68F), a variety of aryl aldehydes and alkyl α-diazo acetates could be converted to the corresponding olefin products with high catalytic efficiency (1,100–4,900 TON) and very good to excellent E-selectivity (94–99.9% de). The Mb-catalyzed aldehyde olefination reported here contributes to expand the growing number of synthetically valuable transformations accessible through catalysis with engineered and artificial metalloproteins.[5, 7–8, 13, 16]
Supplementary Material
Acknowledgments
This work was supported by the U.S. National Institute of Health grant GM098628. MS instrumentation was supported by the U.S. NSF grant CHE-0946653.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Wittig G, Geissler G. Liebigs Ann Chem. 1953;580:44–57. [Google Scholar]
- 2.a) Maryanoff BE, Reitz AB. Chem Rev. 1989;89:863–927. [Google Scholar]; b) Kolodiazhnyi OI. Phosphorus Ylides: Chemistry and Applications in Organic Chemistry. Wiley-VCH; New York: 1999. [Google Scholar]; c) Abell AD, Edmonds MK. Modern Carbonyl Olefination. Wiley-VCH; Weinheim: 2004. [Google Scholar]; d) Nicolaou KC, Harter MW, Gunzner JL, Nadin A. Liebigs Ann/Recl. 1997:1283–1301;. [Google Scholar]; e Cristau HJ. Chem Rev. 1994;94:1299–1313. [Google Scholar]
- 3.Zhang XM, Bordwell FG. J Am Chem Soc. 1994;116:968–972. [Google Scholar]
- 4.a) Lu XY, Fang H, Ni ZJ. J Organomet Chem. 1989;373:77–84. [Google Scholar]; b) Herrmann WA, Wang M. Angew Chem Int Ed. 1991;30:1641–1643. [Google Scholar]; c) Ledford BE, Carreira EM. Tetrahedron Lett. 1997;38:8125–8128. [Google Scholar]; d) Santos AM, Romao CC, Kuhn FE. J Am Chem Soc. 2003;125:2414–2415. doi: 10.1021/ja021315c. [DOI] [PubMed] [Google Scholar]; e) Lebel H, Paquet V. J Am Chem Soc. 2004;126:320–328. doi: 10.1021/ja038112o. [DOI] [PubMed] [Google Scholar]; f) Lebel H, Ladjel C. Organometallics. 2008;27:2676–2678. [Google Scholar]; g) Graban E, Lemke FR. Organometallics. 2002;21:3823–3826. [Google Scholar]; h) Chen Y, Huang L, Ranade MA, Zhang XP. J Org Chem. 2003;68:3714–3717. doi: 10.1021/jo0341158. [DOI] [PubMed] [Google Scholar]; i) Lebel H, Davi M. Adv Synth Catal. 2008;350:2352–2358. [Google Scholar]; j) Mirafzal GA, Cheng GL, Woo LK. J Am Chem Soc. 2002;124:176–177. doi: 10.1021/ja016721v. [DOI] [PubMed] [Google Scholar]; k) Chen Y, Huang LY, Zhang XP. Org Lett. 2003;5:2493–2496. doi: 10.1021/ol0347444. [DOI] [PubMed] [Google Scholar]; l) Cheng GL, Mirafzal GA, Woo LK. Organometallics. 2003;22:1468–1474. [Google Scholar]; m) Aggarwal VK, Fulton JR, Sheldon CG, de Vicente J. J Am Chem Soc. 2003;125:6034–6035. doi: 10.1021/ja029573x. [DOI] [PubMed] [Google Scholar]; n) Cao P, Li CY, Kang YB, Xie ZW, Sun XL, Tang Y. J Org Chem. 2007;72:6628–6630. doi: 10.1021/jo0709899. [DOI] [PubMed] [Google Scholar]; o) Li CY, Wang XB, Sun XL, Tang Y, Zheng JC, Xu ZH, Zhou YG, Dai LX. J Am Chem Soc. 2007;129:1494–1495. doi: 10.1021/ja068642v. [DOI] [PubMed] [Google Scholar]
- 5.a) Heinisch T, Ward TR. Eur J Inorg Chem. 2015:3406–3418. [Google Scholar]; b) Yu FT, Cangelosi VM, Zastrow ML, Tegoni M, Plegaria JS, Tebo AG, Mocny CS, Ruckthong L, Qayyum H, Pecoraro VL. Chem Rev. 2014;114:3495–3578. doi: 10.1021/cr400458x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lewis JC. Acs Catalysis. 2013;3:2954–2975. [Google Scholar]; d) Rosati F, Roelfes G. Chemcatchem. 2010;2:916–927. [Google Scholar]
- 6.a) Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K. Nature. 2012;485:185–194. doi: 10.1038/nature11117. [DOI] [PubMed] [Google Scholar]; b) Reetz MT. J Am Chem Soc. 2013;135:12480–12496. doi: 10.1021/ja405051f. [DOI] [PubMed] [Google Scholar]; c) Fasan R. ACS Catal. 2012;2:647–666. [Google Scholar]; d) Turner NJ. Nat Chem Biol. 2009;5:568–574. doi: 10.1038/nchembio.203. [DOI] [PubMed] [Google Scholar]
- 7.a) Coelho PS, Brustad EM, Kannan A, Arnold FH. Science. 2013;339:307–310. doi: 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]; b) Coelho PS, Wang ZJ, Ener ME, Baril SA, Kannan A, Arnold FH, Brustad EM. Nat Chem Biol. 2013;9:485–487. doi: 10.1038/nchembio.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Wang ZJ, Peck NE, Renata H, Arnold FH. Chem Sci. 2014;5:598–601. doi: 10.1039/C3SC52535J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Bordeaux M, Tyagi V, Fasan R. Angew Chem Int Ed. 2015;54:1744–1748. doi: 10.1002/anie.201409928. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sreenilayam G, Fasan R. Chem Commun. 2015;51:1532–1534. doi: 10.1039/c4cc08753d. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tyagi V, Bonn RB, Fasan R. Chem Sci. 2015;6:2488–2494. doi: 10.1039/c5sc00080g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lionetti C, Guanziroli MG, Frigerio F, Ascenzi P, Bolognesi M. J Mol Biol. 1991;217:409–412. doi: 10.1016/0022-2836(91)90744-q. [DOI] [PubMed] [Google Scholar]
- 10.Mcclelland RA, Coe M. J Am Chem Soc. 1983;105:2718–2725. [Google Scholar]
- 11.Khade RL, Fan WC, Ling Y, Yang L, Oldfield E, Zhang Y. Angew Chem Int Ed. 2014;53:7574–7578. doi: 10.1002/anie.201402472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Vedejs E, Snoble KAJ. J Am Chem Soc. 1973;95:5778–5780. [Google Scholar]; b) Byrne PA, Gilheany DG. Chem Soc Rev. 2013;42:6670–6696. doi: 10.1039/c3cs60105f. [DOI] [PubMed] [Google Scholar]
- 13.Bordeaux M, Singh R, Fasan R. Bioorg Med Chem. 2014;22:5697–5704. doi: 10.1016/j.bmc.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ringe D, Petsko GA, Kerr DE, de Montellano PRO. Biochemistry. 1984;23:2–4. doi: 10.1021/bi00296a001. [DOI] [PubMed] [Google Scholar]
- 15.Vedejs E, Peterson MJ. Topics in Stereochemistry. Vol. 21. Wiley-CH; New York: 1994. [Google Scholar]
- 16.a) Ozaki S, Matsui T, Watanabe Y. J Am Chem Soc. 1997;119:6666–6667. [Google Scholar]; b) Collot J, Gradinaru J, Humbert N, Skander M, Zocchi A, Ward TR. J Am Chem Soc. 2003;125:9030–9031. doi: 10.1021/ja035545i. [DOI] [PubMed] [Google Scholar]; c) Ueno T, Koshiyama T, Ohashi M, Kondo K, Kono M, Suzuki A, Yamane T, Watanabe Y. J Am Chem Soc. 2005;127:6556–6562. doi: 10.1021/ja045995q. [DOI] [PubMed] [Google Scholar]; d) Lin YW, Yeung N, Gao YG, Miner KD, Tian SL, Robinson H, Lu Y. Proc Nat Acad Sci USA. 2010;107:8581–8586. doi: 10.1073/pnas.1000526107. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Mayer C, Gillingham DG, Ward TR, Hilvert D. Chem Commun. 2011;47:12068–12070. doi: 10.1039/c1cc15005g. [DOI] [PubMed] [Google Scholar]; f) Bos J, Fusetti F, Driessen AJM, Roelfes G. Angew Chem Int Ed. 2012;51:7472–7475. doi: 10.1002/anie.201202070. [DOI] [PubMed] [Google Scholar]; g) Hyster TK, Knorr L, Ward TR, Rovis T. Science. 2012;338:500–503. doi: 10.1126/science.1226132. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Liu X, Yu Y, Hu C, Zhang W, Lu Y, Wang J. Angew Chem Int Ed. 2012;51:4312–4316. doi: 10.1002/anie.201108756. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Singh R, Bordeaux M, Fasan R. ACS Catal. 2014;4:546–552. doi: 10.1021/cs400893n. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Singh R, Kolev JN, Sutera PA, Fasan R. ACS Catal. 2015;5:1685–1691. doi: 10.1021/cs5018612. [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Giovani S, Singh R, Fasan R. Chem Sci. 2016 doi: 10.1039/C5SC02857D. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Hyster TK, Farwell CC, Buller AR, McIntosh JA, Arnold FH. J Am Chem Soc. 2014;136:15505–15508;. doi: 10.1021/ja509308v. [DOI] [PMC free article] [PubMed] [Google Scholar]; m) Farwell CC, McIntosh JA, Hyster TK, Wang ZJ, Arnold FH. J Am Chem Soc. 2014;136:8766–8771. doi: 10.1021/ja503593n. [DOI] [PMC free article] [PubMed] [Google Scholar]; n) Sommer DJ, Vaughn MD, Ghirlanda G. Chem Commun. 2014;50:15852–15855. doi: 10.1039/c4cc06700b. [DOI] [PubMed] [Google Scholar]; o) Srivastava P, Yang H, Ellis-Guardiola K, Lewis JC. Nat Commun. 2015;6:7789. doi: 10.1038/ncomms8789. [DOI] [PMC free article] [PubMed] [Google Scholar]; p) Yu Y, Cui C, Liu X, Petrik ID, Wang J, Lu Y. J Am Chem Soc. 2015;137:11570–11573. doi: 10.1021/jacs.5b07119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.