

Abstract
Mitochondrial myopathies belong to a larger group of systemic diseases caused by morphological or biochemical abnormalities of mitochondria. Mitochondrial disorders can be caused by mutations in either the mitochondrial or the nuclear genome. Only 5% of all mitochondrial disorders are autosomal dominant. We analyzed DNA from members of a previously reported Puerto Rican kindred with an autosomal dominant mitochondrial myopathy (Heimann-Patterson et al. 1997). Linkage analysis suggested a putative locus on the pericentric region of the long arm of chromosome 22 (22q11). Using the tools of integrative genomics, we established C22orf16 (later designated as CHCHD10) as the only high scoring mitochondrial candidate gene in our minimal candidate region. Sequence analysis revealed a double missense mutation (R15S; G58R) in cis in CHCHD10 which encodes a coiled-coil helix coiled-coil helix protein of unknown function. These two mutations completely co-segregated with the disease phenotype and were absent in 1481 Caucasian and 80 Hispanic (including 32 Puerto Rican) controls. Expression profiling showed that CHCHD10 is enriched in skeletal muscle. Mitochondrial localization of the CHCHD10 protein was confirmed using immunofluorescence in cells expressing either wild-type or mutant CHCHD10. We found that expression of the G58R, but not the R15S, mutation induced mitochondrial fragmentation. Our findings identify a novel gene causing mitochondrial myopathy, thereby expanding the spectrum of mitochondrial myopathies caused by nuclear genes. Our findings also suggest a role for CHCHD10 in the morphologic remodeling of the mitochondria.
Keywords: Mitochondrial myopathy, genetics, CHCHD10, mitochondria
Introduction
Mitochondria play a vital role in various biological processes including oxidative phosphorylation, calcium buffering, apoptosis, and various metabolic pathways. Mitochondrial diseases of juvenile-onset have an incidence of 1 in 5000 (1). Only 5% of all mitochondrial disorders are autosomal dominant. Mitochondrial diseases are phenotypically and genetically heterogeneous disorders that are due to the dysfunction of the mitochondrial respiratory chain. These disorders can be caused by mutations in either the mitochondrial DNA (mtDNA) or the nuclear DNA (nDNA). The mtDNA is a 16.6 kb double stranded circular DNA that encodes 13 respiratory chain proteins, 22 tRNAs and 2 rRNAs (2). The nDNA is estimated to encode about 1,500 mitochondrial proteins (3). These proteins represent most of the respiratory chain subunits, many nuclear factors needed for the proper assembly and maintenance of the respiratory chain, the mitochondrial import machinery, the mtDNA replication and repair system and the control of mitochondrial dynamics. Only a subset of these nuclear encoded proteins has been identified so far and some have been linked to human diseases. For instance, mutations have been identified in genes encoding subunits of the respiratory chain or in ancillary proteins (4). In addition, genetic defects in intergenomic signaling which leads to multiple mtDNA deletions or depletion syndromes, have also been described (4), as well as, alterations in mitochondrial fusion or fission as seen in cases of mutations in MFN2 and GDAP1 (5).
In this report, using unbiased approaches, such as, linkage analysis and an algorithm that allows for prediction of mitochondrial genes in the nuclear genome, we report a family with an autosomal dominant mitochondrial myopathy due to mutations in CHCHD10 which encodes a novel nuclear-encoded mitochondrial coiled-coil helix coiled-coil helix (CHCH) protein of unknown function (6). Preliminary results have already been published in abstract form (6).
Materials and Methods
Sequencing analysis of the CHCHD10 gene
Genomic DNA was extracted from transformed lymphoblastoid cell lines or whole blood using standard protocols (Qiagen, Valencia, CA). Intronic primers covering the coding sequence were designed at least 50 bp away from the intron/exon boundaries. Primers were designed using Oligo Analyzer (IDT, Coralville, IA), ExonPrimer (Institute of Human Genetics, Germany) and UCSC Genome Bioinformatics Browser. Genomic DNA was amplified according to standard protocols. Unconsumed dNTPs and primers were digested with Exonuclease I and Shrimp Alkaline Phosphatase (ExoSAP-IT) (USB, Cleveland, OH). Fluorescent dye labeled single strand DNA was amplified with Beckman Coulter sequencing reagents (GenomeLab DTCS Quick Start Kit) followed by single pass bi-directional sequencing with CEQ™ 8000 Genetic Analysis System (Beckman Coulter, Fullerton, CA). Forward primer was used for mutation screening and all variations were confirmed by reverse sequencing. When a variant was identified, it was first excluded in the dbSNP, 1000 Genomes, and the Exome Variant Server (NHLBI GO Exome Sequencing Project; http://evs.gs.washington.edu/EVS/) (7, 8) databases, and then a large number of control DNA samples were analyzed to exclude the possibility of a polymorphism.
Expression constructs
A full length human CHCHD10 cDNA clone was used as a template for construction of the expression constructs. Two primers anchored with an EcoRI (FEcoR1: 5′CAGGAATTCATGCCTCGGGGAAGCCGCAGC3′) and BamHI (RBamH1 5′CATGGATCCGGGCAGGGAGCTCAGACCA3′) were used to amplify the full length coding sequence. For the Myc-tagged constructs, the Myc tag and NotI site were added at the end of the CHCHD10 gene by double PCR. The amplified fragment was cloned into plasmid vector pBluescript M13. The CHCHD10 sequence was verified by direct sequencing. The mutations were introduced into the plasmid vector by site-directed mutagenesis using primers containing each respective mutation. The CHCHD10 (WT or mutant) linked to the Myc-tag were sub-cloned into pSPORT6 vector at EcoRI/NotI site for use in mitochondrial colocalization studies. Similarly, a dual expression vector pIRES2-ZsGreen1 was used to create such constructs as WT and mutant CHCHD10-ZsGreen1 (Clontech, Mountain View, CA) for use in studies of mitochondrial morphology. Mitochondrial localization was examined using pDsRed2-Mito (Clontech, Mountain View, CA).
Expression of wild-type and mutant CHCHD10
HEK293 cells were grown on collagen-coated plates in Dulbecco's modified Eagle's medium containing 10% (v/v) human serum, 2 mM L-glutamine, 2 U/ml penicillin, and 2 mg/ml streptomycin at 37°C in a humidity-controlled incubator with 5% CO2. The cells were transiently transfected with different combinations of expression vectors using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to manufacturer's instructions.
Immunocytochemistry and confocal microscopy
After 24 hours of transfection, the cells were fixed with 4 % Paraformaldehyde in PBS. Following permeabilization with PBS containing 0.1 % Triton, 1 % BSA and 2 % Horse serum cells were incubated with anti-Myc primary monoclonal antibody (Covince; CAT#MMS-150P) overnight at 4°C. After washing the cells extensively with PBS, secondary anti-mouse FITC conjugated was added to the cells for 1h at room temperature. The staining was then visualized using a Carl Zeiss LSM510 META or a Nikon A1R confocal microscope. To quantify mitochondrial morphology, a custom ImageJ macro was utilized as previously described (9). Statistical analysis was performed using the unpaired Student's t-test and values of p<0.05 were considered significant.
Western blotting
Western blotting was performed using the protocol previously described (10). We generated the CHCHD10 antibody, which was raised in rabbit using a polypeptide of human CHCHD10 (amino acids 17-31, AAPSAHPPAHPPPSA). The antiserum was affinity-purified.
Bioinformatics
For linkage analysis, individual genotypes were entered into Progeny software. The data were extracted from Progeny and location for the markers was obtained using 36.7/hg18. Mega2 was used to generate a SimWalk2 list. In SimWalk2, the data were run using the non-parametric and parametric methods. MitoCarta which is based on the published Maestro algorithm was used to identify the candidate mitochondrial gene (11). Mitochondrial localization signal prediction was determined using MitoProt (12). Conserved domains were determined with NCBI Conserved Domain Database (13). Sequence cluster alignment was performed with NCBI HomoloGene that uses BlastP to compare related sequences.
Results
Patients
In 1997, Heimann-Patterson and colleagues described a large Puerto Rican kindred, with 15 affected family members presenting with exercise intolerance and a proximal myopathy in the first decade of life, consistent with an autosomal dominant inheritance pattern (8). Weakness also occurred in the shoulder girdle and neck flexor muscles in the second decade, with mild facial weakness developing in the third to fourth decades. None of the patients developed cardiac problems. Some patients had elevated serum lactate and creatine kinase levels. Muscle biopsy showed ragged red fibers and lipid accumulation (Supplementary Figure 1) (8). Electron microscopy showed a combination of the following ultrastructural abnormalities in the presence of normal myofibrillar architecture: increased number of mitochondria containing abnormal circular cristae and globular mitochondrial inclusions, marginalization of mitochondria, as well as, glycogen and lipid accumulation (Figure 1). Biochemical analysis showed reduced muscle activities of mitochondrial NADH–cytochrome c reductase (Complex I + III), succinate–cytochrome c reductase (Complex II + III), and cytochrome c oxidase (Complex IV) (8). After normalization to citrate synthase, the complex IV defect was more severe (6% of mean control value), as compared to the defects in complex I + III (20%) or complex II + III (12%).
Figure 1.
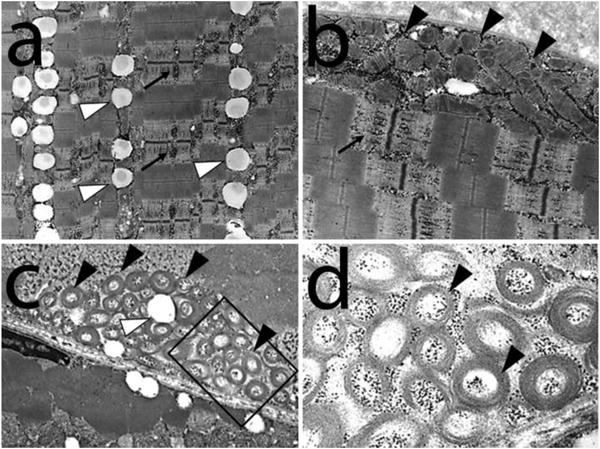
Electron micrographs from the index patient shows a variety of ultrastructural abnormalities in the presence of normal myofibrillar architecture (a-d). Note the accumulation of glycogen (black arrows) and lipids (white arrow heads). There is also an increased number of mitochondria containing abnormal circular cristae (black arrow heads) and marginalization of mitochondria (panels b and c). Selected area (square) from panel (b) is shown in higher power in panel (d).
Sequencing of the muscle mitochondrial genome of one affected individual did not reveal any mutations with pathogenic significance (8). Southern blot with a mtDNA probe did not show any mtDNA deletions (8).
Genetic linkage of autosomal dominant mitochondrial myopathy to 22q11
A genome wide screen was conducted using over 325 fluorescent microsatellites markers with an average interval 10 cM apart. Evidence of linkage was first observed with marker D22S315. Additional microsatellites markers flanking this region were genotyped using PCR, and haplotypes were constructed. All affected individuals shared the same haplotype for markers D22S1174, D22S925, D22S419, D22S315, D22S1148 and D22S1154 (Supplementary Figure 2). The public databases ENSEMBL and NCBI were used to determine the location of these markers on the physical map. Two-point LOD scores over 1.3 were obtained for five consecutive microsatellite markers, with the highest two-point LOD score of 1.8 at θ=0 at D22S925 (Supplementary Table 1). We further conducted SNP genotyping using the Illumina genome wide SNP markers (close to 6000 SNPs) which verified the microsatellite data with a multilocus LOD score close to 2.5 (Supplementary Figure 3), with a minimal candidate region (MCR) of 4.55Mb between markers rs2283792 and rs738402 (Supplementary Figure 3).
Identification of the candidate gene
No other large mitochondrial myopathy families were available to us to further narrow down the MCR. Further efforts were therefore focused on identifying the causative gene in our original family. There were about 200 genes in the MCR. Genes in the MCR were analyzed on the basis of relevance to mitochondrial function and expression profile. Public databases including NCBI, ENSEMBL, UCSC Genome Bioinformatics and Celera were used to identify transcripts mapping to the MCR on chromosome 22. Sequence contigs obtained from public and private databases were also examined using the Genscan gene prediction software. Candidate genes were identified for sequencing based on their putative role in mitochondrial biology. The prediction software PSORT II was used to identify transcripts with mitochondrial translocation signals. Accordingly, 19 known and predicted candidate genes, were sequenced and excluded for mutations co-segregating with disease in this family (Supplementary Table 2).
Since there were no other easily identifiable mitochondrial genes in the MCR, we used MitoCarta which is based on the published Maestro algorithm, to identify the candidate mitochondrial gene (11). MitoCarta program integrates eight different types of genomic data to make predictions about the human mitochondrial proteome and assign every gene a score of likelihood of localization to the mitochondrion. MitoCarta combines predictions of mitochondrial targeting signal, protein-domain enrichment, yeast homology, ancestry, tandem mass spectrometry, coexpression, and transcriptional induction during mitochondrial biogenesis. Chromosome 22 open reading frame 16 (C22orf16) was identified as the only high scoring mitochondrial gene in the candidate region (Figure 2). This gene has since been renamed coiled-coil-helix-coiled-coil-helix domain-containing protein 10, mitochondrial precursor (CHCHD10). CHCHD10 contains a conserved mitochondrial targeting signal, is highly co-expressed with other mitochondrial genes, and is transcriptionally activated during mitochondrial biogenesis. Additionally, the mouse homolog of this gene, Chchd10, is highly expressed in skeletal muscle, consistent with a potential role in myopathy. Interestingly, CHCHD10 was computationally predicted to be a regulator of oxidative phosphorylation ranking 422 out of 18128 genes in the mouse/human genome for their co-expression with oxidative phosphorylation (14). In line with this, knockdown of CHCHD10 in cell culture has already been shown to cause a defect in complex IV function (15). Therefore, our finding of reduced complex IV function in the skeletal muscle tissue from our patients is consistent with the known function of CHCHD10.
Figure 2.

MitoCarta which is based on the published Maestro algorithm was used to identify the candidate mitochondrial gene. Chromosome 22 open reading frame 16 (C22orf16) was identified as the only high scoring mitochondrial gene in the candidate region.
CHCHD10 is mutated in autosomal dominant mitochondrial myopathy
Sequencing of CHCHD10 in the index patient identified two silent variants and a double missense mutation in cis in the coding region (Figure 3). The first, a C to A substitution at position 43 at the level of coding DNA (c.43C>A), is predicted to result in an amino acid substitution of arginine by a serine at codon 15 at the protein level (p.R15S). The second mutation at nucleotide position 172 (c.172G>C) is predicted to cause a substitution of a glycine by an arginine at codon 58 (p.G58R). Both mutations completely co-segregated with disease phenotype in this large pedigree (Figure 3). Both these mutations were not present in the dbSNP, 1000 genomes databases, or the Exome Variant Server (NHLBI GO Exome Sequencing Project). We also did not observe these mutations in samples from 1481 Caucasian and 80 Hispanic (including 32 Puerto Rican) control subjects (a total of 1561 controls, representing 3122 chromosomes). In addition, we analyzed the control data available through the 1000 Genomes Project. In total, there is sequencing data on 55 Puerto Rican controls. Exome sequencing revealed no variants at the positions corresponding to either of our two variants: R15S (lowest depth 2×, highest depth 25×) and G58R (lowest depth 5×, highest depth 35×). Thus, we have excluded these variants in a total of 87 ethnically matched controls (representing 174 chromosomes).
Figure 3.
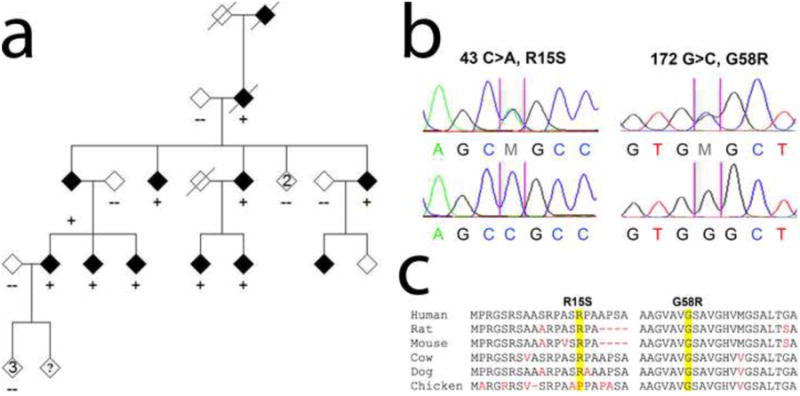
Mutations of CHCHD10 in patients with mitochondrial myopathy. Sequencing of CHCHD10 in the index patient identified a double missense mutation (R15S; G58R) in cis in the coding region (a and b). Both mutations co-segregated with disease phenotype in this large mitochondrial myopathy pedigree (a). The first, a C to A substitution at position 43 at the level of coding DNA (c.43C>A), is predicted to result in an amino acid substitution of arginine by a serine at codon 15 at the protein level (p.R15S) (b). The second mutation at nucleotide position 172 (c.172G>C) is predicted to cause a substitution of a glycine by an arginine at codon 58 (p.G58R) (b). (c) Evolutionary conservation of amino acids in the mutated region of CHCHD10 in various species. Mutated amino acids are highlighted in yellow. Nonconserved residues are shown in red.
Expression profile of CHCHD10
We next explored the expression profile of CHCHD10 by determining its Shanon entropy values as described by Schug et al. (15, 16). These entropy values are indicative for the enrichment of the mRNA in one given tissue when compared with a set of other tissues. The dataset containing the genome-wide expression data across 12 human tissues was obtained from a public accessible dataset via NCBI GEO (16, 17) which showed that CHCHD10 expression is enriched in skeletal muscle (Supplementary Figure 4). Moreover, the Broad Institute Genotype-Tissue Expression Portal (GTEx) dataset containing expression data from various human tissues also showed that CHCHD10 expression is enriched in skeletal muscle (data not shown). Consistent with these data, we observed an enrichment of CHCHD10 protein expression in human skeletal muscle by Western blot analysis (Supplementary Figure 4).
CHCHD10 is a CHCH protein that localizes to the mitochondria
CHCHD10 is encoded by four exons (Figure 4). CHCHD10 is conserved in chicken, mouse, rat, cow, dog, and human (Figure 3). CHCHD10 has a highly conserved CHCH domain at the C-terminus (amino acids 102-133). Both the mutations we identified occur outside this domain (Figure 4). Of the two mutations identified in our pedigree, the G58R substitution is located in a highly conserved region of CHCHD10 and affects a conserved amino acid residue (Figure 3).
Figure 4.
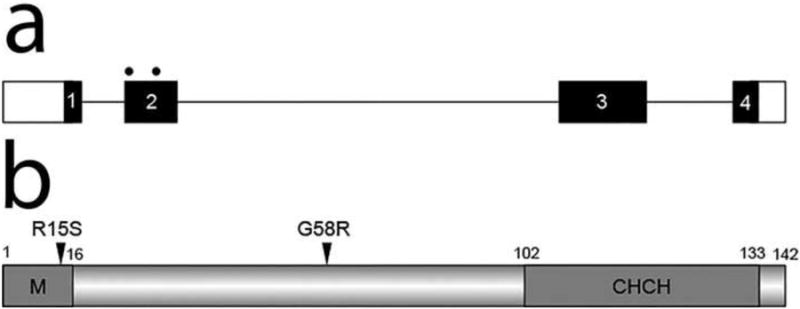
The coiled-coil helix coiled-coil helix protein (CHCHD10) is encoded by four exons. Both mutations occur in exon 2 (a). Predicted structural and functional domains of CHCHD10, a protein of 142 amino acids, are shown (b). The only predicted structural and functional domain is a CHCH domain (102-133). Sequence analysis revealed an N-terminal mitochondrial localization signal (M; amino acids 1-16).
No conserved domains were identified in the N-terminal half of the protein. However, sequence analysis revealed an N-terminal mitochondrial localization signal (amino acids 1-16). The R15S substitution occurs in this potential mitochondrial transit peptide (Figure 4). However, short mitochondrial proteins often do not need such a targeting sequence for effective targeting. In line with this, in silico analysis revealed no effect of CHCHD10 mutations on mitochondrial targeting (data not shown). To test the functionality of the mitochondrial localization signal in vivo, we performed immunolocalization studies which showed Myc-tagged wild-type (WT)-CHCHD10 colocalizing with DsRed-Mito-labeled mitochondria (Figure 5). The mutant CHCHD10 protein containing both the R15S and G58R mutations also targeted to the mitochondria (Figure 5), implying that these mutations have no effect on mitochondrial localization.
Figure 5.
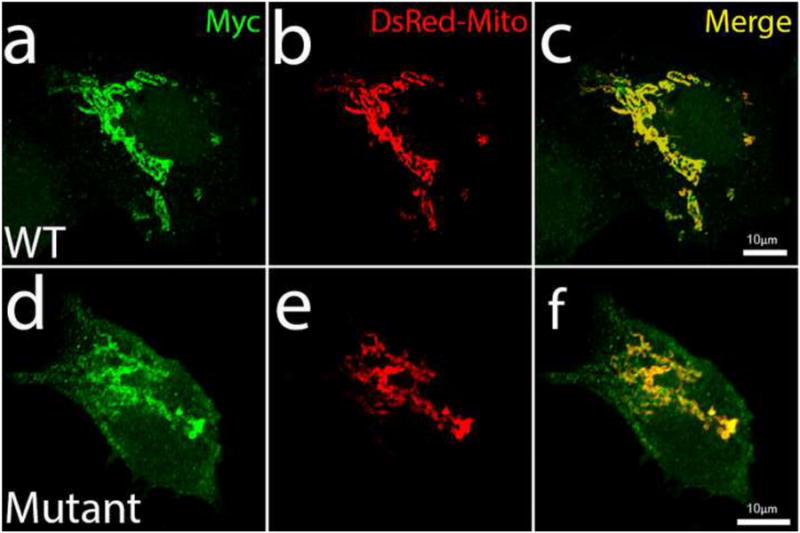
Co-localization of CHCHD10 with mitochondria. HEK-293 cells were transfected with either wild-type (WT) CHCHD10 (a-c), or mutant (R15S/G58R) CHCHD10 (d-f). CHCHD10 is myc-tagged (green). CHCHD10 colocalizes with the mitochondrial marker, DsRed-Mito (red). Merged images are shown in yellow. Scale bars, 10 μm.
Expression of mutant CHCHD10 disrupts mitochondrial morphology
Not much is known about the function of CHCHD10. Depletion of CHCHD3, another CHCH domain-containing protein, has been shown to cause fragmentation of the mitochondrial network (18). To identify the pathogenic mutation, and further explore the role of CHCHD10 in the maintenance of the mitochondrial network, we utilized computer-automated image analysis in cells expressing either WT-CHCHD10 or mutant (R15S, G58R or R15S/G58R). Since we did not have any primary cell lines available from our patients, we studied mitochondrial morphology in transfected cultured cells. Interestingly, cells expressing the R15S/G58R or G58R mutants showed fragmentation of the mitochondria compared with WT-CHCHD10 or R15S-CHCHD10 expressing cells, which displayed elongated mitochondria (Figure 6).
Figure 6.
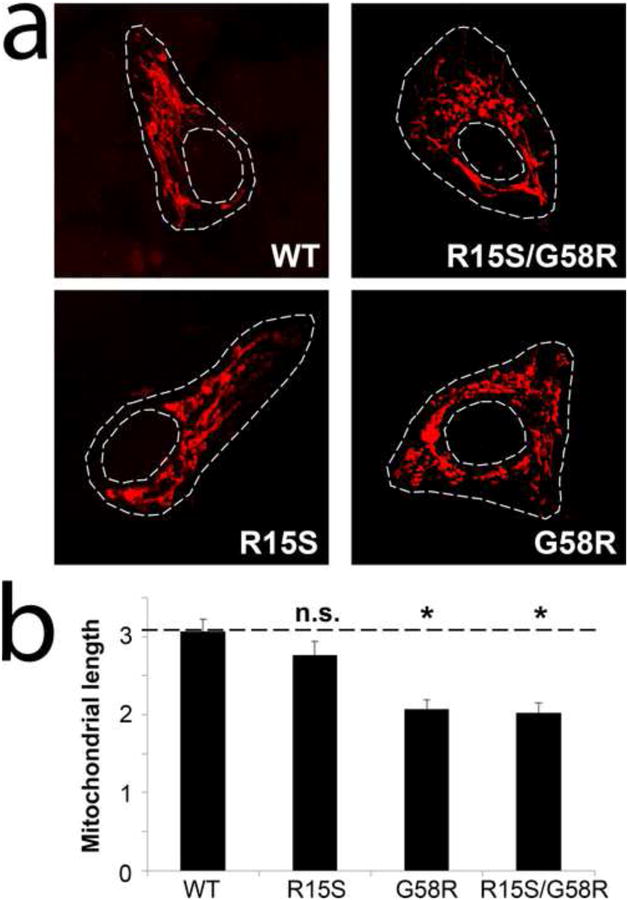
Confocal images of mitochondria labeled by transient transfection of DsRed-Mito in HEK-293 cells expressing either wild-type (WT) or mutant (R15S, G58R or R15S/G58R) CHCHD10 (a). Quantitative image analysis of mitochondrial length is shown in panel (b) (n>10 cells in at least three independent experiments; *, p < 0.05 versus WT; n.s., not significant; error bars represent means ± S.E.)
Discussion
Isolated mitochondrial myopathies are rare disorders and few of them are caused by mutations in nuclear genes. Such mutations are usually described in sporadic cases and no large pedigrees are usually available for study. Of note, are mutations in TK2 and DNA2 which are nuclear genes causing isolated mitochondrial myopathies (19). In this study, we have used two unbiased techniques, linkage analysis and a mitochondrial gene prediction method to identify a nuclear encoded mitochondrial protein (CHCHD10) in a large five generation family with isolated mitochondrial myopathy. Rare mutations in rare diseases can be interpreted as private polymorphisms, but we first detected genetic linkage to disease and predicted the gene in the linkage interval, and then found the mutations. At that time the function of the gene, CHCHD10, was unknown and it was called C22orf16. Using the tools of integrative genomics, such as MitoCarta (11), we identified C22orf16 or CHCHD10 as the highest scoring mitochondrial gene in the minimum candidate region. Moreover, we have excluded these mutations in a large number of controls and in public databases. CHCHD10 is highly expressed in skeletal muscle and has very low expression in other tissues of the body, thereby supporting our conclusions.
While this paper was under review, Bannwarth et al., after obtaining information from us about our work on this gene, reported a large family with multiple phenotypes, including, motor neuron disease, cognitive decline, cerebellar ataxia and myopathy (20). In all patients, muscle biopsy showed ragged-red and cytochrome c oxidase-negative fibers with combined respiratory chain deficiency. Patient fibroblasts had respiratory chain deficiency, mitochondrial ultrastructural alterations and fragmentation of the mitochondrial network. Interestingly, whole-exome sequencing in this family identified a missense mutation (S59L) in CHCHD10. Overexpression of mutant CHCHD10 in HeLa cells also led to fragmentation of the mitochondrial network. This mutation affects a residue adjacent to the Gly58 residue mutated in our mitochondrial myopathy family. Collectively, these findings suggest that CHCHD10 mutations can cause mitochondrial defects and lead to multiple phenotypes by affecting tissues that are highly dependent on oxidative phosphorylation, including the muscle and nervous system.
CHCHD10 has a target mitochondrial signaling peptide of 16 amino acids and a highly conserved C-terminal region that is predicted to contain a coiled coil-helix-coiled-coil-helix (CHCH) domain. Sequence analysis demonstrated two segregating mutations in cis: one in the targeting sequence (R15S) and the other was outside the targeting sequence (G58R). Therefore, we performed experiments to identify which of these two mutations were more likely to be pathogenic. We have two lines of evidence to show that the R15S mutation present in the targeting sequence is probably not pathogenic. Firstly, the presence of this mutation does not affect mitochondrial localization of CHCHD10. It is possible that the presence of the CHCH domain allowed the protein to get into the mitochondrial using the MIA40 pathway and the oxidative folding trap that is particular to the import of such proteins (21). Secondly, the R15S mutation has no effect on mitochondrial morphology. On the other hand, G58R, which affects a highly conserved residue has a significant effect on mitochondrial morphology and is more likely to be the pathogenic one.
Within each helix of the CHCH domain of CHCHD10 there are two highly conserved cysteines separated by 9 amino acids or two cysteine-x9-cysteine also known as twin (Cx9C) motifs (22). The two disulfide bonds formed by the twin Cx9C motif blocks the two helices in an anti-parallel orientation and is essential for the proper folding and structural stabilization of the protein (23). This distinctive cysteine pattern is found in many intermembrane space (IMS) mitochondrial proteins. This pair of cysteines can be separated by 3 residues to form the twin Cx3C motif or by 9 residues to form the twin Cx9C motif. The function of twin Cx9C motif-containing proteins is not completely understood. The yeast contains 14 twin Cx9C proteins. They are all mitochondrial proteins and are crucial for the activity of the respiratory chain and in particular in the function of cytochrome c oxidase (Complex IV) (22). Recently, a systematic search of eukaryotic twin Cx9C motif-containing proteins using bioinformatics tools and literature review revealed that most of these proteins are involved in a limited number of functions that are mostly performed in the IMS and include a possible scaffolding role for mitochondrial protein complexes, participating in COX assembly and maintaining mitochondrial structure (24). Other newly identified proteins with a CHCH domain include, CHCHD2, recently identified as a regulator of oxidative phosphorylation (14), and two inner membrane proteins, CHCHD3 and CHCHD6, recently found to be important for the control of mitochondrial cristae morphology (18, 25). Interestingly, knock down of CHCHD10 in HELA cells cultured on glucose free medium supplemented with galactose causes a significant decrease in ATP levels and complex IV activity suggesting that CHCHD10 may be involved in complex IV function (15). This finding is in concordance with the markedly reduced cytochrome c oxidase (Complex IV) function, as noted in skeletal muscle tissue from affected individuals in our mitochondrial myopathy kindred. Moreover, expression of the G58R mutation of CHCHD10 in cultured cells induces fragmentation of the mitochondrial network.
In conclusion, this is the first report that implicates mutation in a CHCH domain-containing protein in a human disease and widens the spectrum of mitochondrial myopathies to include this family of proteins. It is interesting that mutation in CHCHD10 causes a pure myopathy phenotype rather than a multi-systemic phenotype that is usually seen in mitochondrial myopathies that are linked to nuclear gene mutations. Further work is needed to identify additional CHCHD10 mutations in independent cohorts and to better understand the function of CHCH domain-containing proteins.
Supplementary Material
Acknowledgments
We thank the patients and their family members for participating in this study. We also thank Kreshnik B. Ahmeti, LiJun Cheng and Yi Yang for technical assistance. This work was supported by the National Institute of Neurological Disorders and Stroke (NS050641), the Les Turner ALS Foundation/Herbert C. Wenske Foundation Professorship, the David C. Asselin MD Memorial Fund, the Vena E. Schaff ALS Research Fund, the George Link, Jr. Foundation, Inc. and the Foglia Family Foundation. This work was supported by a grant (R01GM097136) from the National Institutes of Health to VKM. Imaging work was performed at the Northwestern University Cell Imaging Facility generously supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center.
Footnotes
Conflicts of interest None
References
- 1.Debray FG, Lambert M, Mitchell GA. Disorders of mitochondrial function. Curr Opin Pediatr. 2008;20:471–482. doi: 10.1097/MOP.0b013e328306ebb6. [DOI] [PubMed] [Google Scholar]
- 2.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 3.Lopez MF, Kristal BS, Chernokalskaya E, Lazarev A, Shestopalov AI, Bogdanova A, Robinson M. High-throughput profiling of the mitochondrial proteome using affinity fractionation and automation. Electrophoresis. 2000;21:3427–3440. doi: 10.1002/1522-2683(20001001)21:16<3427::AID-ELPS3427>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 4.DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. The New England journal of medicine. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 5.Milone M, Benarroch EE. Mitochondrial dynamics: general concepts and clinical implications. Neurology. 2012;78:1612–1619. doi: 10.1212/WNL.0b013e3182563c46. [DOI] [PubMed] [Google Scholar]
- 6.Ajroud-Driss S, Fecto F, Ajroud K, Siddique T. Mutations in the Nuclear Encoded Novel Mitochondrial Protein CHCHD10 Cause an Autosomal Dominant Mitochondrial Myopathy. Neurology. 2012;78 doi: 10.1007/s10048-014-0421-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heiman-Patterson TD, Argov Z, Chavin JM, Kalman B, Alder H, DiMauro S, Bank W, Tahmoush AJ. Biochemical and genetic studies in a family with mitochondrial myopathy. Muscle Nerve. 1997;20:1219–1224. doi: 10.1002/(sici)1097-4598(199710)20:10<1219::aid-mus2>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 9.Dagda RK, Cherra SJ, 3rd, Kulich SM, Tandon A, Park D, Chu CT. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N, Yang Y, Fecto F, Shi Y, Zhai H, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215. doi: 10.1038/nature10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Claros MG, Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 13.Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, Fong JH, Geer LY, Geer RC, Gonzales NR, et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225–229. doi: 10.1093/nar/gkq1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baughman JM, Nilsson R, Gohil VM, Arlow DH, Gauhar Z, Mootha VK. A computational screen for regulators of oxidative phosphorylation implicates SLIRP in mitochondrial RNA homeostasis. PLoS Genet. 2009;5:e1000590. doi: 10.1371/journal.pgen.1000590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martherus RS, Sluiter W, Timmer ED, VanHerle SJ, Smeets HJ, Ayoubi TA. Functional annotation of heart enriched mitochondrial genes GBAS and CHCHD10 through guilt by association. Biochem Biophys Res Commun. 2010;402:203–208. doi: 10.1016/j.bbrc.2010.09.109. [DOI] [PubMed] [Google Scholar]
- 16.Schug J, Schuller WP, Kappen C, Salbaum JM, Bucan M, Stoeckert CJ., Jr Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol. 2005;6:R33. doi: 10.1186/gb-2005-6-4-r33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roth RB, Hevezi P, Lee J, Willhite D, Lechner SM, Foster AC, Zlotnik A. Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics. 2006;7:67–80. doi: 10.1007/s10048-006-0032-6. [DOI] [PubMed] [Google Scholar]
- 18.Darshi M, Mendiola VL, Mackey MR, Murphy AN, Koller A, Perkins GA, Ellisman MH, Taylor SS. ChChd3, an inner mitochondrial membrane protein, is essential for maintaining crista integrity and mitochondrial function. J Biol Chem. 2011;286:2918–2932. doi: 10.1074/jbc.M110.171975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milone M, Wong LJ. Diagnosis of mitochondrial myopathies. Mol Genet Metab. 2013;110:35–41. doi: 10.1016/j.ymgme.2013.07.007. [DOI] [PubMed] [Google Scholar]
- 20.Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, Fragaki K, Berg-Alonso L, Kageyama Y, Serre V, Moore DG, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain. 2014 doi: 10.1093/brain/awu138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herrmann JM, Kohl R. Catch me if you can! Oxidative protein trapping in the intermembrane space of mitochondria. The Journal of cell biology. 2007;176:559–563. doi: 10.1083/jcb.200611060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Longen S, Bien M, Bihlmaier K, Kloeppel C, Kauff F, Hammermeister M, Westermann B, Herrmann JM, Riemer J. Systematic analysis of the twin cx(9)c protein family. Journal of molecular biology. 2009;393:356–368. doi: 10.1016/j.jmb.2009.08.041. [DOI] [PubMed] [Google Scholar]
- 23.Banci L, Bertini I, Ciofi-Baffoni S, Tokatlidis K. The coiled coil-helix-coiled coil-helix proteins may be redox proteins. FEBS letters. 2009;583:1699–1702. doi: 10.1016/j.febslet.2009.03.061. [DOI] [PubMed] [Google Scholar]
- 24.Cavallaro G. Genome-wide analysis of eukaryotic twin CX9C proteins. Molecular bioSystems. 2010;6:2459–2470. doi: 10.1039/c0mb00058b. [DOI] [PubMed] [Google Scholar]
- 25.An J, Shi J, He Q, Lui K, Liu Y, Huang Y, Sheikh MS. CHCM1/CHCHD6, novel mitochondrial protein linked to regulation of mitofilin and mitochondrial cristae morphology. The Journal of biological chemistry. 2012;287:7411–7426. doi: 10.1074/jbc.M111.277103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.