

This study is the first to demonstrate the essential role for fibroblast growth factor receptors 1 and 2 in endothelial cells for vascular remodeling after cardiac ischemia-reperfusion injury in vivo. Impaired vascular remodeling in mice deficient of endothelial fibroblast growth factor receptors resulted in persistent tissue hypoxia, increased postischemic cardiac dysfunction, and increased infarct size.
Keywords: fibroblast growth factor, endothelium, vascular remodeling, myocardial infarction, cardiac ischemia-reperfusion injury
Abstract
Fibroblast growth factor (FGF) signaling is cardioprotective in various models of myocardial infarction. FGF receptors (FGFRs) are expressed in multiple cell types in the adult heart, but the cell type-specific FGFR signaling that mediates different cardioprotective endpoints is not known. To determine the requirement for FGFR signaling in endothelium in cardiac ischemia-reperfusion injury, we conditionally inactivated the Fgfr1 and Fgfr2 genes in endothelial cells with Tie2-Cre (Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice). Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice had normal baseline cardiac morphometry, function, and vessel density. When subjected to closed-chest, regional cardiac ischemia-reperfusion injury, Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice showed a significantly increased hypokinetic area at 7 days, but not 1 day, after reperfusion. Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice also showed significantly worsened cardiac function compared with controls at 7 days but not 1 day after reperfusion. Pathophysiological analysis showed significantly decreased vessel density, increased endothelial cell apoptosis, and worsened tissue hypoxia in the peri-infarct area at 7 days following reperfusion. Notably, Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice showed no impairment in the cardiac hypertrophic response. These data demonstrate an essential role for FGFR1 and FGFR2 in endothelial cells for cardiac functional recovery and vascular remodeling following in vivo cardiac ischemia-reperfusion injury, without affecting the cardiac hypertrophic response. This study suggests the potential for therapeutic benefit from activation of endothelial FGFR pathways following ischemic injury to the heart.
NEW & NOTEWORTHY
This study is the first to demonstrate the essential role for fibroblast growth factor receptors 1 and 2 in endothelial cells for vascular remodeling after cardiac ischemia-reperfusion injury in vivo. Impaired vascular remodeling in mice deficient of endothelial fibroblast growth factor receptors resulted in persistent tissue hypoxia, increased postischemic cardiac dysfunction, and increased infarct size.
ischemic heart disease remains the leading cause of death worldwide despite advances in providing early reperfusion in the setting of acute myocardial infarction (31). For patients who survive the initial insult of cardiac ischemia-reperfusion (I/R), changes in cardiomyocytes, interstitial cells, and vessels during the repair and remodeling processes can ultimately result in deleterious changes in cardiac morphometry and function progressing to heart failure (13). New therapies are needed to not only provide protection to limit reperfusion injury in the acute setting but also to favorably alter cardiac repair and remodeling to limit the chronic morbidity and mortality caused by ischemic heart disease.
The fibroblast growth factor (FGF) family, consisting of 18 signaling proteins and four receptors (FGFRs), is an attractive therapeutic target due to its known role in cell growth and proliferation, angiogenesis, and tissue repair (25). Multiple FGF ligands induce cardioprotection in various models of cardiac ischemia or I/R injury. The most widely studied of these ligands, FGF2, or basic FGF, is an essential endogenous mediator of protection in ex vivo I/R injury (14), in vivo chronic cardiac ischemia (30), and in vivo I/R injury (16). In a closed-chest model of regional cardiac I/R injury, Fgf2−/− mice had worsened acute outcomes (increased cell death, increased wall motion abnormalities, increased cardiac dysfunction) as well as alterations in cardiac remodeling (impaired cardiac hypertrophy and impaired vascular remodeling in the peri-infarct area) (16). As FGF2 is expressed in multiple cardiac cell types including cardiomyocytes, endothelial cells, smooth muscle cells, and fibroblasts (19), all of which may contribute to these phenotypes, we sought to understand how cell-autonomous FGF signaling in various cardiac cell types contributes to the response to cardiac injury and mediates repair and remodeling. Because of the prominent role of the vasculature in acute ischemic injury and in the repair process, we questioned here whether cardioprotective functions of FGFs could include direct signaling to vascular endothelial cells in vivo.
A major aspect of cardiac repair involves neo-angiogenesis in the damage area, which begins in the peri-infarct or border zone and then expands into the infarct area, reaching peak microvascular density in the infarcted myocardium at 7 days after infarction (33). These newly formed microvessels in the peri-infarct area express high levels of FGF2 (32). Stimulating angiogenesis has been shown to be beneficial in multiple models of ischemic or infarcted hearts and is a possible therapeutic target in the setting of ischemic heart disease (1, 2). Intramyocardial injection of FGF2 increases angiogenesis in infarcted rabbit myocardium (4). In vitro data suggest that endothelial cells may mediate the angiogenic response as activation of FGFR1 or FGFR2 in endothelium by FGF1, FGF2, and FGF4 leads to endothelial cell proliferation (8).
To investigate if cell-autonomous FGF signaling in endothelium altered cardiac injury response and repair processes, we generated mice with Tie2-Cre targeted inactivation of the Fgfr1 and Fgfr2 genes (Tie2-Cre, Fgfr1f/f, Fgfr2f/f). These endothelial cell double conditional knockout (DCKO) mice and control mice were subjected to a closed-chest model of regional, cardiac I/R injury, a model that more closely mimics acute myocardial infarction compared with many small animal models of cardiac I/R injury. Here, we show that inactivation of Fgfr1 and Fgfr2 in endothelium does not affect acute post-I/R cardiac function or contribute to segmental LV dysfunction. However, we find that Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice exhibit impaired vascular remodeling and increased hypoxia in the peri-infarct area during cardiac remodeling and have worsened cardiac dysfunction, increased wall motion abnormalities, and increased infarct size at 7 days after I/R injury.
METHODS
Mice
Mice were housed in a pathogen-free facility and handled in accordance with standard use protocols, animal welfare regulations, and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All protocols were approved by the Washington University Animal Studies Committee. To create a conditional inactivation of Fgfr1 and Fgfr2 in endothelial cells, Tie2-Cre transgenic mice (20) were crossed with mice with both Fgfr1 and Fgfr2 genes flanked by loxP sites (Fgfr1f/f, Fgfr2f/f). This resulted in endothelial-specific ablation of FGFR1 and FGFR2 (Tie2-Cre, Fgfr1f/f, Fgfr2f/f). These mice are referred to as Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice. For endothelial cell lineage analysis, the ROSA26mT/mG reporter allele (23) was also included. Controls for these experiments include Fgfr1f/f, Fgfr2f/f double flox mice (DFF) and Tie2-Cre mice with wild-type Fgfrs. No differences were observed between either control groups for any outcomes, so the presented data include a combination of both types of controls. Mice were maintained on a mixed C57BL/6J;129X1 genetic background. Control and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice (both male and female) at 8 to 10 wk of age were randomly assigned to the present study: baseline (no injury/intervention) group, 90-min ischemia + 1-day reperfusion, sham myocardial infarction surgery + 1-day recovery, 90-min ischemia + 3-day reperfusion, sham myocardial infarction surgery + 3-day recovery, 90-min ischemia + 7-day reperfusion, or sham myocardial infarction surgery + 7-day recovery. We performed a post hoc power calculation for our primary outcome [7-day hypokinetic area of the left ventricle (LV)], which showed a 93.8% power to detect a difference between genotypes with an alpha of 0.05 (http://www.clincalc.com).
FACS Sorting of Endothelium and Quantitative RT-PCR for FGFs
Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO and control mice at 6–8 wk of age were anesthetized, and the lungs were harvested and minced. Minced lungs were dissociated in a solution of 0.1% collagenase type 2 (LS004177; Worthington Biochemical) and 0.002% dispase 1 (D4818; Sigma) made in DMEM (11965118; Life Technologies). Lungs were digested in a 37° shaking water bath for 40 min. Digested tissue was then further dissociated by cannulation utilizing an 18-g needle and 3-ml syringe. Dissociated cells were filtered through successive 100-μm, 70-μm, and finally 40-μm cell strainers to achieve a single cell suspension. Cells were placed in a 1:100 dilution of biotin labeled CD144 (cat. no. 13-1441-82; eBioscience) and allowed to incubate for 25 min at 4°C. Cells were then washed and placed in a solution containing 1:1000 streptavidin (cat. no. 17-4317-82; eBioscience) and 1:150 PE conjugated CD31 (cat. no. 553373; Pharmingen) antibodies and were incubated at 4°C for 25 min. Cells were washed and strained through a 40-μm cell strainer before FACS sorting. FACS sorting was performed on a FACS Aria II flow cytometer (BD Biosciences). RNA from sorted cells was harvested utilizing an RNeasy Plus Micro Kit (74034; Qiagen). RNA was reverse transcribed using an iScript RT kit (170–8841; Bio-Rad). Relative Fgfr1 expression was measured utilizing a Taqman probe targeting the deleted region of the Fgfr1 floxed allele (Mm_00438923_m1; Life Technologies). Relative Fgfr2 expression was measured utilizing a custom-made Taqman probe targeting the deleted region of the Fgfr2 floxed allele (4331348; Life Technologies). All Taqman assays were evaluated using a Life Technology's Step-One-Plus thermocycler running in fast mode. Expression was normalized to GAPDH (Mm_99999915_g1; Life Technologies).
Mouse Model of Closed-Chest Cardiac I/R Injury
The mouse model of closed-chest, regional cardiac I/R injury was performed in the Mouse Cardiovascular Phenotyping Core at Washington University in St. Louis School of Medicine as previously described (16). Mice were subjected to 90 min of ischemia and varying amounts of reperfusion ranging from 1 to 7 days. The surgeon was blinded to mouse genotype for all interventions.
Echocardiography
Mouse echocardiography was performed using a Visual Sonics Vevo2100 High-Resolution In Vivo Imaging System as previously described (16). Echocardiography analysis of cardiac function and wall motion abnormalities was obtained at baseline, during ischemia, after 1 day of reperfusion, and after 7 days of reperfusion. Mice were anesthetized with Avertin (2,2,2-tribromoethanol, 250 mg/kg ip), which was chosen due to its lack of cardiovascular effects at the doses used. Ejection fraction was calculated by measuring the end systolic and diastolic dimensions from long-axis images using the following formula: 100 × (LV end diastolic volume − LV end systolic volume/LV end diastolic volume). Infarct area was calculated by examining serial short-axis images (1 mm apart) spanning from the base of the LV (level of the aortic valve) to the apex. The percent area of the ventricle that was infarcted was calculated by determining the percentage of the ventricle that was hypokinetic. All images were obtained by a single operator with expertise in mouse echocardiography who was blinded to genotype and surgical treatment.
Histology
Assessment of cardiomyocyte cross-sectional area, blood vessel density and size, and capillary density and size was performed as previously described (16). For all histological quantification, multiple random microscopic fields were assessed at three different levels of the LV (base, mid, and apex) by a microscopist blinded to genotype and treatment group.
mTmG reporter gene histology.
Adult Tie2-Cre, Fgfr1f/f, Fgfr2f/f, ROSA26mT/mG and control mice were anesthetized and killed. Hearts were harvested, rinsed in PBS, halted in diastole with KCl-saturated PBS, and fixed in 10% formalin overnight at 4°C. Hearts were washed three times in PBS and placed in increasing concentrations of PBS/sucrose solution overnight at 4°C (3, 10, and 30%) for cryoprotection and in sucrose:OCT (TissueTek 4583; 30% in PBS) until the specimen reached the bottom of the mold. Specimens were then frozen in OCT compound and stored at −80°C. Cryosections were cut at 10-μm thickness using a Cryostat LEICA CM1850. Sections were then washed in distilled water for 30 s followed by PBS three times for 5 min each. Slides were mounted using Vectashield HardSet Mounting Medium with DAPI (H-1500; Vector Laboratories). Fluorescence was visualized and images were captured using Zeiss AxioImager Z1 ApoTome microscope.
CD45, CD31/BrdU, and CD31/TUNEL histology.
Paraffin-imbedded sections (6 μm) were deparaffinized with xylenes and washed with ethanol. Peroxidase activity was inhibited by incubating in methanol:hydrogen peroxide solution (3%) for 40 min and then washed in distilled water for 10 min. Antigen retrieval was performed by boiling the slides in 10 mM citrate buffer pH 6.0 for 15 min. After immunostaining was performed, slides were washed with PBST (0.1% Triton) and PBS twice for 5 min, distilled water for 5 min, and treated with 10 mM copper sulfate, pH 5.0 (made in 50 mM ammonium acetate) to quench any remaining red blood cells. Slides were mounted using Vectashield Hard set mounting medium with DAPI (H-1500). Immunofluorescence was visualized with Zeiss Confocal Microscope Z2 LSM700, and pictures were taken at ×40 using immersion oil.
For detection of CD45, slides were permeabilized with PBST (0.1% Triton) for 30 min and blocked with 10% goat serum in PBST for 1 h. Primary antibody incubation was performed at 4°C overnight (CD45; 550539; BD Biosciences; 1:100 dilution). Secondary antibody was applied for 1 h at room temperature (Alexa Fluor 488; A11006; 1:200 dilution). CD45-positive cells where quantified using National Institutes of Health (NIH) ImageJ cell counter plugin. At least 12–14 fields per mouse were quantified.
For colabeling of CD31 and bromodeoxyuridine (BrdU), mice were injected with BrdU (100 mg/kg of body wt) intraperitoneally 4 h before death. Slides were permeabilized with PBST (0.5% Triton) for 30 min and blocked with 10% goat serum in PBST for 1 h. Primary antibody incubation was performed at 4°C overnight (BrdU; 347580; BD Biosciences; 1:200). Incubation with secondary antibody was for 1 h at room temperature (Alexa Fluor 488; A11029; Invitrogen, Grand Island, NY; 1:200 dilution). CD31 staining was performed as described above. Double BrdU/CD31-positive cells where quantified using NIH ImageJ cell counter plugin. At least 12–14 fields per animal were quantified.
Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL) staining was performed using the DeadEnd Fluorometric System (G3250; Promega) following the manufacturer's instructions.
Measurement of tissue hypoxia.
Mice were given an intraperitoneal injection with hypoxyprobe solution (2.5 mg pimonidazole HCl; NPI, Burlington, MA) 1 h before death, and hearts were collected and fixed with 10% formalin overnight. Paraffin-embedded specimens were cut at 10-μm thickness and deparaffinized. Endogenous peroxidase activity was inhibited with 3.3% hydrogen peroxide:methanol solution for 1 h and heat-induced antigen retrieval in 10 mM citrate buffer, pH 6.0.
Hypoxyprobe Plus Kit (HP2-200) was used to visualize hypoxyprobe staining according to the manufacturer's specifications (primary antibody 1:50, secondary antibody 1:50). Horseradish peroxidase reaction was performed using a Histostain DAB kit (Invitrogen) according to the manufacturer's specifications. Slides were counterstained with hematoxylin to visualize the tissue and mounted. Slides were scanned at ×40 resolution using digital slide scanner (NanoZoomer 2.0-HT; Hamamatsu) microscope. Multiple ×40 microscopic fields in the peri-infarct area of the LV were selected. Images were scored in a blinded fashion according to the scoring template (see Fig. 9). Alternatively, thresholding color intensity with NIH ImageJ software allowed the hypoxia staining to be quantified as percent area per microscopic field.
Fig. 9.

Hypoxyprobe staining scoring template. Representative images are shown to depict the scoring system that was used for blinded assessment of hypoxia. Scale bars = 200 μm in ×10 and 50 μm in ×40.
Analysis of Vascular Perfusion Domains
Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO and control mice at 8–10 wk of age were anesthetized with ketamine/xylazine (100 mg/kg and 10 mg/kg ip) and were ventilated through a tracheostomy. Mice were subjected to a left minithoracotomy and a suture was placed around the left anterior descending artery (LAD) to induce ischemia. After 10 min of ischemia to allow collateral circulation to open, mice received an intravenous (tail vein) injection of FITC dextran (200 μl, 2.5% in PBS). Mice were killed 10 min later and the hearts were removed from the thoracic cavity. Whole mount microscopy of the LV was performed using a Zeiss Discovery V12 microscope. The hypoperfused area was measured using NIH ImageJ Software.
Statistical Analysis
All values are expressed as mean ± SE. Echocardiography data for LV infarct size and ejection fraction were compared using ANOVA with a post hoc Student's t-test. The remaining data were compared using a Student's t-test. Data with a P < 0.05 were considered statistically significant.
RESULTS
As endothelial FGFR signaling may alter cardiac homeostasis and cardiac injury response, endothelial cell-specific double conditional knockout mice (Tie2-Cre, Fgfr1f/f, Fgfr2f/fDCKO) were generated in which a Tie2-Cre transgenic allele was used to inactivate floxed alleles of Fgfr1 and Fgfr2. Control mice included mice homozygous for floxed Fgfr1 and Fgfr2 alleles as well as mice expressing the Tie2-Cre allele with wild-type Fgfr1 and Fgfr2 alleles. Fgfr1 and Fgfr2 were targeted as they are the most predominantly expressed Fgfrs in endothelial cells (3, 10, 17).
To confirm Cre recombinase activation in endothelium in the heart, the ROSA26mT/mG reporter allele (ROSAmT/mG) (23) was bred into the Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mouse line. The ROSAmT/mG reporter gene allows visualization of the lineage of cells exposed to Cre recombinase by Cre-mediated inactivation of the constitutively expressed membrane-targeted Tomato and activation of a membrane-targeted green fluorescent protein (GFP). Immunofluorescent microscopy showed Tomato expression without GFP expression in control ROSAmT/mG mouse heart, whereas Tie2-Cre, ROSAmT/mG mice showed GFP expression in an endothelial pattern in the heart with Tomato expression in other cardiac cell types (Fig. 1A). To confirm that Fgfr1 and Fgfr2 expression was significantly reduced in endothelial cells of Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice, adult lung endothelial cells were obtained through FACS sorting. VE-cadherin/CD31 double-positive endothelial cells were subjected to quantitative RT-PCR. This analysis showed a 90 and 98% decrease in mRNA expression of Fgfr1 and Fgfr2, respectively, in Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO endothelial cells compared with control endothelial cells (Fig. 1B, P < 0.01). FACS sorting was performed in lung endothelium due to the relative abundance of endothelial cells in adult lung compared with adult heart.
Fig. 1.

Endothelial-specific ablation of Fgfr1 and Fgfr2. A: representative images of mTmG reporter gene expression in adult left ventricle (LV) of control and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice. GFP expression is seen in an endothelial pattern in Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice, while Tomato expression is observed in nonendothelial cells of Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice and all cells of control hearts. Green: GFP; red: tomato. Scale bars = 50 μm in ×10 magnification images and 25 μm in ×20 magnification images. B: quantitative RT-PCR of FACS-sorted lung endothelial cells showing significantly reduced expression of Fgfr1 and Fgfr2 in Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice compared with controls. *P < 0.01 vs. control; n = 4.
To determine the effect of endothelial Fgfr1 and Fgfr2 inactivation in the heart under homeostatic conditions, adult (8–10 wk old) Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO and control mice were subjected to echocardiography. No significant differences were observed between genotypes for various aspects of cardiac function and morphometry (Table 1). Histologic analysis also showed no significant difference between Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO and control mice with respect to cardiomyocyte size, vessel number and size, and capillary number and size (Table 2).
Table 1.
Cardiac function and morphometry
| Control | Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO | |
|---|---|---|
| HR, beats/min | 631 ± 11 | 600 ± 12 |
| LVPWd | 0.83 ± 0.03 | 0.77 ± 0.02 |
| LVPWs | 1.43 ± 0.03 | 1.37 ± 0.06 |
| IVSd | 0.84 ± 0.03 | 0.79 ± 0.02 |
| IVSs | 1.48 ± 0.04 | 1.44 ± 0.05 |
| LVIDd | 3.38 ± 0.08 | 3.29 ± 0.13 |
| LVIDs | 1.50 ± 0.07 | 1.48 ± 0.1 |
| LV mass | 93.7 ± 3.8 | 82.1 ± 5.5 |
| RWT | 0.50 ± 0.02 | 0.48 ± 0.03 |
| FS, % | 55.6 ± 1.6 | 55.0 ± 2.3 |
Data are mean ± SE; n = 8.
HR, heart rate; LVPWd, left ventricular posterior wall thickness in diastole; LVPWs, left ventricular posterior wall thickness in systole; IVSd, interventricular septum thickness in diastole; IVSs, interventricular septum thickness in systole; LVIDd, left ventricular internal diameter in diastole; LVIDs, left ventricular internal diameter in systole; LV mass, left ventricular; RWT, relative wall thickness; FS, fractional shortening.
Table 2.
Histologic analysis
| Control | Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO | |
|---|---|---|
| Cardiomyocyte size, μm2 | 215 ± 26 | 200 ± 6 |
| Vessel number, per 10× field | 6.1 ± 0.7 | 6.8 ± 0.3 |
| Vessel diameter, μm | 5.9 ± 0.3 | 6.9 ± 0.7 |
| Capillary density | 0.90 ± 0.26 | 1.03 ± 0.09 |
| Capillary area, μm2 | 92.7 ± 6.3 | 91.7 ± 5.5 |
Data are mean ± SE; n = 4–5. Vessel number and diameter are α-smooth muscle actin-positive vessels. Capillary density is the number of CD31-positive capillaries/number of nuclei to normalize for differences in tissue density.
To determine the effect of inactivation of endothelial Fgfrs in acute cardiac injury and repair, Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO and control mice were subjected to in vivo, closed-chest regional, cardiac I/R injury. Because endothelial FGFR signaling could affect vascular patterning during development, and thus the area at risk following I/R injury in the adult, the perfusion domain of the LAD was evaluated. Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO and control mice received LAD artery occlusion for 10 min (to allow collateral vessels to be perfused), followed by intravenous injection of FITC-labeled dextran to reveal areas of cardiac perfusion (Fig. 2A). Analysis of ischemic regions of the heart showed no significant difference in the perfusion defect created by LAD occlusion between control (24 ± 1% LV) and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO (27 ± 5% LV) hearts (Fig. 2B). The closed-chest I/R injury model allows the use of simultaneously performed echocardiography during the ischemic period to evaluate cardiac ischemia based on the quantitation of segmental wall motion abnormalities. Using two different echocardiographic methods (area at risk or segmental wall motion score index), there was no significant difference in the noninvasively determined area at risk of the LV between control and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice (Fig. 2C). Measures of cardiac function showed significantly decreased ejection fraction during ischemia compared with preischemia values for both genotypes as expected (Fig. 2D, P < 0.01), but no significant difference between genotypes in ejection fraction either before or during cardiac ischemia (Fig. 2D).
Fig. 2.

Removal of endothelial Fgfr1 and Fgfr2 does not significantly alter left anterior descending artery perfusion territory. A: representative whole mount images of FITC-dextran injected control and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice showing similar area at risk with left-anterior descending artery ligation. The hypoperfused area is outlined in white dashed lines. Scale bar = 1 mm. B: quantification of FITC-dextran injected hearts showing no significant difference in the hypoperfused area (%LV) (n = 3). C: echocardiography during the ischemic period showing no significant difference in control and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice for area at risk (AAR) or segmental wall motion score index (SWMSI) (n = 7). D: measurements of cardiac function showed significantly decreased ejection fraction during ischemia for both control and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts but no significant difference between genotypes at either time point (#P < 0.01 vs. preischemia; n = 7).
After I/R injury, echocardiographic analysis of cardiac function and wall motion abnormalities was performed at 1 and 7 days of reperfusion to evaluate the response to I/R injury both acutely and during cardiac repair and remodeling. Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice showed significantly reduced ejection fraction at 7 days after I/R injury compared with control mice (P < 0.05, Fig. 3A), but there was no difference in cardiac function between genotypes at 1 day after I/R injury. Similarly, wall motion abnormalities, expressed as the percentage of the LV that is hypokinetic, were significantly (P < 0.01) increased in the Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice compared with controls at 7 days but not 1 day after I/R injury (Fig. 3B). Representative echocardiography at day seven post-I/R injury is shown in Supplemental Fig. S1 (long axis views of the LV) and Supplemental Fig. S2 (short axis views of the LV; Supplemental Material for this article is available online at the Journal website). Infarct size was determined by measuring fibrotic area of Masson's trichrome stained histological sections at multiple levels through the LV. Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice showed significantly (P < 0.05) increased infarct area compared with controls at 7 days after I/R injury (Fig. 3C).
Fig. 3.
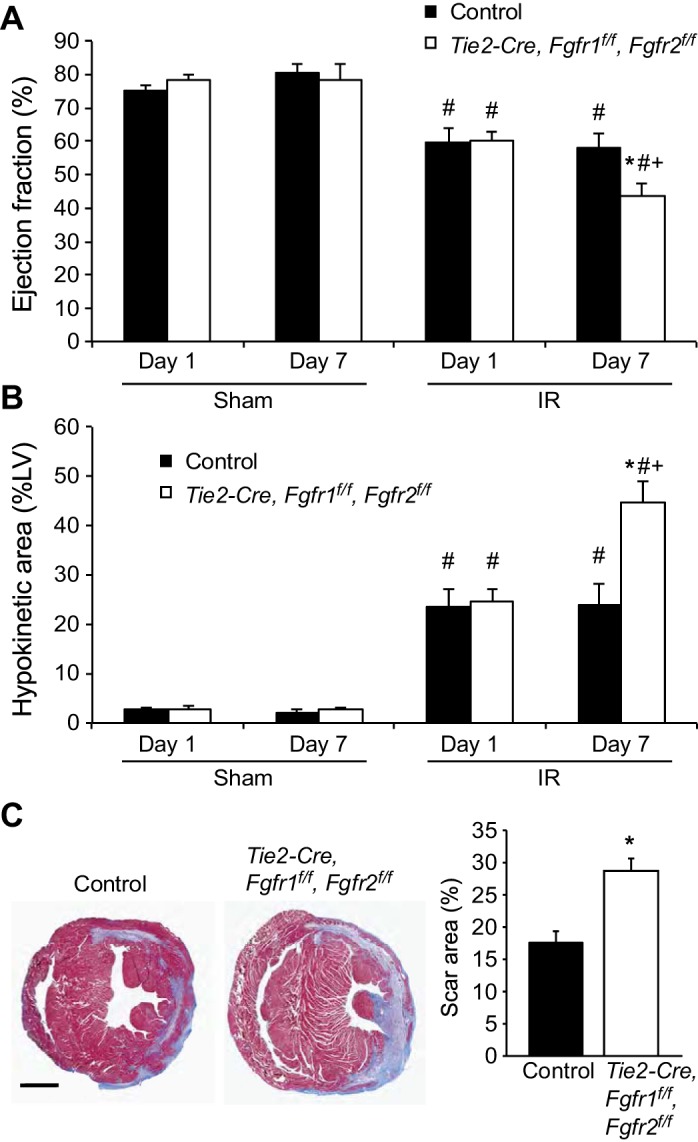
Endothelial Fgfr1 and Fgfr2 ablation results in worsened cardiac functional recovery and increased hypokinetic area at 7 but not 1 day after I/R injury. Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts show reduced ejection fraction (A) and increased hypokinetic area (B) at 7 days but not 1 day after ischemia-reperfusion (I/R) injury. C: representative images of Masson's trichrome stained control and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice 7 days after I/R injury. Scar size (%LV) is significantly larger in Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice 7 days after I/R injury compared with controls. Scale bar = 1 mm. *P < 0.05 vs. control; #P < 0.05 vs. sham; +P < 0.05 vs. 1-day time point; n = 4–12.
As Tie2-Cre targets cells in the hematopoietic lineage in addition to endothelial cells, immune cell infiltration into the infarct area was assessed. Representative images of CD45-positive cells in the infarct area (Fig. 4A) are shown, and quantitation of cell number showed significantly more CD45-positive cells in the infarct area at 3 days and significantly fewer CD45-positive cells at 7 days after I/R injury compared with 1 day after I/R injury for both control and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice (Fig. 4B, P < 0.01). There was no significant difference, however, between control and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice in the number of CD45-positive cells in the infarct area at any time point (Fig. 4B).
Fig. 4.

Endothelial Fgfr1 and Fgfr2 ablation does not affect CD45-positive cell number after I/R injury. A: representative images of CD45-positive cells in the peri-infarct area of control and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice after I/R injury (green: CD45; blue: DAPI). Scale bar = 20 μm. B: quantitation of CD45-positive cells showing significantly increased numbers at 3 days and significantly decreased numbers at 7 days after I/R injury compared with 1 day after I/R injury in both control and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts. There is no significant difference in CD45-positive cell number between control and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts at any time point. #P < 0.01 vs. 1-day time point; n = 4–8.
The worsened post-I/R cardiac dysfunction and wall motion abnormalities observed in Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice compared with controls at 7 days but not 1 day after I/R injury suggest a role for endothelial Fgfr1 and Fgfr2 in the cardiac remodeling and repair process. To investigate various aspects of cardiac remodeling, Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO and control mice were subjected to I/R injury, and histology was performed at either 3 days or 7 days after reperfusion. Histology with FITC-labeled wheat germ agglutinin was performed to evaluate cardiomyocyte size (Fig. 5A). As expected, increased average cardiomyocyte cross-sectional area was observed at both 3 and 7 days after I/R injury in the peri-infarct area compared with sham-treated hearts (P < 0.05, Fig. 5B). However, no significant difference in cardiomyocyte cross-sectional area was observed between Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO and control mice at any time point (Fig. 5B).
Fig. 5.

The cardiac hypertrophic response after cardiac I/R injury is not affected by lack of endothelial Fgfr1 and Fgfr2. A: representative images of wheat germ agglutinin (WGA) staining in the peri-infarct area of control and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO mice at 3 and 7 days after I/R injury or sham treatment (green: WGA; blue: DAPI). Scale bar = 50 μm. B: after I/R injury, there is a significant increase in cardiomyocyte cross-sectional area in the peri-infarct area, but there is no significant difference in the cardiac hypertrophic response between control and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts at any time point. #P < 0.05 vs. sham; n = 4–13.
Vascular remodeling after I/R injury was evaluated using immunostaining for α-smooth muscle actin (Fig. 6A) to label medium and larger vessels and CD31 (Fig. 7A) to identify capillaries. Quantitation of α-smooth muscle actin-positive vessels showed a significant decrease in the number of smooth muscle actin-positive vessels in the peri-infarct area of all genotypes after I/R injury compared with sham operated hearts; however, Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts had significantly (P < 0.05) fewer vessels compared with controls at 7 days after I/R injury (Fig. 6B). No differences in vessel diameter were observed in any treatment group (Fig. 6C). Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts also showed decreased density of capillaries at 7 days after I/R injury compared with control hearts (Fig. 7B) but no difference in capillary area (Fig. 7C). These data suggest a defect in vascular remodeling in the absence of endothelial Fgfr1 and Fgfr2. The vascular remodeling defects present in Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts could be related to increased endothelial cell death or decreased endothelial proliferation. Colabeling of CD31 and TUNEL showed increased endothelial cell death in the peri-infarct area of Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts compared with controls at 3 days after I/R injury (Fig. 8, A and C, P < 0.05). Endothelial cell proliferation (BrdU/CD31 colabeled) was not significantly different between control and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts at 3 days after I/R injury (Fig. 8, B and C).
Fig. 6.

Endothelial Fgfr1 and Fgfr2 are required for vascular remodeling after I/R injury. A: representative images of immunofluorescence for α-smooth muscle actin (red) and nuclei (DAPI: blue) in the peri-infarct area of the LV. Scale bar = 100 μm. B: after I/R injury, there is a significant decrease in the number of vessels in the peri-infarct area and Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts show decreased vessel density compared with controls at 7 days after I/R injury. C: there are not significant differences in vessel diameter in any treatment group. *P < 0.05 vs. control; #P < 0.05 vs. sham; n = 4–8.
Fig. 7.

Endothelial Fgfr1 and Fgfr2 regulate capillary density during postischemic cardiac remodeling. A: representative images of immunofluorescence for CD31 (red) and nuclei (blue) showing capillaries in the peri-infarct area. Scale bar = 100 μm. B: Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts have significantly decreased capillary density in the peri-infarct area at 7 days after I/R injury compared with controls. C: no difference in capillary area was observed in any treatment group. *P < 0.05 vs. control; #P < 0.05 vs. sham; n = 4–8.
Fig. 8.

Endothelial Fgfr1 and Fgfr2 mediate endothelial cell survival after cardiac I/R injury. A: representative images of immunofluorescence of CD31 (red) and Terminal deoxynucleotidyl transferase dUTP-mediated nick-end labeling (TUNEL)-positive cells (green) in the peri-infarct area. Inset: ×3 magnification showing a TUNEL-positive CD31-positive endothelial cell. Scale bar = 20 μm. B: representative images of immunofluorescence for CD31 (red) and bromodeoxyuridine (BrdU)-positive cells (green) in the peri-infarct area. Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts have significantly more TUNEL-positive endothelial cells at 3 days after I/R injury (C) but no significant difference in the number of BrdU-positive endothelial cells (D). *P < 0.05 vs. control; n = 5–9.
We hypothesized that impaired postischemic vascular remodeling in Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts could lead to persistent hypoxia in the peri-infarct area, which could explain the increased infarct size, worsened cardiac function, and increased wall motion abnormalities that were observed in these hearts at 7 days after I/R injury but not acutely (at 1 day after I/R injury). To test this hypothesis, Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO and control mice were given an intraperitoneal injection of pimidazole, which binds to hypoxic cells at Po2 ≤ 10 mmHg (5, 11) 1 h before death. A semiquantitative scoring system was used to evaluate the extent of hypoxia in the peri-infarct area in a blinded fashion (Fig. 9). Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts showed significantly (P < 0.05) increased hypoxia in the peri-infarct region compared with controls at both 3 and 7 days after I/R injury (Fig. 10, A and B). To validate this scoring system and reconfirm these findings, quantification of positive staining for hypoxia was performed by thresholding color intensity with NIH ImageJ software. Similarly, significantly increased hypoxia was observed in the peri-infarct region at both time points for Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts compared with controls (Fig. 10C).
Fig. 10.

Endothelial Fgfr1 and Fgfr2 ablation results in persistent hypoxia in the peri-infarct area. A: representative images of hypoxyprobe labeling of hypoxia in the peri-infarct region of Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO and control hearts 3 and 7 days after I/R injury. (brown: hypoxyprobe; blue: hematoxylin). Scale bar = 50 μm. Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts have a significantly higher average hypoxia score (B) and %hypoxic area (C) in the peri-infarct area of the LV compared with control hearts. *P < 0.05 vs. control; n = 3–4.
DISCUSSION
Activation of FGF signaling remains a promising therapeutic strategy in the context of I/R injury including acute myocardial infarction. Multiple animal models have shown cardioprotective efficacy of FGF ligands, especially FGF2, which has been either genetically induced (2814) or pharmacologically administered (9, 12). In vivo studies have shown that endogenous FGF2 mediates postischemic cardiac repair including cardiac hypertrophy in the peri-infarct area, vascular remodeling, and fibrosis after chronic cardiac ischemia (30), and cardiac I/R injury (16). Understanding the cell-type specific signaling that mediates these effects on various cardioprotective endpoints is essential to the development of targeted therapeutic approaches to improve acute cardiomyocyte survival as well as various aspects of cardiac repair.
This study establishes the cardioprotective function of endogenous FGFR1 and FGFR2 in the endothelium following cardiac I/R injury. Even though multiple FGFRs are expressed in the adult heart, FGFR1 and FGFR2 are the most predominantly expressed FGFRs in endothelial cells (3, 10, 17). FGFR3 has more recently been detected in endothelial cells but is more sparsely expressed (7, 29), and FGFR4 expression has not been observed in endothelial cells (21). There was no significant effect of ablation of FGFR1 and FGFR2 in endothelial cells (Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO) on vessel and capillary number and size in the adult heart. This is consistent with a previous report of normal vascular development in the absence of endothelial FGFR1 and FGFR2 in other noncardiac tissues including embryonic limb buds, adult ear skin, lung, kidney, and retina (24). Similarly, the perfusion territory of the LAD was unchanged in Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts compared with controls, and there was no difference in the area at risk of the LV as determined by echocardiographic measurements of wall motion abnormalities after LAD occlusion. There was also no effect of endothelial FGFR1 and FGFR2 ablation in adult cardiac morphometry or function.
One day after cardiac I/R injury, there was no difference in acute cardiac functional recovery or hypokinetic area between Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO and control mice showing that endothelial FGFR1 and FGFR2 are not essential to normal acute cardiac recovery. This suggests that the acute cardioprotective functions of FGF2 observed at 1 day after I/R injury (16) are mediated through nonendothelial cell types in the heart. At 7 days after I/R injury, however, Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts had worsened cardiac function and increased wall motion abnormalities compared with control hearts. The measurement of wall motion abnormalities via echocardiography used in this study has been shown to correlate well with more traditional histologic methods of infarct size determination (18). Furthermore, scar area measured on histological sections was consistent and showed increased scar size in Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts compared with controls at 7 days after I/R injury. Together these data suggest that the Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts had a defect in the cardiac repair processes that ultimately affected both cardiac function and final infarct size. Using a gain of function approach, Matsunaga et al. (22) showed that chronic overexpression of a constitutively active FGFR2 (Crouzon mutation allowing ligand-independent receptor dimerization) driven by a Tie2 promoter resulted in decreased infarct size and improve cardiac performance 28 days after the onset of cardiac ischemia. The cardioprotective effect observed in this model is likely due in part to autocrine activation of FGFR2 in endothelial cells but also secondarily from increased FGF2 expression and signaling to the myocardium.
In contrast, this study focused on the acute effects of endothelial FGF signaling within the first week after reperfusion to identify the repair defects that resulted in worsened cardiac function and infarct size. Several studies have demonstrated that FGF2 mediates cardiac hypertrophy in various in vivo models of injury including pressure overload (27), pharmacologic induced (15, 26), and postischemic (16, 30). Because ablation of FGFR1 and FGFR2 in endothelial cells had no effect on the cardiac hypertrophic response in the peri-infarct area, we conclude that FGF signaling in other cardiac cell types such as cardiomyocytes or stromal cells mediates FGF2’s prohypertrophic effect.
Analysis of vascular remodeling in Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts showed a reduced number of smooth muscle actin-positive vessels and decreased numbers of capillaries at 7 days after I/R injury compared with controls, with no effect on vessel or capillary size. An increased percentage of TUNEL-positive endothelial cells was observed at 3 days after I/R injury in Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts, which suggests increased susceptibility of endothelial cells to cell death in the absence of FGFR1 and FGFR2 signaling. These data correlate well with endothelial FGFR2 overexpression, which was shown to result in increased numbers of smooth muscle actin-positive vessels and increased capillary density 28 days after chronic cardiac ischemia (22). This group suggested that FGFR2 may preferentially promote arteriogenesis more than capillary formation since they observed a larger percentage increase in vessels vs. capillaries (40 vs. 20% respectively) in the peri-infarct area of FGFR2 transgenic hearts compared with controls after ischemic damage (22). We also observed a larger effect of ablation of FGFR1 and FGFR2 in endothelium on vessels compared with capillaries in the peri-infarct area (49% reduction in vessel number and 22% reduction in capillary density).
We further tested the hypothesis that vascular remodeling defects resulted in persistent tissue hypoxia that ultimately affected final infarct size and cardiac function in the Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts by measuring hypoxia in the peri-infarct region at 3 and 7 days after I/R injury. The observed increase in hypoxia at 3 days postreperfusion preceded the decrease in vessel number that was observed at 7 days. This suggests that even though the number of vessels or capillaries is unchanged in the Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts at 3 days after I/R injury, there is a defect in the functioning of these vessels. We also observed increased endothelial cell death at 3 days after I/R injury in the Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts compared with controls that preceded observed changes in capillary density.
A confounding factor in this study is that Tie2-Cre also targets hematopoietic lineages. Previous studies of ablation of FGFR1 and FGFR2 using Flk1-Cre, which has a similar combined expression pattern in endothelium and hematopoietic lineages, showed no effect on the number of B cells, T cells, macrophages, granulocytes, erythrocytes, or hematopoietic progenitor cells in adult bone marrow (24). Tie2 promoter driven overexpression of a constitutively active FGFR2 also had no effect on neutrophil or monocyte infiltration into the border zone or expression levels of multiple monocyte-induced cytokines after chronic cardiac ischemia (22). While there may be an effect related to hematopoietic lineage reduction in FGFR1 and FGFR2 expression in our model, normal numbers of CD45-positive cells in Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts suggest that the observed phenotype is primarily related to vascular function and remodeling effects of FGFR1 and FGFR2 in endothelial cells.
In conclusion, this study establishes the requirement of endogenous FGFR1 and FGFR2 in endothelium for normal vascular remodeling in a clinically relevant model of in vivo closed-chest, regional cardiac I/R injury but does not decipher whether the effects are mainly through FGFR1 or FGFR2 or both. Abnormal vascular remodeling in Tie2-Cre, Fgfr1f/f, Fgfr2f/f DCKO hearts led to persistent hypoxia in the peri-infarct area compared with controls, which ultimately resulted in worsened cardiac function and increased infarct size. These data suggest that therapeutic targeting of endothelial FGFR signaling may be beneficial for postischemic vascular remodeling and reduce chronic postischemic cardiac dysfunction and maladaptive remodeling.
GRANTS
This work was funded by American Heart Association Grant 11FTF730072 (to S. L. House); National Heart, Lung, and Blood Institute Grant HL-105732 (to D. M. Ornitz); and startup funds from the Washington University Division of Emergency Medicine (to S. L. House).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: S.L.H. and D.M.O. conception and design of research; S.L.H., A.M.C., T.S.L., C.W., C.S., and A.K. performed experiments; S.L.H. analyzed data; S.L.H. and D.M.O. interpreted results of experiments; S.L.H. prepared figures; S.L.H. drafted manuscript; S.L.H., A.M.C., A.K., and D.M.O. edited and revised manuscript; S.L.H., A.M.C., T.S.L., C.W., C.S., A.K., and D.M.O. approved final version of manuscript.
Supplementary Material
REFERENCES
- 1.Ahn A, Frishman WH, Gutwein A, Passeri J, Nelson M. Therapeutic angiogenesis a new treatment approach for ischemic heart disease– Part I. Cardiol Rev 16: 163–171, 2008. [DOI] [PubMed] [Google Scholar]
- 2.Ahn A, Frishman WH, Gutwein A, Passeri J, Nelson M. Therapeutic angiogenesis a new treatment approach for ischemic heart disease–Part II. Cardiol Rev 16: 219–229, 2008. [DOI] [PubMed] [Google Scholar]
- 3.Bastaki M, Nelli EE, Dell'Era P, Rusnati M, Molinari-Tosatti MP, Parolini S, Auerbach R, Ruco LP, Possati L, Presta M. Basic fibroblast growth factor-induced angiogenic phenotype in mouse endothelium. A study of aortic and microvascular endothelial cell lines. Arterioscler Thromb Vasc Biol 17: 454–464, 1997. [DOI] [PubMed] [Google Scholar]
- 4.Bougioukas I, Didilis V, Ypsilantis P, Giatromanolaki A, Sivridis E, Lialiaris T, Mikroulis D, Simopoulos C, Bougioukas G. Intramyocardial injection of low-dose basic fibroblast growth factor or vascular endothelial growth factor induces angiogenesis in the infarcted rabbit myocardium. Cardiovasc Pathol 16: 63–68, 2007. [DOI] [PubMed] [Google Scholar]
- 5.Chou SC, Azuma Y, Varia MA, Raleigh JA. Evidence that involucrin, a marker for differentiation, is oxygen regulated in human squamous cell carcinomas. Br J Cancer 90: 728–735, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Claus P, Grothe C. Molecular cloning and developmental expression of rat fibroblast growth factor receptor 3. Histochem Cell Biol 115: 147–155, 2001. [DOI] [PubMed] [Google Scholar]
- 8.Cross MJ, Claesson-Welsh L. FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci 22: 201–207, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Cuevas P, Carceller F, Lozano RM, Crespo A, Zazo M, Gimenez-Gallego G. Protection of rat myocardium by mitogenic and non-mitogenic fibroblast growth factor during postischemic reperfusion. Growth Factors 15: 29–40, 1997. [DOI] [PubMed] [Google Scholar]
- 10.Dell'Era P, Belleri M, Stabile H, Massardi ML, Ribatti D, Presta M. Paracrine and autocrine effects of fibroblast growth factor-4 in endothelial cells. Oncogene 20: 2655–2663, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Gross MW, Karbach U, Groebe K, Franko AJ, Mueller-Klieser W. Calibration of misonidazole labeling by simultaneous measurement of oxygen tension and labeling density in multicellular spheroids. Int J Cancer 61: 567–573, 1995. [DOI] [PubMed] [Google Scholar]
- 12.Hampton TG, Amende I, Fong J, Laubach VE, Li J, Metais C, Simons M. Basic FGF reduces stunning via a NOS2-dependent pathway in coronary-perfused mouse hearts. Am J Physiol Heart Circ Physiol 279: H260–H268, 2000. [DOI] [PubMed] [Google Scholar]
- 13.Heusch G, Libby P, Gersh B, Yellon D, Bohm M, Lopaschuk G, Opie L. Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 383: 1933–1943, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.House SL, Bolte C, Zhou M, Doetschman T, Klevitsky R, Newman G, Schultz Jel J. Cardiac-specific overexpression of fibroblast growth factor-2 protects against myocardial dysfunction and infarction in a murine model of low-flow ischemia. Circulation 108: 3140–3148, 2003. [DOI] [PubMed] [Google Scholar]
- 15.House SL, House BE, Glascock B, Kimball T, Nusayr E, Schultz JE, Doetschman T. Fibroblast growth factor 2 mediates isoproterenol-induced cardiac hypertrophy through activation of the extracellular regulated kinase. Mol Cell Pharmacol 2: 143–154, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.House SL, Wang J, Castro AM, Weinheimer C, Kovacs A, Ornitz DM. Fibroblast growth factor 2 is an essential cardioprotective factor in a closed-chest model of cardiac ischemia-reperfusion injury. Physiol Rep 3: pii: e12278, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Javerzat S, Auguste P, Bikfalvi A. The role of fibroblast growth factors in vascular development. Trends Mol Med 8: 483–489, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Kanno S, Lerner DL, Schuessler RB, Betsuyaku T, Yamada KA, Saffitz JE, Kovacs A. Echocardiographic evaluation of ventricular remodeling in a mouse model of myocardial infarction. J Am Soc Echocardiogr 15: 601–609, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Kardami E, Detillieux K, Ma X, Jiang Z, Santiago JJ, Jimenez SK, Cattini PA. Fibroblast growth factor-2 and cardioprotection. Heart Fail Rev 12: 267–277, 2007. [DOI] [PubMed] [Google Scholar]
- 20.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol 230: 230–242, 2001. [DOI] [PubMed] [Google Scholar]
- 21.Lieu C, Heymach J, Overman M, Tran H, Kopetz S. Beyond VEGF: inhibition of the fibroblast growth factor pathway and antiangiogenesis. Clin Cancer Res 17: 6130–6139, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsunaga S, Okigaki M, Takeda M, Matsui A, Honsho S, Katsume A, Kishita E, Che J, Kurihara T, Adachi Y, Mansukhani A, Kobara M, Matoba S, Tatsumi T, Matsubara H. Endothelium-targeted overexpression of constitutively active FGF receptor induces cardioprotection in mice myocardial infarction. J Mol Cell Cardiol 46: 663–673, 2009. [DOI] [PubMed] [Google Scholar]
- 23.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis 45: 593–605, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Oladipupo SS, Smith C, Santeford A, Park C, Sene A, Wiley LA, Osei-Owusu P, Hsu J, Zapata N, Liu F, Nakamura R, Lavine KJ, Blumer KJ, Choi K, Apte RS, Ornitz DM. Endothelial cell FGF signaling is required for injury response but not for vascular homeostasis. Proc Natl Acad Sci USA 111: 13379–13384, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol 4: 215–266, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pellieux C, Foletti A, Peduto G, Aubert JF, Nussberger J, Beermann F, Brunner HR, Pedrazzini T. Dilated cardiomyopathy and impaired cardiac hypertrophic response to angiotensin II in mice lacking FGF-2. J Clin Invest 108: 1843–1851, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schultz JE, Witt SA, Nieman ML, Reiser PJ, Engle SJ, Zhou M, Pawlowski SA, Lorenz JN, Kimball TR, Doetschman T. Fibroblast growth factor-2 mediates pressure-induced hypertrophic response. J Clin Invest 104: 709–719, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sheikh F, Sontag DP, Fandrich RR, Kardami E, Cattini PA. Overexpression of FGF-2 increases cardiac myocyte viability after injury in isolated mouse hearts. Am J Physiol Heart Circ Physiol 280: H1039–H1050, 2001. [DOI] [PubMed] [Google Scholar]
- 29.Shin JW, Min M, Larrieu-Lahargue F, Canron X, Kunstfeld R, Nguyen L, Henderson JE, Bikfalvi A, Detmar M, Hong YK. Prox1 promotes lineage-specific expression of fibroblast growth factor (FGF) receptor-3 in lymphatic endothelium: a role for FGF signaling in lymphangiogenesis. Mol Biol Cell 17: 576–584, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Virag JA, Rolle ML, Reece J, Hardouin S, Feigl EO, Murry CE. Fibroblast growth factor-2 regulates myocardial infarct repair: effects on cell proliferation, scar contraction, and ventricular function. Am J Pathol 171: 1431–1440, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.World Health Organization. The Top Ten Causes of Death. Geneva, Switzerland: World Health Organization, 2014, p. 1–5. [Google Scholar]
- 32.Zhao T, Zhao W, Chen Y, Ahokas RA, Sun Y. Acidic and basic fibroblast growth factors involved in cardiac angiogenesis following infarction. Int J Cardiol 152: 307–313, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao W, Zhao T, Chen Y, Ahokas RA, Sun Y. Reactive oxygen species promote angiogenesis in the infarcted rat heart. Int J Exp Pathol 90: 621–629, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.