

Abstract
While the neural control of glucoregulatory responses to insulin-induced hypoglycemia is beginning to be elucidated, brain sites responsible for behavioral responses to hypoglycemia are relatively poorly understood. To help elucidate central control mechanisms associated with hypoglycemia unawareness, we first evaluated the effect of recurrent hypoglycemia on a simple behavioral measure, the robust feeding response to hypoglycemia, in rats. First, food intake was significantly, and similarly, increased above baseline saline-induced intake (1.1 ± 0.2 g; n = 8) in rats experiencing a first (4.4 ± 0.3; n = 8) or third daily episode of recurrent insulin-induced hypoglycemia (IIH, 3.7 ± 0.3 g; n = 9; P < 0.05). Because food intake was not impaired as a result of prior IIH, we next developed an alternative animal model of hypoglycemia-induced behavioral arousal using a conditioned place preference (CPP) model. We found that hypoglycemia severely blunted previously acquired CPP in rats and that recurrent hypoglycemia prevented this blunting. Pretreatment with a brain penetrant, selective orexin receptor-1 antagonist, SB-334867A, blocked hypoglycemia-induced blunting of CPP. Recurrently hypoglycemic rats also showed decreased preproorexin expression in the perifornical hypothalamus (50%) but not in the adjacent lateral hypothalamus. Pretreatment with sertraline, previously shown to prevent hypoglycemia-associated glucoregulatory failure, did not prevent blunting of hypoglycemia-induced CPP prevention by recurrent hypoglycemia. This work describes the first behavioral model of hypoglycemia unawareness and suggests a role for orexin neurons in mediating behavioral responses to hypoglycemia.
Keywords: diabetes, perifornical hypothalamus, serotonin, awareness
hypoglycemia unawareness and concomitant failure to activate behavioral defenses against hypoglycemia (such as feeding) contribute to the problem of hypoglycemia-associated autonomic failure and the vicious cycle of insulin-induced hypoglycemia (IIH) in Type 1 diabetes mellitus and advanced Type 2 diabetes mellitus patients undergoing intensive insulin therapy for tight glycemic control (8, 9). The neurogenic symptoms generated by adrenergic and cholinergic activation (such as palpitations and diaphoresis) and neuroglycopenic symptoms generated by central glucopenia per se (such as diminished cognitive function) result in awareness of hypoglycemia and stimulate corrective behavioral responses (such as feeding or glucose administration). However, the central neural substrates for awareness of the hypoglycemic state and the neuronal mechanisms underlying the blunting of this awareness following repeated bouts of IIH are not well understood. More importantly, no animal model of blunted awareness following repeated bouts of IIH exists.
In this series of studies, we attempted to develop a behavioral measure by which hypoglycemia unawareness could be modeled in animals. We first evaluated whether or not the robust feeding response to IIH is blunted by antecedent hypoglycemic episodes, as occurs for hormonal responses [epinephrine (Epi), norepinephrine, glucagon, corticosterone, etc. (16, 39, 45, 53)]. Based on the postulate that activation of behavioral responses to hypoglycemia is contingent upon centrally mediated awareness of hypoglycemic symptoms, we reasoned that putative blunting of feeding by recurrent IIH (RH) could serve as a surrogate for hypoglycemia unawareness. We used a rat model of hypoglycemia-associated autonomic failure (33, 45, 46), involving three bouts of IIH within a 24-h period, which results in significant impairment of glucagon, Epi, norepinephrine, and corticosterone responses to the third IIH exposure and found that the feeding counterregulatory response (CRR) was preserved despite prior IIH exposure.
We next used the well-characterized behavioral paradigm of palatable food-induced conditioned place preference (CPP) (17–19, 51, 55) as a surrogate to assess the aversive effects of IIH and the blunting of those effects following recurrent bouts of IIH seen in humans (8, 9). We propose that awareness of these aversive effects of hypoglycemia in humans and rats requires a state of heightened arousal to recognize both the peripheral effects associated with Epi release [tremor, tachycardia, diaphoresis (27)] and a central component modulated by perifornical hypothalamic (PFH) orexin/hypocretin (here referred to as “orexin”) neurons. These neurons are glucosensing (4, 35, 56) and are involved in the autonomic responses to glucoprivation (29, 37), feeding (14, 43), reward-based learning, and behavioral arousal (23, 24, 28, 40).
We found that, while a single bout of IIH severely blunted a learned CPP for palatable food, this blunting was prevented by both three bouts of RH and by SB-334867-A, a brain-penetrant orexin receptor-1 antagonist (25, 42). It was not prevented using sertraline, a selective serotonin reuptake inhibitor, which we (37) and others (37, 46) have shown to amplify sympathoadrenal responses to acute hypoglycemia and prevent counterregulatory failure after RH. Our results provide the first animal model for assessing the behavioral response to hypoglycemia arousal (“awareness”) after a single bout, its blunting following RH, and the first suggestion that PFH orexin neurons, but not serotonin, play an integral part in mediating these responses.
MATERIALS AND METHODS
Animals
Adult (7–8 wk old, 250–400 g) male Sprague-Dawley rats were used in all studies. Rats for feeding and blood glucose studies were obtained from Simonsen Laboratories (Gilroy, CA), and those for CPP studies were from Charles River Laboratories (Kingston, NY). Rats were maintained at 23–24°C on a conventional 12:12-h light-dark cycle (lights on at 0800) at the animal research facility of the Veterans Affairs Puget Sound Health Care System (Seattle, WA) (feeding and blood glucose studies) and East Orange Veterans Affairs Medical Center (CPP studies). Food (Purina rat chow no. 5001) and water were available ad libitum unless otherwise specified. Experimental groups contained 4–8 rats each for CPP studies and 8–10 rats for feeding studies. All animal protocols and procedures were approved by the Institutional Animal Care and Use Committees of the Veterans Affairs Puget Sound Health Care System and the East Orange Veterans Affairs Medical Center.
Feeding Study: Surgical Procedure
All surgical procedures were performed using ketamine-xylazine anesthesia [60 mg/kg ketamine (Abbott Laboratories) and 7.8 mg/kg xylazine (Phoenix Pharmaceutical)]. Silastic intravenous catheters for blood drawing were surgically implanted using previously described methods (16, 44). The catheter was tunneled subcutaneously, exteriorized through a midline scalp incision, and secured by acrylic cement (Lang Dental) and four skull screws (Small Parts). To maintain patency, the catheter tubing was filled with a mixture of 60% polyvinylpyrrolidone (Sigma) and heparin (1,000 U/ml; Elkins-Sinn) before use. All rats were allowed to recover presurgical weight before onset of the experimental procedure.
Food Intake Study: Experimental Procedure
Feeding was assessed to a first or third bout of IIH using our established model of hypoglycemia-associated autonomic failure (45, 46). All experimental procedures began at lights on and were conducted in square acrylic testing chambers. Rats were familiarized with the testing chamber and habituated to injection (using saline) during the week before experimentation. Nonfasted rats were subjected to one of the following protocols: 1) repeated IIH, H/H/H, n = 7: on day 1, rats were subjected to two bouts of IIH (2.5 U/kg sc regular human insulin, recombinant DNA origin; Novo Nordisk) separated by 1 h during which time food was available; on day 2, rats were subjected to a single bout of IIH (2.5 U/kg sc); 2) single IIH, S/S/H, n = 7: 2 injections of saline (1 ml/kg sc) on day 1 followed by one episode of IIH (2.5 U/kg sc) on day 2; and 3) control, S/S/S, n = 6: two injections of saline (1 ml/kg sc) on day 1 followed by one injection of saline (1 ml/kg sc) on day 2. Blood glucose levels were assayed in response to each 2-h episode of IIH or after saline injection on days 1 and 2. Blood samples (0.1 ml) were drawn from indwelling intravenous catheters 0, 60, and 120 min after injection of insulin or saline for immediate assessment of plasma glucose concentrations (Beckman Glucose Analyzer). Cumulative 3-h food intake was measured on day 2.
IIH Procedure for CPP Studies
On the day before experimental induction of IIH, Charles River Sprague-Dawley rats were semifasted overnight (access to chow restricted to ∼13 g). On the morning of the experiment, all remaining food was removed, and hypoglycemia was initiated by bolus insulin (4.5 U/kg sc unless otherwise specified; Humulin; Lilly). Control animals received equal-volume saline injections. After the period of hypoglycemia (1 h for hypoglycemia in the CPP chamber and 2 h for home cage hypoglycemia), food was returned, and animals were allowed to feed ad libitum.
Single Bout IIH CPP Protocol
The CPP apparatus consisted of a modified open-field test box with transparent Plexiglas walls and two chambers distinguished by differences in the patterns on the floor (checkered or striped). Passage between chambers was controlled by a moveable barrier made of the same material as the rest of the apparatus. On test days (see below), video recordings (from overhead cameras) of 30-min trials were obtained, and time spent on either side of the CPP apparatus was scored by an observer blind to the experimental design. The CPP paradigm was chosen due to ease of implementation, prior work demonstrating its utility in studying the interface between metabolic neurocircuitry and motivated behavior (17–19, 51), and because neurons localized to the lateral hypothalamus have been previously implicated in reward-based learning (23, 24, 40) (Fig. 1A).
Fig. 1.

Timeline of conditioned place preference (CPP) protocol for single (A) and recurrent (B) hypoglycemia studies. A: intrinsic place preference was assessed on day 1 (pretest), CPP training was done on days 2-7, testing for CPP acquisition was done on day 8, rats underwent insulin-induced hypoglycemia (IIH) in the CPP apparatus on day 9, and CPP was again assessed on day 10. B: protocol for days 1-8 was identical to above, but on each of days 9-11 rats underwent single daily 2-h home cage bouts of IIH or saline injections followed by hypoglycemia in the CPP apparatus on day 12 and CPP assessment on day 13 (see materials and methods for details).
On day 1, rats were placed in the CPP box for 30 min with no barrier in place between sides to determine intrinsic place preference. Next, over six contiguous days, rats were placed on alternating sides of the CPP box for 30 min with a barrier in place. On alternating days (days 2, 4, and 6 or days 3, 5, and 7), rats received ∼3 g chocolate (Nestle Chocolate Drops) on the nonpreferred side (as determined from the pretest on day 1), and their intake was measured. All rats received <1 g chocolate in their home cage before training to minimize neophobia. The CPP box was cleaned with 70% ethanol between animals. Training was conducted between 0800 and 1200 (same time of day for each rat). On day 8, rats were placed in the CPP box for 30 min without a barrier in place, and place preference was measured as time spent on each side of the two-chamber apparatus. On day 9, rats underwent IIH (4.5 U/kg sc bolus insulin injection) for 1 h on the preferred side (as determined from testing on day 8) with a barrier in place. On day 10, rats were placed in the CPP box for 30 min without a barrier, and place preference was measured as before. In additional studies designed to assess the role of orexin in this paradigm, SB-334867A (20 mg/kg ip; Tocris) (25) or vehicle (DMSO) were administered immediately before insulin. Food intake data for SH CPP and RH CPP (described below) are summarized in Table 1.
Table 1.
Food intake data during CPP training
| Experiment | Group | n | Days 2/3 | Days 4/5 | Days 6/7 |
|---|---|---|---|---|---|
| Experiment 2 | |||||
| CPP SH | 4 | 1.1 ± 0.3 | 2.2 ± 0.4 | 2.8 ± 0.4 | |
| Experiment 3 | |||||
| CPP RH | SH | 6 | 1.8 ± 0.4 | 2.4 ± 0.5 | 3.2 ± 0.3 |
| RH | 6 | 1.8 ± 0.5 | 2.8 ± 0.5 | 3.3 ± 0.4 | |
| Experiment 4 | |||||
| CPP SH SB-334867A | SAL + VEH | 4 | 1.0 ± 0.2 | 1.7 ± 0.6 | 2.6 ± 0.6 |
| SAL + SB-334867A | 4 | 1.6 ± 0.4 | 2.7 ± 0.5 | 3.0 ± 0.4 | |
| HYPO + VEH | 4 | 1.5 ± 0.3 | 1.9 ± 0.4 | 3.2 ± 0.5 | |
| HYPO + SB-334867A | 4 | 1.3 ± 0.3 | 2.5 ± 0.3 | 3.2 ± 0.2 | |
| Experiment 5 | |||||
| CPP SERT | SH | 7 | 1.4 ± 0.4 | 2.2 ± 0.5 | 2.8 ± 0.6 |
| RH | 8 | 1.5 ± 0.4 | 1.9 ± 0.4 | 2.7 ± 0.6 |
Data are means ± SE; n, no. of rats. CPP, conditioned place preference; SH, saline hypoglycemia; RH, recurrent insulin-induced hypoglycemia; SERT, sertraline; SAL, saline; VEH, vehicle; HYPO, hypothalamic. On alternating days (days 2, 4, and 6 or days 3, 5, and 7), rats received approximately 3 g chocolate (Nestle Chocolate Drops) on the nonpreferred side (as determined from the pretest on day 1), and their intake was measured. There were no significant differences between groups.
RH CPP Protocol
For these studies, we used a different RH protocol than that used for the feeding studies to more closely temporally parallel the multiday period of training required for rats to acquire a CPP. We have used this protocol successfully in the past to produce a blunted neurohumoral response to RH (36). Charles River Sprague-Dawley rats were assessed for initial place preference and trained for CPP as described above (days 1-8). On days 9-11, in their home cages, rats received either a single 2-h bout (at lights on) of IIH (RH group; 4 U/kg bolus insulin sc) or an equal volume saline injection (single IIH group) on each of three consecutive days (20, 33, 37). Animals were semifasted overnight (as described above) before each hypoglycemic episode or saline injection, and then food was returned ad libitum after 2 h of hypoglycemia. On day 12, all rats received IIH (4.0 U/kg bolus insulin sc) for 1 h on the preferred side (as determined from testing on day 8) with a barrier in place. On day 13, rats were placed in the CPP box for 30 min without a barrier, and CPP was measured as before. In a separate set of studies, sertraline was administered beginning 1 wk before and continued throughout the CPP protocol (Fig. 1B).
Minipump Implantation for Systemic Sertraline Infusion
Osmotic minipumps (model 2001; Alzet) were implanted subcutaneously via interscapular incision under ketamine-xylazine anesthesia. Minipumps were loaded with sertraline (Toronto Research Chemicals) at a concentration calculated to deliver 7.5 mg·kg−1·day−1 sertraline or vehicle (50% ethanol) as per the manufacturer's instructions (37, 46).
mRNA Determination by Quantitative Real-Time PCR
Frozen brains from the RH study groups were cut on a cryostat to obtain 300-μm coronal sections and stored in RNAlater (Ambion) until micropunching of the PFH and lateral hypothalamic area (LHA) was performed under stereotaxic guidance as previously described (57). Quantification of mRNA in micropunched brain areas was carried out as previously described (57). Standard curves were generated from serially diluted pooled samples for each probe and for constitutively expressed mRNA (cyclophilin) to control for differences in amplification efficiency and micropunch size. Results were calculated from the standard curves relative to cyclophilin mRNA levels in the same samples.
Statistical Analysis
All statistical analyses were carried out using commercially available software (Systat 8.0). One-way or two-way ANOVA were used for determination of significance depending on experimental design, and post hoc corrections were made using Bonferroni's test. Results are reported as means ± SE.
RESULTS
Experiment 1: Feeding and Glucose Responses to Acute Hypoglycemia and RH
Blood glucose responses to acute and repeated IIH.
Blood glucose reliably decreased in response to both day 1 insulin injections in the H/H/H group [30 ± 3 vs. 27 ± 3 mg/dl, (at 60 min), respectively; Fig. 2C]. Blood glucose levels remained unchanged relative to baseline in response to saline injections in both the S/S/S and S/S/H groups on day 1 (Fig. 2, A and B). Blood glucose levels declined to similar values [45 ± 3 vs. 48 ± 3 mg/dl (at 60 min) (Fig. 2D) in the S/S/H and H/H/H groups, respectively, on day 2, demonstrating quantitatively comparable hypoglycemic stimuli]. Because food was available during day 2 and not during day 1 IIH exposures, the blood glucose nadir was significantly higher in the H/H/H group during day 2 (48 ± 3 mg/dl) vs. day 1 (23 ± 1 mg/dl) (P = 0.01).
Fig. 2.

Day 1 and 2 blood glucose values during IIH. A: day 1 blood glucose values in animals exposed to 3 saline injections in a 2-day protocol (S/S/S). B: day 1 blood glucose values in animals exposed to 2 saline injections on day 1 followed by IIH on day 2 (S/S/H). C: day 1 blood glucose values in animals exposed to 3 insulin injections in a 2-day protocol (H/H/H). D: day 2 blood glucose values in animals exposed to 3 saline injections (S/S/S), 2 saline injections followed by a single bout of IIH (S/S/H), or 3 bouts of IIH (H/H/H) in a 2-day protocol. C: *P < 0.05, day 1 vs. day 2. D: *P < 0.05 S/S/S vs. S/S/H and S/S/S vs. H/H/H. Error bars indicate ± SE throughout.
Feeding responses to acute and repeated hypoglycemia.
A single exposure to IIH on day 2 (S/S/H) significantly increased 3-h cumulative food intake above that induced by saline in the S/S/S group (4.4 ± 0.3 vs. 1.1 ± 0.2 g, respectively; P = 0.001; Fig. 3). Similarly, food intake in response to a third exposure to IIH (H/H/H) (3.7 ± 0.3 g) was also significantly greater than food intake in the S/S/S group (P = 0.05) but was not significantly different from food intake in rats experiencing their first bout of IIH (S/S/H).
Fig. 3.
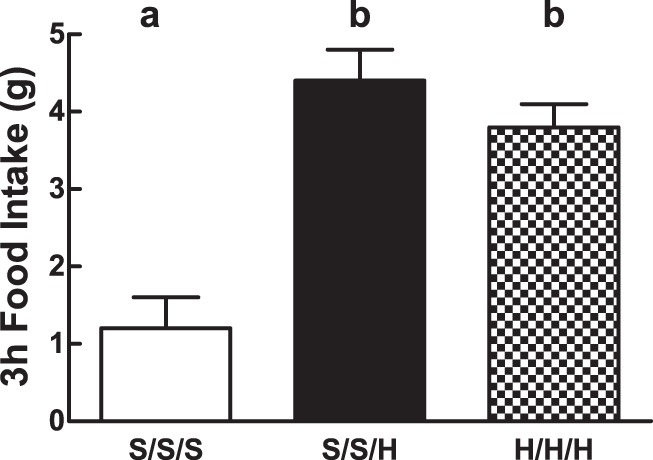
Day 2 cumulative food intake during IIH. Day 2 cumulative (3-h) food intake in animals exposed to S/S/S, S/S/H, or H/H/H in a 2-day protocol. Letters denote significant differences in treatment × time by 2-way repeated-measures ANOVA. Error bars indicate ± SE. F(2, 17) = 80.3 (S/S/S vs. S/S/H vs. H/H/H).
Experiment 2: Effect of Acute IIH on CPP
An animal's responsiveness (here referred to as awareness) of the negative symptoms of hypoglycemia (i.e., the aversive salience of hypoglycemia) can potentially be measured as the ability of hypoglycemia to prevent or attenuate a previously learned preference. We tested the hypothesis that a single bout of IIH would attenuate an animal's learned preference for one side of a two-sided CPP chamber using our palatable food reward CPP paradigm. Before training, rats (n = 4/group) spent 69% more time on one striped side of the CPP apparatus (P = 0.03, side 1 vs. side 2). This intrinsic place preference was completely prevented by the 6-day training regimen with a palatable food reward, resulting in a marked CPP (∼79% of time spent on previously nonpreferred side; P = 0.003, side 1 vs. side 2). A single bout of IIH, on the side conditioned to be preferred, attenuated CPP (39% of time spent on previously preferred side; P = 0.09, side 1 vs. side 2) such that neither side of the two-chamber apparatus was preferred (Fig. 4).
Fig. 4.
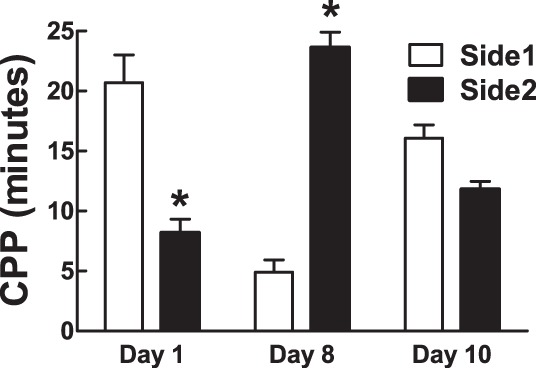
CPP in rats before (day 1) and after (day 8) training with a palatable food reward and 1 day after IIH (day 10). CPP is expressed as minutes spent on either side of a 2-sided CPP apparatus. Data are means ± SE; *P ≤ 0.05 between groups. F(3,3) = 2.7 (day 10, side 1 vs. side 2).
Experiment 3: Effect of RH on CPP Responding to Additional IIH
Because a single bout of IIH severely blunted food reward CPP in rats (above) while repeated bouts of hypoglycemia blunt or prevent awareness of hypoglycemic symptoms in humans (7, 9, 10, 26), we tested the hypothesis that antecedent RH would attenuate the hypoglycemia-induced blunting of reward-based CPP. The CPP protocol was altered (Fig. 1B) to include three sequential days of home cage IIH [RH (4 U/kg bolus insulin sc); n = 4] or saline (single IIH, SH; n = 4) between the posttraining test (day 8) and a fourth bout of hypoglycemia in the CPP chamber (day 12). CPP was then assessed as before on day 13. SH rats, which experienced hypoglycemia for the first time on day 12 (after 3 days of home cage saline injections), showed the expected blunting of CPP (Fig. 5A). However, rats that experienced three prior days of sequential home cage bouts of hypoglycemia failed to blunt a CPP when assessed after a fourth bout of hypoglycemia in the CPP apparatus on day 12 (P = 0.036, SH vs. RH, day 13 CPP; Fig. 5A). Furthermore, three sequential bouts of IIH caused a 50% decrease in the expression of preproorexin mRNA in the PFH, but not in the adjacent LHA, relative to a single bout of IIH (P = 0.003, SH vs. RH; Fig. 5B). This suggested that PFH orexin neurons and their reduced expression of orexin mRNA might be involved in the failure to prevent CPP after RH.
Fig. 5.

CPP in rats before (day 1) and after training with a palatable food reward (day 8) and after IIH (day 13) preceded by 3 previous bouts of home cage hypoglycemia (RH) or saline injections (SH). A: CPP is expressed as the difference in time spent on either side of a 2-sided CPP apparatus. Change in the sign of the CPP value reflects a change in preference relative to the previous time point. B: preproorexin mRNA relative abundance in the PFH and LHA in recurrent hypoglycemic or single hypoglycemic rats. Data are means ± SE; *P ≤ 0.05 between groups. Letters denote significant differences in treatment × time by 2-way repeated-measures ANOVA. F(5,4) = 5.3 (day 13, SH vs. RH).
Experiment 4: Effect of SB-334867A on Acute IIH-Induced Prevention of Food Reward CPP
To test the hypothesis that orexin neurons are involved in the CPP responses to a single bout of IIH, rats (n = 4/group) underwent the standard CPP protocol but received either IIH (4.5 U/kg bolus insulin sc) + SB-334867A (20 mg/kg ip), IIH + DMSO (vehicle), saline + SB-334867A (20 mg/kg ip), or saline + DMSO on day 9. While IIH completely prevented the CPP in vehicle-treated rats, it failed to do so when paired with SB-334867A (P = 0.001, hypoglycemia + vehicle vs. hypoglycemia + SB-334867A; Fig. 6). Because no significant difference was detected between the saline + vehicle and saline + SB-334867A groups, these data were pooled during analysis. There were no significant differences in place preference before training or CPP acquisition between groups.
Fig. 6.
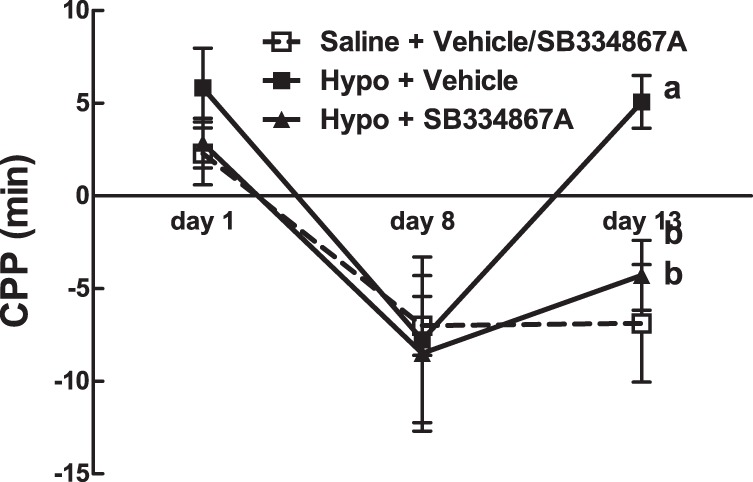
CPP in rats before (day 1) and after (day 8) training with a palatable food reward and after IIH (day 10) or saline paired with SB-334867A (20 mg/kg ip) or vehicle. CPP is expressed as the difference in time spent on either side of a 2-sided CPP apparatus. Change in the sign of the CPP value reflects a change in preference relative to the previous time point. Data are means ± SE. Letters denote significant differences in treatment × time by 2-way repeated-measures ANOVA. F(3,3) = 2.8 (day 13, Hypo-SB vs. Sal-SB/Sal-DMSO); F(3,2) = 6.6 (day 13, Hypo-DMSO vs. Sal-SB/Sal-DMSO).
Experiment 5: Effect of Chronic Sertraline Administration on CPP Responding to Single vs. Recurrent Bouts of IIH
Although 21 days of pretreatment with sertraline (7.5 mg·kg−1·day−1 sc) had previously been shown to amplify the counterregulatory response to acute hypoglycemia and prevent blunting of the CRR after RH (46), treatment with fluoxetine, another selective serotonin reuptake inhibitor, failed to amplify the behavioral symptoms associated with a single bout of IIH in humans (3). Therefore, we hypothesized that sertraline pretreatment would not prevent the IIH prevention of CPP as seen after recurrent bouts of IIH (Fig. 5A). In fact, sertraline pretreatment affected neither acquisition of CPP nor the failure of a fourth bout of IIH to prevent the CPP after three previous bouts of IIH (P = 0.012, sertraline SH vs. sertraline RH, day 13 CPP; Fig. 7).
Fig. 7.
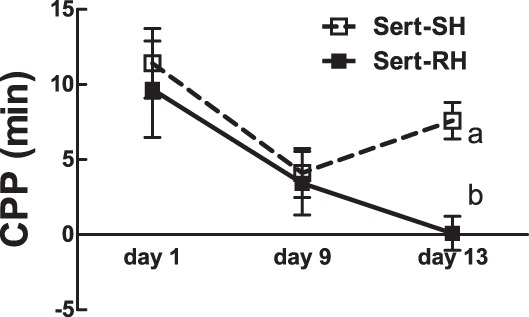
CPP with (RH) and without (SH) 3 bouts of prior home cage hypoglycemia in rats pretreated with 21 days of 7.5 mg·kg−1·day−1 sertraline and assessed for CPP on day 13. CPP is expressed as the difference in time spent on either side of a 2-sided CPP apparatus. Change in the sign of the CPP value reflects a change in preference relative to the previous time point. Data are means ± SE. Letters denote significant differences in treatment × time by 2-way repeated-measures ANOVA. F(3,5) = 3.1 (day 13, SH vs. RH).
DISCUSSION
While numerous animal models exist that model the blunting of the neurohumoral CRR to RH that occurs in humans exposed to repeated bouts of IIH (32, 39, 50), no animal model currently exists to mimic the behavioral unawareness that occurs in humans following RH (11). To create such an animal model, we first simultaneously evaluated blood glucose and food intake in response to a first or third bout of IIH. Previous animal studies (15, 47, 48, 53) consistently show significant impairment of glucagon, Epi, and norepinephrine responses as a result of two prior bouts of IIH. However, food intake, a behavioral CRR to hypoglycemia requiring arousal and intact motor activity, was not blunted after RH. On the other hand, although the RH protocol differed, three prior bouts of IIH blocked the blunting of a CPP seen after a single bout of IIH. These results suggest that the CPP model, but not feeding, is a reasonable one to mimic the blunting of hypoglycemia awareness seen in humans following bouts of RH (9, 10, 26, 31).
Acute reductions in blood glucose increase feelings of hunger (49) and potently stimulate food intake (13, 52). Several studies in humans reported that antecedent hypoglycemia impairs hunger perception during subsequent episodes of hypoglycemia (11, 26, 38). However, hunger ratings were often combined with other symptoms of hypoglycemia, many of which are mediated by autonomic nervous system activation (54) and thus subject to impairment by RH. More recently, however, Schultes et al. evaluated feelings of hunger independent from other symptomatic responses to one or multiple bouts of hypoglycemia (49). They demonstrated that, whereas hypoglycemic symptoms (tremor, palpitations, and weakness, for example) were significantly reduced in response to a third bout of hypoglycemia, hunger ratings remained significantly elevated. These results are in keeping with our findings in rats suggesting that stimulation of food intake may not be subject to the same pathological mechanism(s) that impair hormonal CRR or hypoglycemia awareness. This may be due to differential control of neuroendocrine vs. feeding responses to hypoglycemia by distinct neural substrates. Such a dissociation is supported by the experiments of Ritter et al. (41), which demonstrated that, while selective destruction of the rostral-projecting hindbrain catecholamine neurons permanently eliminated glucoprivic feeding, the sympathoadrenal response remained intact. Similarly, we found that, although depletion of PFH serotonin innervation impairs the sympathoadrenal response to local glucoprivation and systemic IIH, it does not impair the feeding response to these manipulations (37).
Given the preservation of the feeding response to IIH after RH, we also attempted to use a food reward-based CPP as a surrogate for the arousal or awareness associated with acute hypoglycemia and its blunting after recurrent bouts of hypoglycemia in humans. We demonstrate here that a single bout of IIH prevented a learned CPP and that this prevention was blunted following antecedent bouts of RH. We postulate that CPP prevention following a single bout of IIH is due to the aversive effects of both peripheral Epi-mediated and central neuronal activation-associated hypoglycemia symptoms. The fact that CPP blunting was prevented by concurrent systemic administration of a brain-penetrant orexin-1 receptor antagonist (25, 42) strongly suggests that orexin neurons, which are found only in the PFH and LHA (12, 43), are critical components of this response. In fact, we have shown that this same antagonist blunts the Epi response to IIH (37) in keeping with the known connections of PFH orexin neurons with brain stem sympathetic outputs (29) and polysynaptic inputs to the adrenal gland (21). Our results demonstrating a selective decrease in PFH, but not LHA preproorexin expression following RH, further potentially implicate PFH orexin neurons in the blunting of the behavioral response to RH. On the other hand, although we previously demonstrated a critical requirement for PFH serotonin release for the full Epi response to both PFH glucoprivation and IIH (37), systemic pretreatment with sertraline failed to block the inhibitory effect of RH on hypoglycemia-induced CPP blunting. These findings are in keeping with those in humans where chronic fluoxetine, another selective serotonin selective reuptake inhibitor, enhanced the counterregulatory neurohumoral responses but failed to enhance the symptoms (awareness) associated with hypoglycemia (3).
In our behavioral model, we used the aversive salience of hypoglycemia (i.e., the ability of hypoglycemia to blunt a previously acquired preference) as a surrogate for the arousal or awareness of hypoglycemia experienced by humans (1, 2, 27, 30). A reward-based CPP was used as opposed to using hypoglycemia as the primary aversive stimulus (i.e., conditioned place aversion) because reward-based learning has been shown to involve orexin neurons (23, 24, 40) and to facilitate the interpretation of experimental results. We found in preliminary studies (data not shown) that, while intrinsic (nonrewarded) place preference was highly variable among subjects and experiments, training for reward-based CPP allowed for reliable baselines for all animals.
We postulate that the aversive effects of hypoglycemia require behavioral arousal to allow for awareness of these aversive effects to be experienced by rats. Of course, we cannot directly measure awareness in rats. However, our use of the CPP paradigm appears to provide the first behavioral surrogate of hypoglycemia unawareness in rats in that RH completely blunts the behavioral arousal and aversion required for a single bout of IIH to prevent reward-based CPP. The present studies show that, in an animal experiencing hypoglycemia for the first time, a single bout of IIH is sufficient to significantly blunt a CPP learned by using highly palatable chocolate drops but that antecedent RH completely prevents hypoglycemia-induced blunting of this CPP. This phenomenon is distinct from simple extinction since rats that experienced three bouts of home cage saline injections still displayed a robust hypoglycemia-induced blunting of CPP when then exposed to a single bout of IIH in the CPP apparatus on the fourth testing day. Thus, although we did not directly assess extinction of the food reward CPP, our RH studies suggest that, within the time frames used in our experiments, significant extinction of learning did not occur.
We propose that the loss of a previously entrained CPP after a single bout of IIH is dependent on the association between the negative consequences of hypoglycemia and the preferred side of a testing apparatus. The loss of this association did not occur after prior recurrent bouts of home cage hypoglycemia, which we interpret to reflect an absence of awareness of the aversive qualities of IIH. However, the absence of insulin-induced CPP loss following recurrent home cage hypoglycemia may, alternatively, be due to habituation of animals to the hypoglycemic stimulus, which might preclude the formation of an association between hypoglycemia and the test chamber. Further studies will be necessary to determine whether or not prevention of insulin-induced CPP blunting by RH is part of a more general habituation phenomenon.
Hypoglycemia-induced prevention of food-reward CPP appears to be mediated at least in part by orexin signaling through the orexin receptor-1 because concurrent administration of SB-334867A with systemic hypoglycemia significantly attenuated CPP prevention. In addition, repeated bouts of hypoglycemia selectively attenuated preproorexin expression in the PFH but not the adjacent LHA. This suggests a potential mechanism of hypoglycemia unawareness, i.e., diminished ability of hypoglycemia to stimulate arousal-promoting orexin neurons of the PFH, which are well described to be activated by hypoglycemia (5, 6, 22, 34). Because PFH orexin neurons are implicated by the present findings in awareness of hypoglycemia, and by previous work in neurohumoral CRR to glucoprivation (29, 37), they constitute a potentially important central neural substrate for mediating multiple components of hypoglycemia-associated autonomic failure. Orexin neurons, especially of the PFH, may contribute to behavioral responses during hypoglycemia, which are due to their activation during lowered glucose levels and which are independent of the symptoms caused by elevated Epi levels.
We (37) and others (3, 46) have shown that serotonergic signaling promotes the autonomic counterregulatory responses to acute IIH. Sanders et al. (46) demonstrated that a 21-day pretreatment with sertraline prevented attenuation of the adrenomedullary response by RH. We therefore reasoned that this regimen would diminish hypoglycemia unawareness, modeled by attenuation of hypoglycemia-induced CPP prevention by repeated antecedent bouts of hypoglycemia. However, sertraline pretreatment failed to prevent RH-induced attenuation of hypoglycemia-induced CPP prevention. These results, and those in humans, where a selective serotonin reuptake inhibitor failed to enhance the behavioral awareness of hypoglycemia (3), suggest that separate neural pathways may mediate behavioral as opposed to neurohumoral and autonomic responses to hypoglycemia.
In summary, we propose a novel rat model for studying hypoglycemia awareness using hypoglycemia-induced prevention of a food reward CPP. We found that expression of this behavioral phenomenon is attenuated by recent antecedent hypoglycemia and contingent upon intact orexin signaling through the orexin-1 receptor. We also provide evidence against a role for selective serotonin reuptake inhibitors in preventing hypoglycemia unawareness.
GRANTS
This work was supported by the Research Service of the Department of Veterans Affairs (B. E. Levin) and the National Institute for Diabetes and Digestive and Kidney Diseases [RO1-DK-53181 (B. E. Levin)].
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: O.O. and B.E.L. conception and design of research; O.O., N.M.S., and A.A.D.-M. performed experiments; O.O. and B.E.L. analyzed data; O.O. and B.E.L. interpreted results of experiments; O.O. prepared figures; O.O. drafted manuscript; O.O. and B.E.L. edited and revised manuscript; O.O. and B.E.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Sunny Lee, Antoinette Moralishvili, and Charlie Salter (all from the Veterans Affairs Medical Center) for technical assistance.
REFERENCES
- 1.Berlin I, Grimaldi A, Payan C, Sachon C, Bosquet F, Thervet F, Puech AJ. Hypoglycemic symptoms and decreased beta-adrenergic sensitivity in insulin-dependent diabetic patients. Diabetes Care 10: 742–747, 1987. [DOI] [PubMed] [Google Scholar]
- 2.Berlin I, Grimaldi A, Thervet F, Puech AJ. Hypoglycaemia unawareness. Lancet 2: 966, 1987. [DOI] [PubMed] [Google Scholar]
- 3.Briscoe VJ, Ertl AC, Tate DB, Davis SN. Effects of the selective serotonin reuptake inhibitor fluoxetine on counterregulatory responses to hypoglycemia in individuals with type 1 diabetes. Diabetes 57: 3315–3322, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burdakov D, Gerasimenko O, Verkhratsky A. Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J Neurosci 25: 2429–2433, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai XJ, Evans ML, Lister CA, Leslie RA, Arch JR, Wilson S, Williams G. Hypoglycemia activates orexin neurons and selectively increases hypothalamic orexin-B levels: responses inhibited by feeding and possibly mediated by the nucleus of the solitary tract. Diabetes 50: 105–112, 2001. [DOI] [PubMed] [Google Scholar]
- 6.Cai XJ, Widdowson PS, Harrold J, Wilson S, Buckingham RE, Arch JR, Tadayyon M, Clapham JC, Wilding J, Williams G. Hypothalamic orexin expression: modulation by blood glucose and feeding. Diabetes 48: 2132–2137, 1999. [DOI] [PubMed] [Google Scholar]
- 7.Clarke WL, Gonder-Frederick LA, Richards FE, Cryer PE. Multifactorial origin of hypoglycemic symptom unawareness in IDDM. Association with defective glucose counterregulation and better glycemic control. Diabetes 40: 680–685, 1991. [DOI] [PubMed] [Google Scholar]
- 8.Cryer PE. Hypoglycemia-associated autonomic failure in diabetes. Am J Physiol Endocrinol Metab 281: E1115–E1121, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Cryer PE. Hypoglycemia unawareness in IDDM. Diabetes Care 16, Suppl 3: 40–47, 1993. [DOI] [PubMed] [Google Scholar]
- 10.Cryer PE. Iatrogenic hypoglycemia as a cause of hypoglycemia-associated autonomic failure in IDDM. A vicious cycle. Diabetes 41: 255–260, 1992. [DOI] [PubMed] [Google Scholar]
- 11.Cryer PE. Symptoms of hypoglycemia, thresholds for their occurrence, and hypoglycemia unawareness. Endocrinol Metab Clin N Am 28: 495–500, 1999. [DOI] [PubMed] [Google Scholar]
- 12.de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS 2nd Frankel WN, van den Pol AN, Bloom FE, Gautvik KM, Sutcliffe JG. The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA 95: 322–327, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dewan S, Gillett A, Mugarza JA, Dovey TM, Halford JC, Wilding JP. Effects of insulin-induced hypoglycaemia on energy intake and food choice at a subsequent test meal. Diabetes/Metab Res Rev 20: 405–410, 2004. [DOI] [PubMed] [Google Scholar]
- 14.Dube MG, Kalra SP, Kalra PS. Food intake elicited by central administration of orexins/hypocretins: identification of hypothalamic sites of action. Brain Res 842: 473–477, 1999. [DOI] [PubMed] [Google Scholar]
- 15.Evans SB, Wilkinson CW, Bentson K, Gronbeck P, Zavosh A, Figlewicz DP. PVN activation is suppressed by repeated hypoglycemia but not antecedent corticosterone in the rat. Am J Physiol Regul Integr Comp Physiol 281: R1426–R1436, 2001. [DOI] [PubMed] [Google Scholar]
- 16.Evans SB, Wilkinson CW, Gronbeck P, Bennett JL, Taborsky GJ Jr, Figlewicz DP. Inactivation of the PVN during hypoglycemia partially simulates hypoglycemia-associated autonomic failure. Am J Physiol Regul Integr Comp Physiol 284: R57–R65, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Figlewicz DP, Bennett J, Evans SB, Kaiyala K, Sipols AJ, Benoit SC. Intraventricular insulin and leptin reverse place preference conditioned with high-fat diet in rats. Behav Neurosci 118: 479–487, 2004. [DOI] [PubMed] [Google Scholar]
- 18.Figlewicz DP, Bennett JL, Naleid AM, Davis C, Grimm JW. Intraventricular insulin and leptin decrease sucrose self-administration in rats. Physiol Behav 89: 611–616, 2006. [DOI] [PubMed] [Google Scholar]
- 19.Figlewicz DP, Higgins MS, Ng-Evans SB, Havel PJ. Leptin reverses sucrose-conditioned place preference in food-restricted rats. Physiol Behav 73: 229–234, 2001. [DOI] [PubMed] [Google Scholar]
- 20.Flanagan DE, Keshavarz T, Evans ML, Flanagan S, Fan X, Jacob RJ, Sherwin RS. Role of corticotrophin-releasing hormone in the impairment of counterregulatory responses to hypoglycemia. Diabetes 52: 605–613, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Geerling JC, Mettenleiter TC, Loewy AD. Orexin neurons project to diverse sympathetic outflow systems. Neuroscience 122: 541–550, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Griffond B, Risold PY, Jacquemard C, Colard C, Fellmann D. Insulin-induced hypoglycemia increases preprohypocretin (orexin) mRNA in the rat lateral hypothalamic area. Neurosci Lett 262: 77–80, 1999. [DOI] [PubMed] [Google Scholar]
- 23.Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature 437: 556–559, 2005. [DOI] [PubMed] [Google Scholar]
- 24.Harris GC, Wimmer M, Randall-Thompson JF, Aston-Jones G. Lateral hypothalamic orexin neurons are critically involved in learning to associate an environment with morphine reward. Behav Brain Res 183: 43–51, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haynes AC, Jackson B, Chapman H, Tadayyon M, Johns A, Porter RA, Arch JR. A selective orexin-1 receptor antagonist reduces food consumption in male and female rats. Regul Pept 96: 45–51, 2000. [DOI] [PubMed] [Google Scholar]
- 26.Heller SR, Cryer PE. Reduced neuroendocrine and symptomatic responses to subsequent hypoglycemia after 1 episode of hypoglycemia in nondiabetic humans. Diabetes 40: 223–226, 1991. [DOI] [PubMed] [Google Scholar]
- 27.Heller SR, Macdonald IA, Herbert M, Tattersall RB. Influence of sympathetic nervous system on hypoglycaemic warning symptoms. Lancet 2: 359–363, 1987. [DOI] [PubMed] [Google Scholar]
- 28.Jones BE. Modulation of cortical activation and behavioral arousal by cholinergic and orexinergic systems. Ann NY Acad Sci 1129: 26–34, 2008. [DOI] [PubMed] [Google Scholar]
- 29.Korim WS, Bou-Farah L, McMullan S, Verberne AJ. Orexinergic activation of medullary premotor neurons modulates the adrenal sympathoexcitation to hypothalamic glucoprivation. Diabetes 63: 1895–1906, 2014. [DOI] [PubMed] [Google Scholar]
- 30.Lager I, Attvall S, Blohme G, Smith U. Altered recognition of hypoglycaemic symptoms in type I diabetes during intensified control with continuous subcutaneous insulin infusion. Diabet Med 3: 322–325, 1986. [DOI] [PubMed] [Google Scholar]
- 31.Lingenfelser T, Renn W, Sommerwerck U, Jung MF, Buettner UW, Zaiser-Kaschel H, Kaschel R, Eggstein M, Jakober B. Compromised hormonal counterregulation, symptom awareness, and neurophysiological function after recurrent short-term episodes of insulin-induced hypoglycemia in IDDM patients. Diabetes 42: 610–618, 1993. [DOI] [PubMed] [Google Scholar]
- 32.McCrimmon RJ, Evans ML, Fan X, McNay EC, Chan O, Ding Y, Zhu W, Gram DX, Sherwin RS. Activation of ATP-sensitive K+ channels in the ventromedial hypothalamus amplifies counterregulatory hormone responses to hypoglycemia in normal and recurrently hypoglycemic rats. Diabetes 54: 3169–3174, 2005. [DOI] [PubMed] [Google Scholar]
- 33.McCrimmon RJ, Fan X, Cheng H, McNay E, Chan O, Shaw M, Ding Y, Zhu W, Sherwin RS. Activation of AMP-activated protein kinase within the ventromedial hypothalamus amplifies counterregulatory hormone responses in rats with defective counterregulation. Diabetes 55: 1755–1760, 2006. [DOI] [PubMed] [Google Scholar]
- 34.Moriguchi T, Sakurai T, Nambu T, Yanagisawa M, Goto K. Neurons containing orexin in the lateral hypothalamic area of the adult rat brain are activated by insulin-induced acute hypoglycemia. Neurosci Lett 264: 101–104, 1999. [DOI] [PubMed] [Google Scholar]
- 35.Muroya S, Uramura K, Sakurai T, Takigawa M, Yada T. Lowering glucose concentrations increases cytosolic Ca2+ in orexin neurons of the rat lateral hypothalamus. Neurosci Lett 309: 165–168, 2001. [DOI] [PubMed] [Google Scholar]
- 36.Orban BO, Routh VH, Levin BE, Berlin JR. Direct effects of recurrent hypoglycaemia on adrenal catecholamine release. Diabet Vasc Dis Res 12: 2–12, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Otlivanchik O, Le Foll C, Levin BE. Perifornical hypothalamic orexin and serotonin modulate the counterregulatory response to hypoglycemic and glucoprivic stimuli. Diabetes 64: 226–235, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ovalle F, Fanelli CG, Paramore DS, Hershey T, Craft S, Cryer PE. Brief twice-weekly episodes of hypoglycemia reduce detection of clinical hypoglycemia in type 1 diabetes mellitus. Diabetes 47: 1472–1479, 1998. [DOI] [PubMed] [Google Scholar]
- 39.Powell AM, Sherwin RS, Shulman GI. Impaired hormonal responses to hypoglycemia in spontaneously diabetic and recurrently hypoglycemic rats. Reversibility and stimulus specificity of the deficits. J Clin Invest 92: 2667–2674, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richardson KA, Aston-Jones G. Lateral hypothalamic orexin/hypocretin neurons that project to ventral tegmental area are differentially activated with morphine preference. J Neurosci 32: 3809–3817, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ritter S, Bugarith K, Dinh TT. Immunotoxic destruction of distinct catecholamine subgroups produces selective impairment of glucoregulatory responses and neuronal activation. J Comp Neurol 432: 197–216, 2001. [DOI] [PubMed] [Google Scholar]
- 42.Rodgers RJ, Halford JC, Nunes de Souza RL, Canto de Souza AL, Piper DC, Arch JR, Upton N, Porter RA, Johns A, Blundell JE. SB-334867, a selective orexin-1 receptor antagonist, enhances behavioural satiety and blocks the hyperphagic effect of orexin-A in rats. Eur J Neurosci 13: 1444–1452, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ, Yanagisawa M. Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92: 573–585, 1998. [DOI] [PubMed] [Google Scholar]
- 44.Sanders NM, Dunn-Meynell AA, Levin BE. Third ventricular alloxan reversibly impairs glucose counterregulatory responses. Diabetes 53: 1230–1236, 2004. [DOI] [PubMed] [Google Scholar]
- 45.Sanders NM, Figlewicz DP, Taborsky GJ Jr, Wilkinson CW, Daumen W, Levin BE. Feeding and neuroendocrine responses after recurrent insulin-induced hypoglycemia. Physiol Behav 87: 700–706, 2006. [DOI] [PubMed] [Google Scholar]
- 46.Sanders NM, Wilkinson CW, Taborsky GJ Jr, Al-Noori S, Daumen W, Zavosh A, Figlewicz DP. The selective serotonin reuptake inhibitor sertraline enhances counterregulatory responses to hypoglycemia. Am J Physiol Endocrinol Metab 294: E853–E860, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sandoval DA, Ertl AC, Richardson MA, Tate DB, Davis SN. Estrogen blunts neuroendocrine and metabolic responses to hypoglycemia. Diabetes 52: 1749–1755, 2003. [DOI] [PubMed] [Google Scholar]
- 48.Sandoval DA, Ping L, Neill RA, Gong B, Walsh K, Davis SN. Brain region-dependent effects of dexamethasone on counterregulatory responses to hypoglycemia in conscious rats. Am J Physiol Regul Integr Comp Physiol 288: R413–R419, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Schultes B, Oltmanns KM, Kern W, Fehm HL, Born J, Peters A. Modulation of hunger by plasma glucose and metformin. J Clin eEdocrinol Metab 88: 1133–1141, 2003. [DOI] [PubMed] [Google Scholar]
- 50.Song Z, Routh VH. Recurrent hypoglycemia reduces the glucose sensitivity of glucose-inhibited neurons in the ventromedial hypothalamus nucleus. Am J Physiol Regul Integr Comp Physiol 291: R1283–R1287, 2006. [DOI] [PubMed] [Google Scholar]
- 51.Stuber GD, Evans SB, Higgins MS, Pu Y, Figlewicz DP. Food restriction modulates amphetamine-conditioned place preference and nucleus accumbens dopamine release in the rat. Synapse 46: 83–90, 2002. [DOI] [PubMed] [Google Scholar]
- 52.Thompson DA, Campbell RG. Hunger in humans induced by 2-deoxy-d-glucose: glucoprivic control of taste preference and food intake. Science 198: 1065–1068, 1977. [DOI] [PubMed] [Google Scholar]
- 53.Tkacs NC, Dunn-Meynell AA, Levin BE. Presumed apoptosis and reduced arcuate nucleus neuropeptide Y and pro-opiomelanocortin mRNA in non-coma hypoglycemia. Diabetes 49: 820–826, 2000. [DOI] [PubMed] [Google Scholar]
- 54.Towler DA, Havlin CE, Craft S, Cryer P. Mechanism of awareness of hypoglycemia. Perception of neurogenic (predominantly cholinergic) rather than neuroglycopenic symptoms. Diabetes 42: 1791–1798, 1993. [DOI] [PubMed] [Google Scholar]
- 55.Tzschentke TM. Measuring reward with the conditioned place preference paradigm: a comprehensive review of drug effects, recent progress and new issues. Prog Neurobiol 56: 613–672, 1998. [DOI] [PubMed] [Google Scholar]
- 56.Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami K, Sugiyama F, Goto K, Yanagisawa M, Sakurai T. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron 38: 701–713, 2003. [DOI] [PubMed] [Google Scholar]
- 57.Zhou L, Podolsky N, Sang Z, Ding Y, Fan X, Tong Q, Levin BE, McCrimmon RJ. The medial amygdalar nucleus: a novel glucose-sensing region that modulates the counterregulatory response to hypoglycemia. Diabetes 59: 2646–2652, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]