

Abstract
At least 120 non-olfactory G protein-coupled receptors in the human genome are ”orphans” for which endogenous ligands are unknown, and many have no selective ligands, hindering elucidation of their biological functions and clinical relevance. Among these is GPR68, a proton receptor that lacks small molecule modulators for probing its biology. Yeast-based screens against GPR68 identified the benzodiazepine drug lorazepam as a non-selective GPR68 positive allosteric modulator. Over 3000 GPR68 homology models were refined to recognize lorazepam in a putative allosteric site. Docking 3.1 million molecules predicted new GPR68 modulators many of which were confirmed in functional assays. One potent GPR68 modulator—ogerin– suppressed recall in fear conditioning in wild-type, but not in GPR68 knockout mice. The same approach led to the discovery of allosteric agonists and negative allosteric modulators for GPR65. Combining physical and structure-based screening may be broadly useful for ligand discovery for understudied and orphan GPCRs.
Keywords: GPR65, GPR68, OGR1, virtual screening, allosteric modulator, positive allosteric modulator (PAM), functional selectivity, proton-sensing
G protein-coupled receptors (GPCRs)–the largest family of proteins encoded in the human genome–transduce signals for the most diverse endogenous ligands of any receptor family. Correspondingly, GPCRs are the most productive drug targets, with over 26% of FDA-approved drugs acting primarily through them. Astonishingly, of the 356 non-olfactory GPCRs, about 38% are understudied or “orphan” receptors whose physiological roles, and often endogenous ligands, remain unknown1. Given the central role of GPCRs in physiology and disease, and the high conservation of orphan GPCRs among organisms from worms to humans, understudied and orphan GPCRs are likely functionally and therapeutically important. Indeed, for the few GPCRs deorphanized since 20031,2 (http://www.iuphar-db.org/latestPairings.jsp/), most have newly approved and investigational drugs1,3. As with kinases4, epigenetic proteins5, and proteases6, ligands specific for orphan GPCRs will illuminate their biology and provide new areas for therapeutic intervention.
A key impediment to GPCR deorphanization is uncertainty about the proteins through which they signal, making functional assays problematic1. This difficulty is increased by the diverse ligands that GPCRs recognize, which range from protons and photons, small neurotransmitters, lipids, to peptides and folded proteins. Thus, generic functional screens are difficult for orphan GPCRs–one neither knows what class of compounds to screen, nor how to screen for it, much less how to demonstrate relevance–thereby explaining the slow progress in determining their roles in signaling and in physiology3.
GPR68 (OGR1) exemplifies both the important roles these understudied and orphan receptors are thought to serve, and our difficulties in illuminating them. GPR68, together with GPR4, GPR65, and GPR132, form a family of proton-sensing GPCRs7. GPR68 couples to multiple signaling pathways through Gq, Gs, G12/13, or Gi/o proteins7–10. GPR68 is expressed in many tissues, and has been implicated in many processes11–16, but is most abundant in mouse cerebellum17 and hippocampus11 (http://www.brain-map.org/), implying yet-to-be identified roles in brain function. In acidic microenvironments, GPR68 appears to regulate inflammatory processes in airway smooth muscle (ASM) and other cells18–20. Confoundingly, studies with GPR68 knockout (KO) mice uncovered only modest changes in these functions21,16,22. Although GPR68 has been reported to be activated by a family of isoxazoles15, their weak activity appears to be non-specific23,24 and could not be reproduced (Results). Thus, though GPR68 may have multiple roles, few of them are well-characterized by knockout and none are known in the CNS, where it is most highly expressed. Like other targets lacking small molecule reagents, GPR68 remains “pharmacological dark matter”1.
Here we describe an integrated experimental and computational approach to discover ligands that modulate GPR68. A lead compound that functions as a positive allosteric modulator (PAM) is demonstrated in vitro and in vivo, providing insights into GPR68 physiology. Application of the same approach found allosteric agonists and negative allosteric modulators (NAMs) for a second understudied GPCR, GPR65, suggesting the approach may be broadly useful.
Yeast-based screen reveals GPR68 actives
In an initial campaign with selected orphan and understudied GPCRs, we modified an yeast assay system25 and screened a small library of approved drugs (http://www.nihclinicalcollection.com/) (Supplementary Figure 1). We confirmed the known activity of short-chain carboxylic acids at the GPR41 and the GPR43 free fatty acid receptors (Extended Data Fig 1a–d), and that of zinc (Extended Data Fig 1e) and several other metals (Extended Data Fig 1f–k) on GPR39. The most striking result was the finding that lorazepam was an agonist at GPR68 (Figure 1).
Figure 1. Lorazepam is a GPR68 positive allosteric modulator.
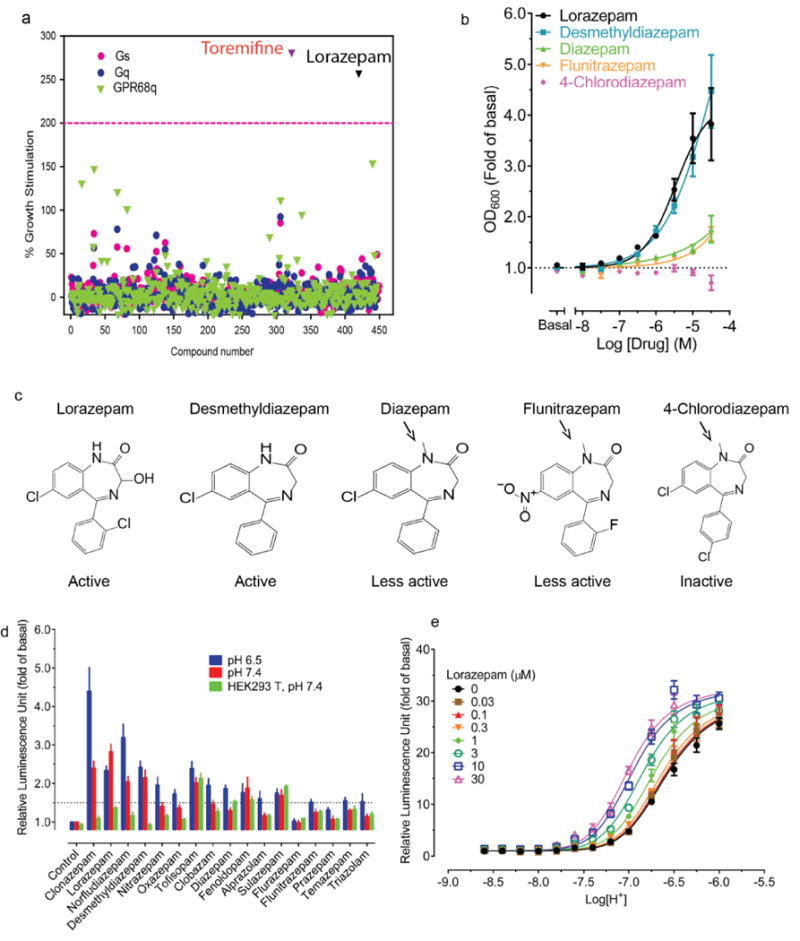
(a) A library of approved drugs (10 μM) screened at yeast expressing chimeric Gs, Gq, or GPR68 and chimeric Gq revealed lorazepam as a true and toremifine as a false positive. (b) Concentration-dependent stimulation of GPR68 Gq-yeast growth by lorazepam and analogues. (c) Structures of representative benzodiazepines (arrows denote methyl substituents with reduced activity at GPR68). (d) Lorazepam is a GPR68 positive allosteric modulator for the agonist proton. Data are mean ± SEM of normalized results (a, b, d, n=3) and concentration-response curves (b, d) were fit via a 4-parameter logistic function (see Methods).
Lorazepam–a benzodiazepine anxiolytic–activated GPR68 signaling, stimulating yeast growth by more than two-fold (Figure 1a). N-unsubstituted benzodiazepines were more efficacious than N-substituted benzodiazepines (Figure 1b–c, Supplementary Table 1) and activated the receptor at both pH 6.5 and 7.4 (Extended Data Fig 1l), with lorazepam most potently shifting the H+ concentration-response profile (Figure 1d and Extended Data Fig 1m–p). The pH dependence of lorazepam’s activity suggested that it functions as a PAM of GPR68; lorazepam had no activity at the related receptors GPR4 or GPR65 (Extended Data Fig 2a–b). When profiled against a panel of CNS targets, lorazepam had substantial activity only at the GABAA receptor, its therapeutic target (Extended Data Fig 3).
Modeling the GPR68-lorazepam complex
Little improvement in activity or selectivity was achieved by testing lorazepam analogs. This observation, and the drug’s potent GABA-A receptor activity, led us to seek specific, optimizable molecules from computational docking screens of multi-million molecule libraries (Figure 2).
Figure 2. Virtual screening workflow and predicted location of GPR68 allosteric site.
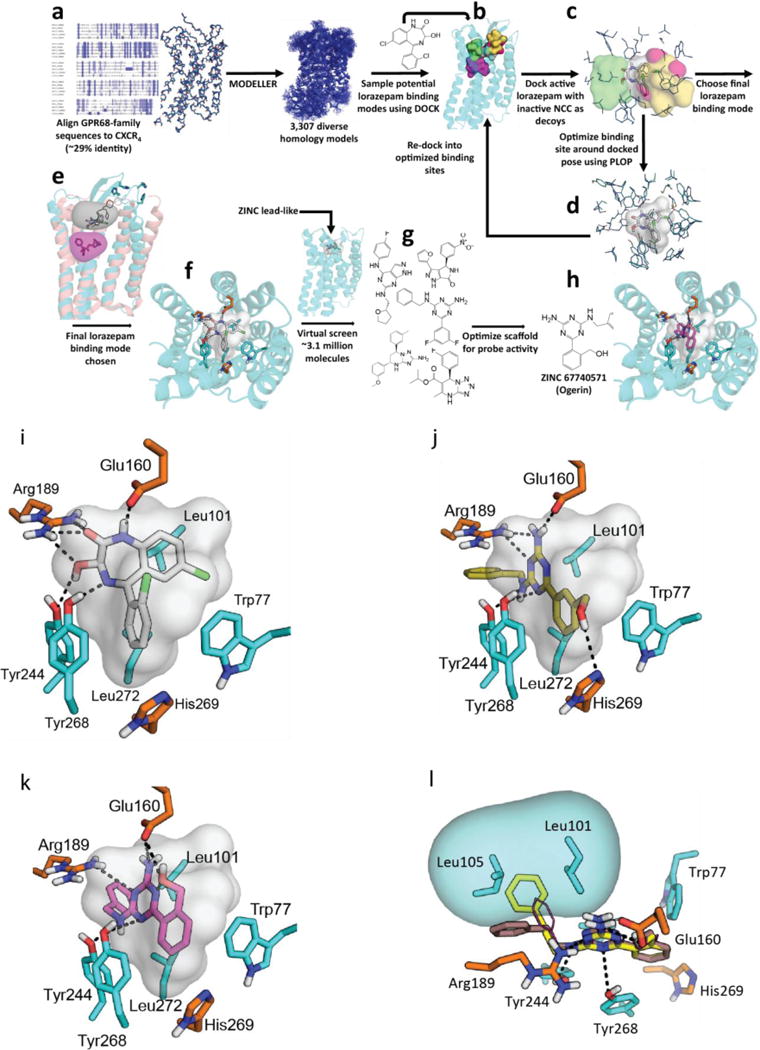
(a) Sequence alignment of GPR68, GPR4, GPR65, and GPR132 to CXCR4 (details in Extended Data Fig 2e). (b) Docking of lorazepam and NCC library to five distinct binding sites (details in Extended Data Fig 2f). (c) Models evaluated by their favorable ranking of lorazepam vs. decoy molecules. (d) Optimizing the most favorable lorazepam binding mode. (e) Optimized lorazepam orientation (gray stick) in GPR68 (cyan ribbon), and M2 muscarinic receptor (salmon ribbon, PDB 4MQT) with allosteric site (grey) and orthosteric site (QNB, magenta). (f) Lorazepam in its predicted orientation and interactions. (g) Virtual screen of ZINC subset (~3.1 million molecules) to identify predicted hits. (h) ZINC67740571 (magenta stick) in its predicted orientation and interactions in GPR68.
We generated 407 homology 3D models for GPR68 templated on the CXCR4 structure (29% sequence identity, Extended Data Fig 2f) and these were expanded by another 2,900 models using elastic network modeling, which sampled backbone and loop conformations. Against each of the 3,307 models we computationally docked the active benzodiazepines, the over 446 inactives from the NCC library, and 176 property-matched decoy molecules26. In each model, five candidate allosteric sites were docked against (Extended Data Fig 2g) based on the binding regions of aminergic GPCRs, the peptide and antagonist sites of CXCR4, and the muscarinic receptor allosteric site. Iterative cycles of modeling and optimization (Figure 2b–e) attempted to capture two aspects of ligand binding. First, the activity of the benzodiazepines as PAMs, and second the role of histidines 17, 84, 169 and 269, which are thought to interact with one another in the inactive state, and move apart on protonation at lower pH values7. This cycle converged to a stable lorazepam docking pose (Figure 2f), and to its ranking first among the 622 decoy molecules. This strategy resembles previous ligand-guided docking27–29, although here the binding site was unknown. In its docked geometry, lorazepam hydrogen bonds with Glu160, Arg189, Tyr244, and Tyr268, and forms non-polar contacts with Trp77, Leu101, Phe173, His269, and Leu272 (Figure 2f).
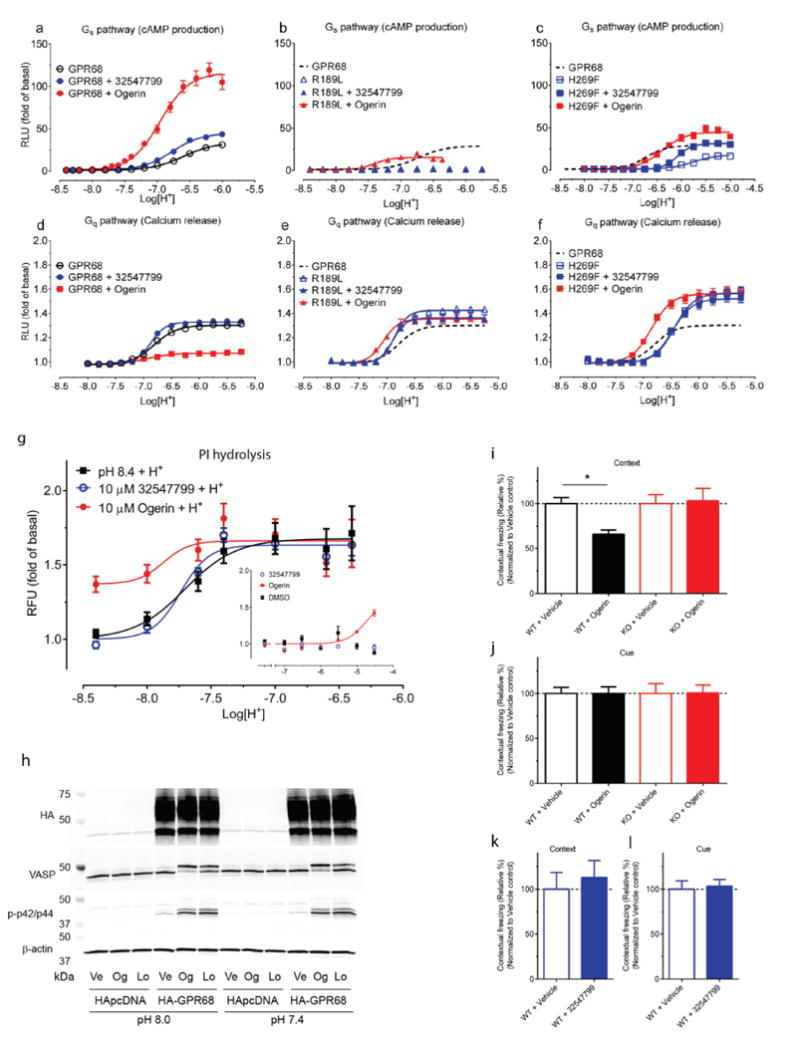
To test the modeled lorazepam site, we mutated residues lining it (Figure 2f, Extended Data Fig 2e–f), Glu160, Arg189, and His269, and determined their roles in proton-mediated cAMP production and calcium release (Extended Data Fig 4). The H269F mutant right-shifted proton-response curves in both assays7, while substitutions at Arg189 selectively abolished cAMP production. Different substitutions at Glu160 had varying effects at downstream signaling pathways – E160A left-shifted the proton-response curve and reduced cAMP production, but was inactive in calcium release, while the E160K and E160Q mutations had modest effects in both pathways (the mutants had little effect on expression, Extended Data Fig 4c). These substantial and differential effects on downstream coupling support a role for these residues in GPR68’s functions, and are consistent with the modeled binding site for lorazepam.
Seeking optimized PAMs, we computationally docked 3.1 million available lead-like molecules against the putative lorazepam site in GPR68. Overall, more than 3.3 trillion complexes were calculated and scored. From among the top 0.1% of the docking-ranked molecules, 17 were purchased for testing; along with their high docking ranks, these compounds recapitulated key interactions made by lorazepam in its docked model, were chemically diverse, and had high-scoring analogs (Supplementary Table 2).
Four of the docking hits increased cAMP production by about 1.5 fold over basal at pH 6.5 (Figure 3a). Though none were as active as lorazepam, two compounds, ZINC4929116 and ZINC32587282 had hundreds of available analogs. These were docked against the GPR68 model, and 25 were chosen for testing (Figure 3a, Supplementary Table 3). Thirteen had greater activity than lorazepam, and their pH-dependent potentiation activity clearly indicates allostery. Though dissimilar, lorazepam and ZINC67740571 dock to form many of the same interactions, with the addition of a new predicted hydrogen-bond to Glu160 from the hydroxyl of ZINC67740571 (Figure 2f, h, Extended Data Fig 2h).
Figure 3. Identification, characterization and optimization of GPR68 positive allosteric modulators.
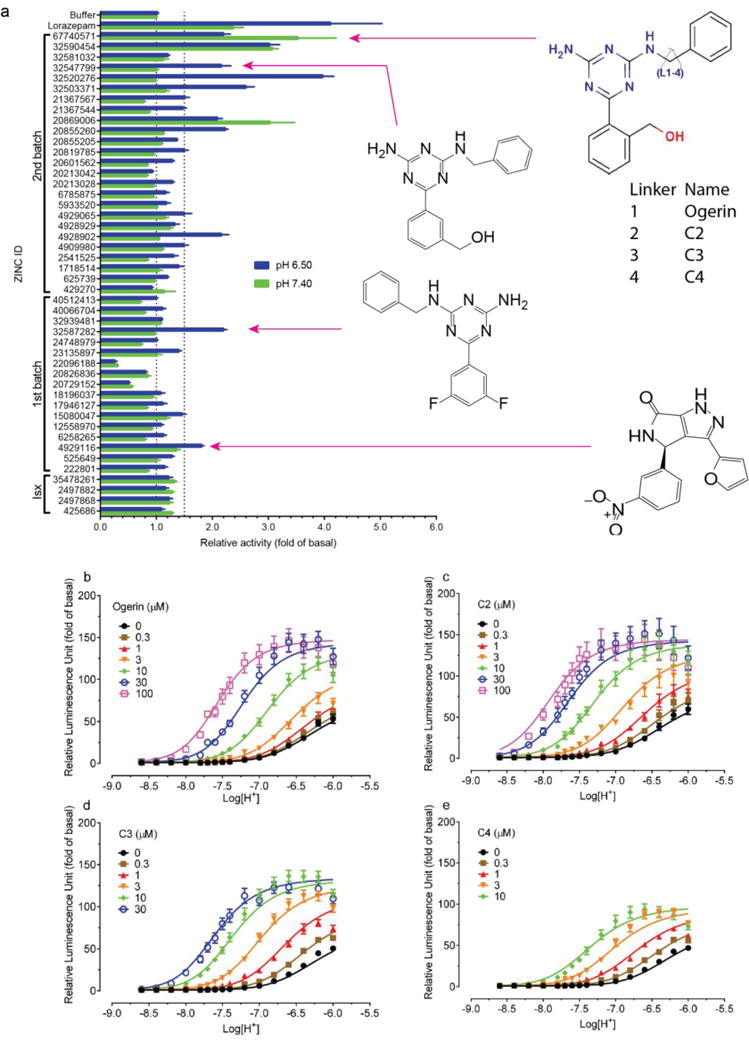
(a) Normalized results of GPR68-mediated cAMP production for selected compounds (ZINC database numbers) are shown; data represent mean ± SEM (n=4–34 measurements) at 10 μM for pH 7.40 and 6.50. Compounds were grouped into a 1st batch from the 1st round of virtual docking, and a 2nd batch from the 2nd round of docking. Compounds labeled Isx are isoxazole analogues. Lead compounds ZINC32587282, ZINC4929116, ZINC67740571 (ogerin), its isomer (ZINC32547799) and analogues (C2, C3 and C4) with different lengths of linkers, are highlighted. Concentration-response curves of normalized data (mean ± SEM; n=4) for ogerin (b), C2 (c), C3 (d), and C4 (e) are shown to illustrate the allosteric potentiation of proton and analyzed using a standard operational allosteric model. Allosteric parameters are summarized in Supplementary Table 8 and curve-fitting details are in Methods.
Ogerin as a selective GPR68 PAM
Ten selected compounds were studied further in functional assays. According to the standard allosteric operational model30, all were GPR68 PAMs, lacking intrinsic activity but increasing agonist potency (α factor) for cAMP production by 1.9 to 8.2-fold, and increasing efficacy (β factor) from 1.1 to 5.6-fold (Supplementary Table 5). It is this ability to shift concentration-response curves leftward and upward (Extended Data Fig 4b) that are the key characteristics of a PAM. ZINC67740571 had a much higher allosteric effect than lorazepam (Figure 3b vs Figure 1d, and Supplemental Table 8); we dubbed it “ogerin” (for OGR1 ligand).
Though ogerin and ZINC32547799 are close analogs (Figure 3a), each had distinct functional activities (Figure 4a, Extended Data Fig 4f–g) and docking poses (Figure 2f, Extended Data Fig 2h). Thus, the ortho-hydroxylmethyl group, which differentiates them, may play a key role in determining PAM activity, perhaps because of its ability to hydrogen-bond with Glu160, which the meta-positioned hydroxylmethyl in ZINC32547799 cannot reach. The structure-guided mutants H269F and R189L responded to ogerin and ZINC32547799 differently (Figure 4a, Extended Data Fig 4f–g, and Supplementary Table 6), supporting the modeled interactions with these residues. Strikingly, rather than activating, ogerin inhibited proton-mediated calcium release–a pathway-specific function rescued in R189L and H269F (Figure 4b, Extended Data Fig 4h–I, and Supplementary Table 7). Meanwhile, ZINC32547799 had little effect on calcium release. To determine if fast kinetics affect the difference between cAMP measurement (under equilibrium) and calcium release (non-equilibrium), we also conducted phosphatidylinositol hydrolysis assays under equilibrium. Ogerin slightly potentiated proton activity here (Extended Data Fig 4j–k) while ZINC32547799 did not. Finally, ogerin had minimal PAM activity at the related proton-sensing GPCRs, GPR4 and GPR65 (Extended Data Fig 2c–d). Ogerin appears to be a functionally selective GPR68 PAM for the agonist proton.
Figure 4. Ogerin modulates signaling and memory.
Ogerin and ZINC32547799 (10 μM) modulate proton-mediated cAMP production (a, n=4), calcium mobilization (d, n=5). Data in a–b are mean ± SEM. Ogerin but not its isomer (ZINC32547799) decreased contextual memory retrieval (c) in wild-type (n=7) but not GPR68 KO mice (n=8) (i, F(1.27)=4.71, p<0.05 for drug × genotype effect, p<0.05 for ogerin at wild-type mice, two-way ANOVA, Bonferroni’s post-hoc test); both had no effect on cued memory retrieval (d) in either wild-type (n=6) or KO mice (n=7). Results (c–d) were normalized to vehicle control; see also Extended Data Fig 8d–i.
If the ogerin/GPR68 model is relevant, we should be able to leverage it for optimization. We designed a virtual library of over 600 ogerin analogs and docked each into the GPR68 model (Extended Data Fig 2h–i). Thirteen high-scoring analogs were synthesized, and three were more active than ogerin (Supplementary Table 9 and Extended Data Fig 6), including 1st and 7th ranked compounds, the latter of which, C2, had the greatest allosteric effect, shifting proton response 3-fold further to the left than does ogerin, for an α-factor of 22 (Figure 3a–c, and Supplementary Table 8). C2 differs from ogerin by addition of a methylene to the benzylamine side chain, which places the phenyl ring deeper into a modeled apolar pocket (Extended Data Fig 2i). The addition of one or two further methylenes in compounds C3 and C4 (Figure 3a), conversely, reduced allostery (Supplementary Table 8 and Extended Data Fig 6f), consistent with reduced complementarity to the apolar pocket in the modeled complex.
To investigate ogerin specificity for GPR68 over unrelated targets, which might affect its usefulness as a biological probe, we first computationally screened ogerin and its analogs for off-targets using the SEA program31 against a panel of 2800 targets. These calculations revealed similarity between the GPR68 ligands and those of only three other GPCRs: the ghrelin and the adenosine A1 and A2A receptors. Subsequent physical profiling against 58 GPCRs, ion channels, and transporters (Extended Data Fig 3) revealed that ogerin had moderate affinity at two GPCRs, 5-HT2B and the A2A receptor (Extended Data Fig 5h–i), the latter consistent with the SEA prediction.
Intrigued by the association between the GPR68 PAMs and adenosine receptor antagonists, we computationally screened a library (http://www.tocris.com/dispprod.php?ItemId=5386#.U_s5ZMVdUrU) of 1,120 reagents and drugs against the GPR68 ligands, again using SEA. SLV320, a selective adenosine A1 antagonist32, was predicted to be a GPR68 PAM and confirmed by a physical screen of the full library (SLV320 αβ=2.8) (Extended Data Fig 7 and Supplementary Table 8), as was a second adenosine receptor antagonist, CGH2466 (αβ=2.9), and tracazolate (αβ=3.4), a GABA-ergic drug that also antagonizes adenosine receptors33. Although CGH2466 has the lowest apparent binding constant (KB) of any GPR68 PAM (48 nM), its allostery is much lower than that of ogerin; additionally, like SLV320 and tracazolate, CGH2466 is a potent phosphodieseterase inhibitor (Extended Data Fig. 7) and had minimal activity in the presence of Ro 20-1724. This previously unknown cross-talk among the GPR68, adenosine and GABA receptor ligands (Extended Data Fig. 7d), along with their activities at phosphodiesterases, should be considered when evaluating the pharmacology of what have been considered specific probes and drugs.
Ogerin as a GPR68 probe
Given its activity and specificity, we sought to explore ogerin’s downstream signaling and its in vivo activity. In GPR68-expressing HEK293 cells we found that both ogerin and lorazepam activate the PKA and MAP kinase pathways (Extended Data Fig 8a), mimicking the low pH-induced signaling observed with GPR68 receptors in human ASM cells19. The activation of GPR68 in smooth muscle cells by extracellular acidification is linked to multiple downstream pathways and biological responses18,19,22,34–37, which a selective allosteric modulator, such as ogerin, may help to disentangle.
To investigate effects in behavior associated with modulation of the hippocampus, where GPR68 is highly expressed17, we evaluated GPR68 KO and WT mice in a learning and memory test, fear conditioning, in which the hippocampus plays important roles (Extended Data Fig 8, 11). In WT mice, ogerin attenuated contextual-based fear memory without effects on cue-based memory (Figure 4c–d). The magnitude of these effects is comparable to those of compounds targeting other hippocampus-expressed GPCRs38,39, and larger effects are rarely observed without surgical lesion of the hippocampus40. Crucially, administration of ogerin had no effect on memory retrieval in GPR68 KO mice (Figure 4c–d), indicating that ogerin’s in vivo effects are GPR68-dependent. Furthermore, the less active ogerin isomer, ZINC32547799, had no measurable effect on learning and memory in wild-type mice (Figure 4c–d and Extended Data Fig 8d–h). Ogerin’s effects thus support a role for GPR68 in hippocampal-associated memory.
General applicability of the approach
To explore the broader usefulness of this approach, we sought ligands for GPR65, another understudied pH-sensing receptor, which shares 37% sequence identity to GPR68. We found that a recently reported GPR65 agonist BTB0908941 is an allosteric agonist of GPR65 (Figure 5d–e, and Extended Data Fig 10a). We used BTB09089 to anchor modeling of GPR65, generating 500 homology models templated on GPR68. The final docked GPR65-BTB09089 model resembles that of GPR68-ogerin, with several side-chain substitutions in the putative binding site (Figure 5a).
Figure 5. Discovery of GPR65 allosteric agonist and negative allosteric modulator.
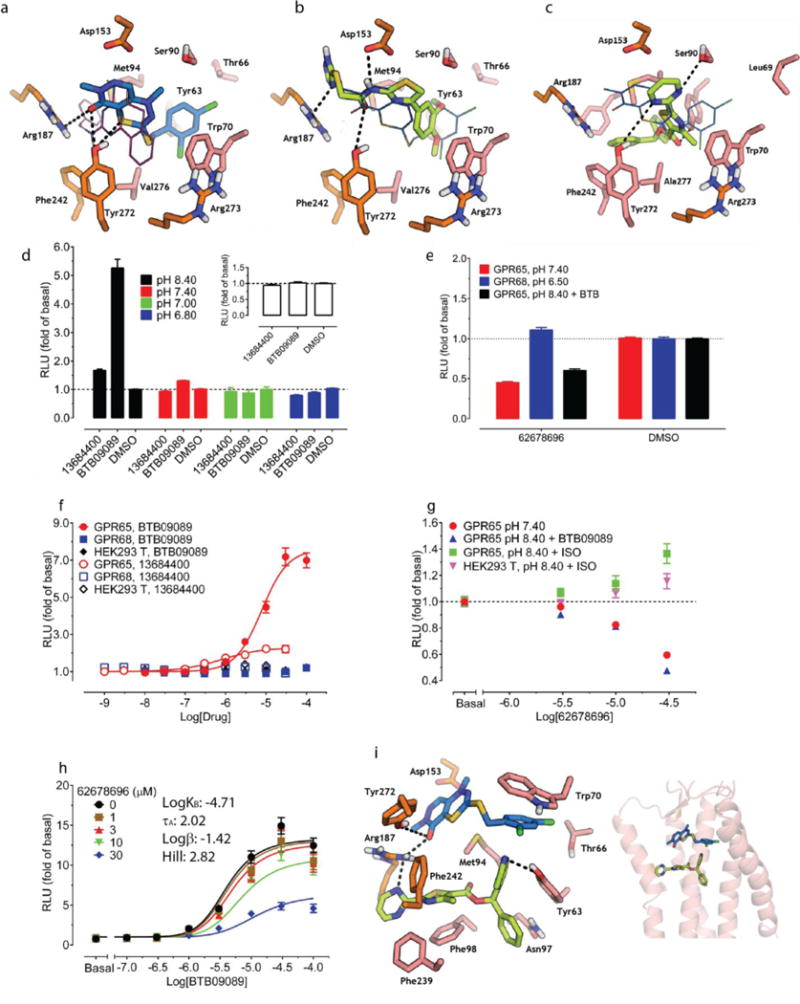
Predicted interactions of BTB09089 (a), ZINC13684400 (b), ZINC62678696 (c) at GPR65. Overlaid ogerin (thin magenta lines) (a) or BTB09089 (thin blue lines) docking poses at GPR68 or GPR65 resectively (b–c). ZINC13684400 (30 μM) displayed GPR65 allosteric agonist activity at pH 8.40 but not at lower pH or in control cells (d, n=32 measurements). (e) ZINC13684400 as a GPR65 agonist at pH 8.40 (n=3). (f) ZINC62678696 shifts BTB09089 curves downward at pH 8.40 (n=4). Normalized results (d–f) are mean ± SEM and curves were analyzed using a 4-prarmeter logistic function (e) or a standard operational allosteric model (f). (g) Predicted ternary complex between GPR656, ZINC62678696 and BTB09089, detailed interactions (left) and overall orientation in the GPR65 structure (right).
We docked the same 3.1 million compounds against the GPR65 model, purchasing 45 novel molecules for testing (Figure 5a–c, Supplementary Table 10). ZINC13684400 showed agonist activity of over 2 fold of basal at GPR65, with a potency of 500 nM, without measurable activity at control cells (Figure 5e, and Extended Data Fig 9). As with BTB09089, ZINC13684400 did not potentiate proton efficacy at GPR65 (Figure 5d), but acted as an allosteric agonist. To test the model, three residues modeled to interact with both BTB09089 and ZINC13684400, Arg187, Phe242, and Tyr272, were mutated, as was Asp153, which appears to only hydrogen-bond with ZINC13684400 (Figure 5b). R187L, F242A, Y272A reduced the activity of both compounds (Extended Data Fig 10f–g), while D153A had no effect on BTB09089 but much reduced the activity of ZINC13684400, consistent with the model. Several other docking hits inhibited GPR65 when the receptors were activated by protons or by BTB09089, including ZINC62678696 (Extended Data Fig 10b–d). Unexpectedly, ZINC62678696 does not compete with BTB09089, as predicted, but rather acts as a BTB09089 NAM (Figure 5f), suggesting that the two molecules can bind to GPR65 simultaneously (Figure 5g).
Discussion
A combined empirical and structure-based approach discovered potent PAMs at the understudied receptor GPR68, and an allosteric agonist and NAMs for the understudied GPR65. This supports the approach’s usefulness for illuminating the “dark matter” of the GPCR-ome–the 38% of non-olfactory GPCR targets whose ligands and function are understudied or unknown1. Whereas truly high-throughput screens are impractical for targets of unknown function, lower-throughput screens are often feasible. Although the hits from such a screen may be unsuitable as probes, they can anchor computational screens for more optimized compounds. Correspondingly, we would not ordinarily expect docking to succeed against models of a target that shares only 29% sequence identity with its nearest template. By calculating several thousand models, and insisting that the relevant ones are those that prioritize active over inactive molecules, functionally relevant models are prioritized. The new ligands that emerged are specific for the target and one is active in vivo, supporting their use as chemical probe for the function of GPR68.
Pharmacologically, the most unexpected observation was the activity of GPR68 in learning and memory. Previous studies in GPR68 KO mice revealed only modest phenotypic changes21,16,22, none in higher brain function, even though GPR68 is most highly expressed in the brain. Ogerin transiently and reversibly reduced contextual-based fear memory in wild-type but not GPR68 KO mice, consistent with on-target activity in vivo. In hindsight, this is perhaps only accessible to chemical modulators, which can have PAM activities. Inhibitory genetic perturbations, such as knockouts or knockdowns, though crucial to demonstrating on-target activity through chemical genetic epistasis, cannot on their own reveal such activation-based modulation.
Deorphanizing a receptor can also illuminate its off-target roles for known drugs. The observation that lorazepam and its primary metabolite, desmethyldiazepam, are GPR68 PAMs may clarify several of the idiosyncratic effects of this widely-used anxiolytic. Lorazepam, uniquely among benzodiazepines, can treat catatonia, an effect hypothesized to involve an unknown secondary target42. GPR68 may have a role in this efficacy, as both drug and metabolite reach micromolar concentrations in plasma during treatment43.
Certain caveats bear airing. The combination of empirical and computational screens will not work for all orphan receptors. GPCRs that are poorly expressed or non-functional in yeast or transfected cells will be problematic, and some orphans will simply not recognize any of the molecules screened in the small empirical libraries. Also, some orphans will bear too little similarity to templates of known structure to support accurate modeling. Even those that do work will demand cycles of testing and optimization, which was crucial for both GPR65 and GPR68.
These cautions should not obscure the key observations from this study–that combining empirical and structure-based screening led to a probe molecule that reveals some of the functions of GPR68. The finding that ogerin potentiates GPR68 activation and downstream MAP kinase pathways, and previous observations that the receptor mediates airway inflammation, enables campaigns for GPR68 PAMs that may regulate respiratory inflammatory responses. Uniquely as PAMs, these compounds would have fidelity to the natural spatial and temporal activation of GPR68. Correspondingly, the role of GPR68 in anxiety offers a new route to treating this condition, and related CNS disorders, an area in need of new therapeutic modalities44. Methodologically, this approach may have broad application to illuminating the function of the “dark matter” of the genome, that still large area of pharmacology where targets are known, but function is hidden.
Online Methods
Chemicals, reagents, and cells lines
Chemicals and reagents used in this study, if not specified otherwise, were purchased from commercial sources (Sigma, Tocris, Fisher Scientific, or specified in Supplementary Tables of chemical structures) or synthesized as outlined in the supplementary information. HEK293 and HEK293 T cells were from ATCC.
Homology Modeling
The alignment for the construction of the GPR68 models was generated using PROMALS3D, and homology models were built with MODELLER 9v845, using the crystal structure of the chemokine CXCR4 receptor (PDB ID: 3ODU) as the template (Extended Data Fig 2f). This alignment was also used to generate 500 models of GPR65 directly from the final GPR68 model. The initial alignment included both human and mouse sequences of GPR68, as well as those of its closest homolog, GPR4. These were aligned against the whole human C-X-C chemokine receptor family. The alignment was manually edited to: remove the N- and C-termini that extended past the template structure, remove the engineered T4 lysozyme, and create different alignments of the flexible and non-conserved second extracellular loop (the final result is given in the provided alignment, Extended Data Fig 2f). 407 models were built directly based on the CXCR4 crystal structure, using MODELER-9v845, while five more were built from each of 580 elastic network models (ENMs), produced by the program 3K-ENM46, for a total of 3,307 models built during each iterative round of model refinement. Models with constraints between pairs of extracellular His residues (His17-His169, His17-His269, His17-His84, His84-His169) to mimic the inactive state of the protein were generated by enforcing a distance constraint of 2.7 Å between the imidazole nitrogens, with a standard deviation of 0.1 Å. Confirmed actives and analogs using CXCR4-based model had neither agonist nor antagonist activity at CXCR4 receptors (Extended Data Fig 5j–k).
Model Evaluation
Prior to docking, the second extracellular loop (EL2), between residues 161–177, was removed from each GPR68 model. Models were ranked on the basis of prioritizing active benzodiazepines (lorazepam and desmethyldiazepam) over the rest of the inactive NCC library that was used in the yeast screen, as well as over property-matched decoys. In addition, the docked pose of lorazepam had to form a hydrogen bond from its N-H group to a polar side-chain in GPR68. Five different sites were sampled for possible lorazepam binding, based on the locations of the co-crystallized CXCR4 small molecule antagonist 1T1t (in PDB code: 3ODU), cyclic peptide CVX15 (in PDB code 3OE0), and the positions of the biogenic amines crystallized with the β2-adrenergic receptor (PDB code: 2RH1) and the dopamine D3 receptor (PDB code: 3PBL). The entire NCC library was docked to each of the five sub-sites for several rounds of iterative binding site refinement. In each round, the top-ranked models were examined for a binding pose that made hydrophobic and electrostatic interactions with the receptor, including the key N-H hydrogen bond. Residues within 6 Å of the lorazepam pose were minimized around the docked ligand with PLOP47. The NCC library was then re-docked into this optimized binding site for each model. This refinement continued for several cycles until the top-ranked models all converged to the same lorazepam pose. Once the final model was chosen, we built the EL2 back onto the receptor using MODELLER-9v845 and optimized 1,000 different EL2 conformations around the lorazepam pose with PLOP. Finally, we docked the NCC library back into these 1,000 different EL2-GPR68 structures and chose a final model that retained the previous pose and prioritized the active compounds over the inactives. The GPR65 model was generated similarly, using the pose of BTB09089 as the primary selection criterion, although in this case the EL2 was always present. To determine the ternary complex model of ZINC 62678696 and BTB09089, ZINC 62678696 was docked to the putative binding site in the GPR65 model with BTB09089 present. Then, both ligands were minimized with PLOP. Next, the side chains of the GPR65 binding pocket were allowed to relax, and, finally, BTB09089 and ZINC 62678696 were simultaneously minimized again with PLOP. Structural models (PDB files) of characteristic GPR68 modeled complexes (with ogerin or lorazepam) and GPR65 modeled complexes (with BTB09089 or BTB09089 and ZINC62678696) are shown in the Supplementary Data.
Virtual Screens
We used DOCK 3.6 to screen the ZINC database (Results). The flexible ligand sampling algorithm in DOCK 3.6 superimposes atoms of the docked molecule onto binding site matching spheres, which represent favorable positions for individual ligand atoms. Forty-five matching spheres were used, using the previous refinement round’s pose of lorazepam. The degree of ligand sampling is determined by the bin size, bin size overlap, and distance tolerance, set at 0.4 Å, 0.1 Å, and 1.5 Å, respectively, for both the matching spheres and the docked molecules. The complementarity of each ligand pose was scored as the sum of the receptor-ligand electrostatic and van der Waals’ interaction energies, and corrected for context-dependent ligand desolvation. Partial charges from the united-atom AMBER force field were used for all receptor atoms; ligand charges and initial solvation energies were calculated using AMSOL48,49 (http://comp.chem.umn.edu/amsol/). The best-scoring conformation of each docked molecule was then subjected to 100 steps of rigid-body minimization.
Selection of Potential Ligands for Testing
We docked the approximately 3.1 million commercially available molecules of the lead-like subset of the ZINC database to the final GPR68 and GPR65 models. The full hit list was automatically filtered to remove molecules that possess high-internal-energy, non-physical conformations, which are not well-modeled by our scoring function. The reported rankings reflect this filtering. From the top 0.1% (~3000 molecules) of the docked ranking list, 17 compounds were chosen for testing, based on complementarity to the binding site and presence of predicted electrostatic interactions with Glu160, Arg189, Tyr244, Tyr268, and His269, mimicking those predicted for lorazepam. For GPR65, compounds were chosen based on complementarity to the binding site and similarity to the predicted binding pose of BTB09089, modeled to interact with Asp153, Arg187, Tyr272, and by aromatic stacking with Trp70.
In silico lead profiling
To examine specificity and to discover other potential GPCR targets for the newly discovered GPR68 positive allosteric modulators (PAMs), we used the Similarity Ensemble Approach (SEA)31,50, which compares individual ligands, and sets of ligands, to the ligand sets for multiple targets; two targets are related, or a particular ligand is predicted to modulate a target, if the sets of ligands are related to one another. Here, the query set was all of the new GPR68 PAMs, which was screened against either the 2,512 ligand-target set with activity of 10 μM or better from the ChEMBL12 database51, or against the Tocris Mini library.
Receptor constructs and Yeast growth assays
24 human GPCR plasmids (GPR1, GPR4, GPR15, GPR31, GPR39, GPR41, GR43, GPR45, GPR55, GPR57, GPR58, GPR62, GPR65, GPR68, GPR83, GPR84, GPR87, GPR88, GPR123, GPR132, GPR133, GPR157, GPR161, ADCYAP1R1) were obtained from cdna.org, subcloned into the multiple cloning site of the yeast high copy number plasmid p426GPD52 and were confirmed by full-length sequencing (Eton Bioscience, Durham, NC). The yeast strains used were kindly provided by Mark Pausch (Merck) and have been previously described53 and used by us25,54 - MPY578t (Gi yeast), MPY578q5 (Gq yeast) and MPY578s5 (Gs yeast) express chimeric G proteins in which the last five amino acids of the yeast G-alpha protein are replaced with their mammalian Gi, Gq or Gs homologues, respectively. These strains contain the HIS3 gene under the control of the FUS1 promoter. GPCR transformants in yeast were selected and maintained on synthetic defined (SD) media lacking uracil (Clontech). GPR68q indicates the GPR68 paired with Gq-yeast; while GPR4s indicates GPR4 paired with Gs-yeast, and similarly for the other GPCRs. The yeast screening assays were carried out as described previously25. Assays were set up in 96-well flat-bottom clear assay plates that contained 50 μl of test compound at 40 μM (final concentration of 10 μM, in triplicate) diluted in SD-His-Ura medium (Clontech), 50 μl of 3-amino-1,2,4-triazole (3-AT) at 4× concentration diluted in SD-His-Ura medium (pH 5.4), and 100 μl of yeast cell suspension diluted in SD-His-Ura medium to a final OD600 of 0.02. Growth was at 30°C for 2 to 5 days. Before measurement of cell growth, cells were re-suspended by repeated gentle pipetting to ensure uniform suspension of cells. Cell growth was measured by absorbance at 600 nm in a microplate reader (POLARstar Omega, BMG Biotech). After culling of data from obviously contaminated wells, the OD600 values of each individual well were adjusted as follows: 100 × (OD600 of test well – OD600 of plate median value) to give % growth stimulation (positive values), or % growth inhibition (negative value) in the form of means ± SEM of three wells.
To measure and control constitutive activity or leaky HIS expression, each receptor-yeast combination was plated as above in the absence of ligand over a range of concentrations of 3-AT. Concentrations of 3-AT that showed moderate yeast growth (i.e., OD values of 0.2 to 0.6) after 2 days at 30°C were used in assays for drug screening. To measure concentration-dependent activity, various concentrations of cognate ligands diluted in SD-His-Ura medium were incubated with transformed yeast and appropriate concentrations of 3-AT for 2 days at 30°C.
Site-directed mutagenesis
The GPR68 plasmid was obtained from cdna.org. Mutation of E160A, E160K, E160Q, R189L, R189M, and H269F in the GPR68 and mutation of D153A, R187L, F242A, and Y272A in the GPR65 were introduced with Agilent’s QuikChange II site-directed mutagenesis kit and confirmed by sequencing. To tag the receptors for comparing receptor expression levels with immunoblotting, FLAG epitope tag was inserted at the C-terminus of the GPR68 wild-type and mutant receptors, also using the QuikChange II site-directed mutagenesis kit. Insertion was confirmed by sequencing.
Split-luciferase based cAMP reporter assays with proton receptors
GPR4, GPR65, and GPR68 plasmids were obtained from cdna.org. GPR68 mutations were made and confirmed as above. Receptor-mediated Gs activation was measured using a split-luciferase reporter assay (GloSensor cAMP assay, Promega). Briefly, HEK293-T cells were transiently co-transfected with receptor DNA and the GloSensor cAMP reporter plasmid (GloSensor 7A). Transfected cells were plated in poly-L-Lys coated 384-well white clear bottom cell culture plates in DMEM supplemented with 1% dialyzed FBS at a density of 15,000 cells per well in a total volume of 40 μl for a minimum of 6 hours. Before assays, culture medium was removed and cells were incubated with Luciferin (4 mM prepared in drug buffer, pH 8.4) for 90 min at 37°C. The drug buffer was made with 1× HBSS supplemented with 10 mM HEPES and 10 mM MES modified from19. TAPS was added to accommodate higher pH values for some assays; no difference was observed between different buffers under the same pH conditions. Cells plated at pH 8.4 for 6 hours generated the same H+ concentration-response curves as those plated at pH 7.4. To make individual pH solutions, the pH was adjusted with NaOH and measured at room temperature with a pH 211 Microprocessor pH meter (Hanna Instruments). To measure modulator activity under different pH conditions, modulator was mixed with pH solutions before adding to cells. To achieve the goal that drug solutions were delivered at the correct pH values, luciferin solution was removed from cell plates before addition of drug solutions at predetermined pH values. To improve solubility for some hydrophobic compounds, 1 mg/ml BSA was added to drug solutions, and it had no effect on H+ concentration-response curves. For Gs-protein activity (cAMP production), the cell plate was usually incubated at room temperature for 20 minutes before being counted in a luminescence counter. Results were analyzed using GraphPad Prism.
Allosteric operational model and data analysis
To estimate allosteric parameters, results were fitted to the allosteric operational model 30,55 as shown in the following equation:
Where
Response is the measured activity in the form of RLUs (Relative Luminescence Units) for measurement of cAMP production. If the results were normalized, the ‘Response’ is RLU in fold of basal (with buffer control as basal).
Emax is a system parameter, representing the maximal possible response of the system, and this value was normally constrained to the maximal reading of the corresponding experiment.
Basal is the baseline in the absence of test ligand, and is constrained to the baseline of the corresponding experiment. If results were normalized to fold of basal, the ‘Basal’ was usually 1.0.
[A] and [B] represent concentrations of the orthosteric and allosteric ligands, respectively. In the case of GPR68, A is proton.
KA and KB are the equilibrium dissociation constants (i.e., binding affinities) of the orthosteric agonist proton (A) and allosteric modulator (B), respectively. To facilitate curve-fitting with the model, KA is usually fixed to the binding affinity determined from traditional radioligand binding assays under the assumption that the experimentally derived binding affinity is not significantly different from the functional affinity under the condition for corresponding functional assay. Since proton binding affinity is not a measurable parameter in this assay system, the proton KA is therefore constrained to the corresponding proton EC50 value in the absence of the allosteric ligand, under the assumption that the proton potency is not significantly different from its binding affinity when the cAMP production assay is carried out. Since protons are present at relevant concentrations at physiological pH values, for a proton receptor KB is largely a fitting parameter without a clear physical meaning.
The term τA is the orthosteric agonist proton efficacy parameter. Since allosteric modulators in this study showed no agonist activity, the allosteric modulator efficacy τB is therefore 0 and not included in the function.
The term n is the slope factor linking receptor occupancy to response. Steep slopes in this study indicated high cooperativity between proton binding and receptor activation, probably reflecting the fact that the proton receptors operate within a narrow physiological pH range.
The allosteric parameter α defines the mutual effect between the orthosteric agonist A and the allosteric modulator B (α > 1 for increased affinity and α < 1 for reduced affinity); while β defines the allosteric effect on agonist efficacy (β > 1 for increased efficacy and β < 1 for reduced efficacy).
With KA, Basal, and Emax constrained to their corresponding values, the parameters KB, τA, α, β, and n are globally shared fitting parameters for a family of proton concentration-response curves in the absence and presence of increasing concentrations of a test allosteric modulator. With the above settings, most curves could be easily fitted to generate reasonable parameters. If Prism could not fit the curves, but generated ‘ambiguous fitting’ results, the α value was then manually constrained to an initial fitting value and systematically changed with small increments or decrements until the highest stable high affinity value (KB) was reached. For GPR65 and GPR68, KB represents the allosteric binding affinity in the absence of protons, which is unmeasurable and thus has little physical meaning. The value KB/α represents the binding affinity of an allosteric ligand in the presence of protons, which could be estimated experimentally. For convenience, we call KB/α the “Biochemical binding affinity, KbB” (Supplementary Table 8) for an allosteric ligand in the presence of an orthosteric agonist (in this case, H+).
Calcium mobilization assays
HEK293-T cells were transfected and plated into poly-L-Lys coated 384-well black clear bottom cell culture plates in DMEM supplemented with 1% dialyzed FBS, at a density of 15,000 cells in 40 μl per well for overnight. Before the assay, medium was removed and cells were loaded with Fluo-4 Direct calcium dye (Invitrogen, CA) for 60 min at 37 C in a 5% CO2 atmosphere. The calcium dye was prepared in drug buffer supplemented with 2.5 mM probenecid (pH 8.0). Proton solutions were made with 1× HBSS, 7 mM HEPES, 7 mM HEPPS, and 7 mM MES and pH was adjusted with NaOH. Drug additions and fluorescence intensity measurement were carried out in a FLIPRTETRA, which was programmed to add drug solutions to cells while recording fluorescence intensity. To measure proton concentration-responses, 10 μl of pH pre-determined solutions were added to each well (with 20 μl Calcium dye) while fluorescence intensity was recorded during and after addition for 4 minutes (one reading per second). The addition procedure was configured in such a way (30 μl per second at height of 10 μl above cells) that local proton concentrations for cells were essentially the same as in the pH working solutions at the moment of addition. Fluorescence intensities reached peak values within 30 seconds after drug addition. To determine the effects of modulators on proton responses, the protocol was modified slightly. In brief, cells were loaded with calcium dye as above, but only at 15 μl per well. The FLIPRTETRA was programmed to first add 5 μl of 4× test compound (final concentration of 10 μM before addition of 10 μl of pH solutions) prepared with the same drug buffer at pH 8.0 (buffer alone served as a control). After a total of 10 min of reading and incubation, 10 μl of the pH solutions were added and the fluorescence intensity was recorded exactly the same way as above. Results (fluorescence intensity in fold of basal) were exported and analyzed in GraphPad Prism. For calcium mobilization assays with 5-HT2B receptors, HEK293 cells stably expressing human 5-HT2B receptors were used instead of transiently-transfected cells. Cells were set up and tested in the same way as above, with 5-HT serving as an agonist control (3 pM – 30 μM) and with 1 nM 5-HT being used in the second addition to determine the antagonist activity of ogerin.
PI hydrolysis assay
HEK293-T cells were transfected for 24 hours and plated in poly-L-Lys coated 96-well black clear bottom cell culture plates in DMEM supplemented with 10% FBS, at a density of 60,000 cells in 100 μl per well. After 5 hours, cells were washed with inositol-free DMEM once and labeled with 3H-inositol (1 μCi/well, PerkinElmer) in inositol-free DMEM supplemented with 5% dialyzed FBS overnight. On the assay day, labeling medium was removed and cells were washed once with assay buffer (1× HBSS, 10 mM HEPES, 10 mM MES, 20 mM LiCl, pH 8.4). To measure drug concentration responses, then cells were then incubated with drug solutions at pH 8.4 for 20 min. To measure proton concentration responses, the assay buffer was pre-adjusted to desired pH values and supplemented with 20 mM LiCl. To measure the effect of ogerin or its isomer ZINC 32547799 on proton concentration –response curves, pH solutions were supplemented with 20 mM LiCl and 10 μM ogerin or ZINC 32547799. The premixed drug solutions were added to cells for 20 min. At the end of incubation, drug solutions were removed and 40 μl per well of 50 mM ice-cold formic acid was added. After incubation at 4°C for 30 min, the acid extracts were transferred to polyethylene terephthalate 96-well sample plates (#1450-401, Perkin Elmer) and mixed with 75 μl (200 μg) YSi RNA binding beads (RPNQ0013, Perkin Elmer). The plate was sealed and further incubated at 4°C for 30 min before being counted on a TriLux MicroBeta counter. Results (cpm/well) were analyzed using Graphpad Prism.
Functional assays with A2A and CXCR4 receptors
Functional assays with A2A adenosine and CXCR4 chemokine receptors were carried out using a slightly different protocol from that previously described for Gs (above) and Gi receptors56. Specifically, HEK293-T cells were transfected and plated using regular DMEM supplemented with 1% dialyzed FBS. Before assays, culture medium was removed, and cells were incubated with 20 μl drug solution (prepared in drug buffer 20 mM HEPES, 1× HBSS, pH 7.4) for 15 min at room temperature. To measure agonist activity, 5 μl of 5× luciferin solution (4 mM final concentration) for A2A (Gs coupled GPCRs) or a mixture of luciferin and isoproterenol at a final concentration of 200 nM for CXCR4 (Gi coupled GPCRs) was added and cells were incubated for another 20 min. To measure antagonist activity, test compound was added first for 10 min before a reference agonist at a final of EC80 concentration for another 10 min, and then followed by addition of luciferin for A2A or a mixture of luciferin and isoproterenol for CXCR4 as above. Luminescence was measured in a luminescence counter. Results were analyzed in GraphPad Prism.
Radioligand binding assays
Radioligand binding assays with selected CNS targets were carried out as described56,57 and as detailed in the PDSP protocol book available online (http://pdsp.med.unc.edu/pdspw/binding.php). Briefly, receptor membrane preparations were made from either animal brain tissues, or stable cell lines, or transiently transfected HEK293-T cells. Receptor expression levels and radioligand binding affinities were determined with saturation binding assays. Competition binding assays were performed with membrane aliquots and a fixed concentration of radioligand in 96-well plates in a final volume of 125 μl. Reactions were incubated in the dark and at room temperature (22°C), and terminated by vacuum filtration onto 96-well formatted GF/B filters. Radioactivity on the filters was counted in a beta counter. Results were analyzed in GraphPad Prism.
Anti-HA immunoblots
HEK293 cells were transfected with either pcDNA3 vector containing an HA cassette within the multiple cloning site, or pcDNA3HA-GPR68 encoding human GPR68 with an N-terminal HA tag. Stable lines were generated by selection with 250 μg/ml G418, with >90% of cells expressing HA after 2 weeks as assessed by immunocytochemistry (not shown). Cells were plated into 12 well plates, grown to confluence, and media switched to Hams-F12 media, with pH adjusted to pH 8.0 or 7.4, for 1 hour. Cells were then stimulated with either vehicle, 50 μM ogerin, or 50 μM lorazepam for 10 min. Lysates were harvested and subjected to immunoblotting, with blots probed using primary antibodies against HA (Sigma cat# H3663), total vasodilator-stimulated phosphoprotein (VASP, BD Biosciences, cat# 610448), p-p42/p44 (Cell Signaling, cat# 5726S), and β-actin (Sigma, cat# A1978), and secondary antibodies (Licor, cat# 926-32213 and 926-32210) conjugated with infrared fluorophores as per58.
Anti-Flag immunoblots
HEK293-T cells were transiently transfected in 10-cm dishes with Flag-tagged GPR68 wild-type and mutant receptors. Untransfected HEK293-T cells served as a negative control. After 48 hours, cells were collected, lysed, and sonicated to shear chromatin before being subjected to immunoblotting. Blots were probed with monoclonal anti-Flag M2-peroxidase antibody (Sigma, cat# A8952). Bands were quantified and normalized to GPR68 wild-type receptor (fold) for graphing.
Data analysis and reporting
Other than in vivo studies (below), no statistical analysis was applied to yeast- or cell-based screening assays. Sample size (number of assays for each compound or receptor) was predetermined to be in triplicate or quadruplicate for primary screening assays at a single concentration. Some samples were repeated more than the others in the primary screening assays and the number of measurements were specified as a range in corresponding figure legends. For concentration-response assays, the sample size (number of assays for each compound at selected receptors) was also predetermined to be tested for a minimum of 3 assays, each in triplicate or quadruplicate. Samples or receptors were tested not randomly but in an alphabetic order or numeric order according to their coded names for easy organization. For each batch of assays, a control assay with isoproterenol and proton concentration-responses were included. If potency values for either isoproterenol or proton was >0.5 log unit away from established averages, assays with the batch of transfected cells were excluded. For structure-activity relationship (SAR) studies, only the assays in which all related compounds were tested side by side were included. None of the functional assays were blinded to investigators.
Generation of GPR68 knockout (KO) mice
To generate GPR68 KO mice, a probe specific for the human GPR68 transcript was generated by PCR amplification of a 450 bp segment of the coding sequence of the final exon of GPR68 using total placental RNA. The probe was used to identify a clone from a 129 mouse genomic lambda library. The genomic insert was subcloned and a restriction map generated using a panel of enzymes. The targeting construct for the GPR68 locus consists of a PGK-1 promoter driven neomycin resistance cassette flanked by two arms of homology with the mouse GPR68 locus. The longer arm of homology was generated using a 7,266 bp PstI fragment extending from the last intron to the beginning of the last exon. This exon contains the entire coding sequence of the GPR68 gene. The 1335 bp shorter arm was generated by PCR amplification and extends from the downstream end of the long arm into the 3′ untranslated region of the gene. Homologous recombination of the targeting construct with the GPR68 locus inserts the neomycin resistance cassette into codon 78 of the gene, thereby disrupting expression. Correctly targeted cell lines were identified by Southern blot analysis using a probe consisting of a 1496 bp PstI fragment immediately upstream of the long arm. This probe recognizes a 14,290 bp EcoRV fragment in the endogenous locus and a 7,855 bp fragment in the targeted locus. Genotyping was carried out by PCR with 3 primers. The common (5′GCA GAG GAA GCC CAC GCT GAT GTA3′) and endogenous (5′TAA ACG GTA GCT GTG ATT ATT CAA3′) primers generate a 516 bp PCR product from the endogenous locus, while the common and targeted (5′AAA TGC CTG CTC TTT ACT GAA GG3′) primers generate a 465 bp product from the targeted locus. The chimeras were bred to C57BL/6J mice and pups carrying the mutant allele identified. After ten successive crosses of heterozygous animals to C57BL/6J mice, heterozygous mice were intercrossed and a congenic GPR68−/− and C57BL/6J breeding colony established. The GPR68 KO mice were profiled in multiple behavioral tests as described below in detail and results are summarized in Extended Data Fig 11 and Supplementary Tables 11 and 12.
In vivo behavioral profiles of GPR68 KO mice
Mice were maintained and handled according to the Guide for the Care and Use of Laboratory Animals approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill. The goal of this study was to determine whether targeted deletion of GPR68 alters behavioral function in mice.
Timeline for behavioral tests
| Age of mice | Tests |
| 6–7 weeks | Elevated plus maze test for anxiety-like behavior. |
| 7–8 weeks | Activity in an open field. Accelerating rotarod (2 tests, 48 hours apart). |
| 8–9 weeks | Three-chamber social approach test. Activity in an open field (re-test). |
| 9–10 weeks | Marble-burying assay. |
| 10–11 weeks | Acoustic startle test. Buried food test for olfactory ability. |
| 11–12 weeks | Visual cue test in the Morris water maze. |
| 12–14 weeks | Hidden platform test for spatial learning. |
| 14–16 weeks | Reversal learning in the Morris water maze. |
| 16–17 weeks | Second acoustic startle test. Hotplate test for thermal sensitivity. |
Summary of results
Mice with deletion of GPR68 had normal performance in most of the behavioral tests. No effects of genotype were observed for body weights, activity and anxiety-like behavior in an elevated plus maze or an open field, motor coordination, sociability, prepulse inhibition of acoustic startle responses, or acquisition in the water maze. However, both male and female GPR68 knockout mice had small, significant decreases in acoustic startle responses, suggesting a reduced responsivity to environmental stimuli. Male GPR68 knockout mice also showed significant decreases in marble burying, a test for anxiety-like phenotypes. Overall, the findings indicate that GPR68 might play a role in specific domains of behavior.
Elevated plus maze
This test is used to assess anxiety–like behavior in rodents. The procedure is based on a natural tendency of mice to actively explore a new environment, versus a fear of being in an open area. In the present study, mice were given one five-minute trial on the plus maze, which had two walled arms (the closed arms, 20 cm in height) and two open arms. The maze was elevated 50 cm from the floor, and the arms were 30 cm long. Animals were placed on the center section (8 cm × 8 cm), and allowed to freely explore the maze. Measures were taken of time on, and number of entries into, the open and closed arms. All of the experimental groups showed a strong preference for the closed arms, in comparison to the open arms, of the elevated plus maze. As shown in Supplementary Table 11, there were no significant differences between the wild type and GPR68 KO mice for percent time or percent entries on the open arms, or for total entries during the task.
Activity in an open field
Exploratory activity in a novel environment was assessed in an open field chamber (41 cm × 41 cm × 30 cm) crossed by a grid of photobeams (VersaMax system, AccuScan Instruments). Counts were taken of the number of photobeams broken during the trial in five-minute intervals, with separate measures for ambulation (total distance traveled) and rearing movements. Time spent in the center region of the open field was measured as an index of anxiety-like behavior. Unfortunately, an equipment malfunction led to the loss of data for 8 mice during the first activity test, conducted when mice were 7–8 weeks in age. Therefore, a second activity test was given, when mice were 8–9 weeks in age. As depicted in Extended Data Fig 11a and 11b, there were no significant differences between the WT and GPR68 KO mice for distance traveled, or for rearing or center time (data not shown), during the second activity test. A significant sex × time interaction was found for the distance measure [F(11,385) = 2.68, p = 0.0025], reflecting higher levels of activity in the female groups at the beginning of the session.
Accelerating rotarod test
Subjects were tested for motor coordination and learning on an accelerating rotarod (Ugo Basile, Stoelting Co., Wood Dale, IL). For the first test session, animals were given three trials, with 45 seconds between each trial. Two additional trials were given 48 hours later. Rpm (revolutions per minute) was set at an initial value of 3, with a progressive increase to a maximum of 30 rpm. across five minutes (the maximum trial length). Measures were taken for latency to fall from the top of the rotating barrel. As shown in Extended Data Fig 11c and 11d, deletion of GPR68 did not lead to deficits in motor coordination on the rotarod. In fact, during the first three acquisition trials, there was a non-significant trend for enhanced performance in the male knockout group [repeated measures ANOVA, genotype × sex interaction, F(1,35) = 3.58, p = 0.0668].
Marble-burying assay
This procedure is used to evaluate anxiety-like behavior and repetitive responses. Mice were tested in a Plexiglas cage located in a sound-attenuating chamber with ceiling light and fan. The cage contained 5 cm of corncob bedding, with 20 black glass marbles (14 mm diameter) arranged in an equidistant 5 × 4 grid on top of the bedding. Animals were given access to the marbles for 30 min. Measures were taken of the number of buried marbles (two thirds of the marble covered by the bedding). A two-way ANOVA indicated a significant genotype × sex interaction [F(1,35) = 7.37, p = 0.0102] (Supplementary Table 11). Post-hoc comparisons revealed that the male GPR68 KO mice buried significantly fewer marbles than both male WT mice and female KO mice in this task.
Buried food test for olfactory function
Several days before the olfactory test, an unfamiliar food (Froot Loops, Kellogg Co., Battle Creek, MI) was placed overnight in the home cages of the mice. Observations of consumption were taken to ensure that the novel food was palatable. Sixteen to twenty hours before the test, all food was removed from the home cage. On the day of the test, each mouse was placed in a large, clean tub cage (46 cm L × 23.5 cm W × 20 cm H), containing paper chip bedding (3 cm deep), and allowed to explore for five minutes. The animal was removed from the cage, and one Froot Loop was buried in the cage bedding. The animal was then returned to the cage and given fifteen minutes to locate the buried food. Measures were taken of latency to find the food reward. As shown in Supplementary Table 11, there were no significant differences between the groups in latency to find the buried food.
Hotplate test for thermal sensitivity
Individual mice were placed in a tall plastic cylinder located on a hotplate, with a surface heated to 55°C (IITC Life Science, Inc., Woodland Hills, CA). Reactions to the heated surface, including hindpaw lick, vocalization, or jumping, led to immediate removal from the hotplate. Measures were taken of latency to respond. The maximum test length was 30 sec, to avoid paw damage. A two-way ANOVA indicated a significant main effect of sex [F(1,1) = 8.83, p = 0.0053], and genotype × sex interaction [F(1,35) = 4.3, p = 0.0455] (Supplementary Table 11). Post-hoc comparisons revealed that the male GPR68 KO mice had significantly lower latencies to respond than female KO mice.
Acoustic startle method
The acoustic startle test can be used to assess auditory function and sensorimotor gating. The test is based on the measurement of the reflexive whole-body flinch, or startle response, that follows exposure to a sudden noise. Mice can be evaluated for levels of startle magnitude and prepulse inhibition, which occurs when a weak prestimulus leads to a reduced startle in response to a subsequent louder noise. For this study, animals were tested with a San Diego Instruments SR-Lab system. Briefly, mice were placed in a small Plexiglas cylinder within a larger, sound-attenuating chamber. The cylinder was seated upon a piezoelectric transducer, which allowed vibrations to be quantified and displayed on a computer. The chamber included a house light, fan, and a loudspeaker for the acoustic stimuli. Background sound levels (70 dB) and calibration of the acoustic stimuli were confirmed with a digital sound level meter (San Diego Instruments). Each session consisted of 42 trials, that began with a five-minute habituation period. There were 7 different types of trials: the no-stimulus trials, trials with the acoustic startle stimulus (40 msec; 120 dB) alone, and trials in which a prepulse stimulus (20 msec; either 74, 78, 82, 86, or 90 dB) occurred 100 ms before the onset of the startle stimulus. Measures were taken of the startle amplitude for each trial across a 65-msec sampling window, and an overall analysis was performed for each subject’s data for levels of prepulse inhibition at each prepulse sound level (calculated as 100 − [(response amplitude for prepulse stimulus and startle stimulus together/response amplitude for startle stimulus alone) × 100].
Results from acoustic startle test
The GPR68 KO mice had decreased startle responses following presentation of acoustic stimuli, in comparison to the WT mice (Extended Data Fig 11e, f). A repeated measures ANOVA, conducted on startle response amplitudes, indicated significant main effects of genotype [F(1,35)=7.22, p=0.011] and sex [F(1,35)=16.61, p=0.0003], and a genotype × decibel level interaction [F(6,210)=5.77, p<0.0001]. Separate comparisons confirmed that both male and female KO mice showed significant reductions in startle responses [genotype × decibel level interaction, males, F(6,84)=2.57, p=0.0245; and females, F(6,126)=3.48, p=0.0032]. The decreased startle responses and overt sex differences were not associated with changes in prepulse inhibition (Extended Data Fig 11g, h). The significant main effects of genotype on startle were no longer evident during a second acoustic startle test, conducted when mice were 16–17 weeks in age.
Morris water maze - visible platform test
The Morris water maze task was used to assess spatial learning and visual function in the mice. The water maze consisted of a large circular pool (diameter = 122 cm) partially filled with water (45 cm deep, 24–26° C), located in a room with numerous visual cues. Mice were first tested using a visible platform. In this case, each animal was given four trials per day, across two days, to swim to an escape platform cued by a patterned cylinder extending above the surface of the water. For each trial, the mouse was placed in the pool at one of four possible locations (randomly ordered), and then given 60 seconds to find the visible platform. If the mouse found the platform, the trial ended, and the animal was allowed to remain 10 seconds on the platform before the next trial began. If the platform was not found, the mouse was placed on the platform for 10 seconds, and then given the next trial. Measures were taken of latency to find the platform via an automated tracking system (Noldus Ethovision). As shown in Supplementary Table 12, all groups of mice demonstrated a high degree of proficiency in the visual cue task.
Acquisition and reversal learning in the hidden platform test (Extended Data Fig 11i–l)
Three days following the visual cue task, mice were tested for their ability to find a submerged, hidden escape platform (diameter = 12 cm). As in the procedure for visual cue learning, each animal was given four trials per day, with one minute per trial, to swim to the hidden platform. The criterion for learning was an average latency of 15 seconds or less to locate the platform on one day. Mice were tested until the criterion was reached, with a maximum of nine days of testing. When criterion was reached, mice were given a one-minute probe trial in the pool with the platform removed. In this case, selective quadrant search was evaluated by measuring number of crosses over the location where the platform (the target) had been placed during training, and the corresponding areas in the other three quadrants. Following the acquisition phase, mice were tested for reversal learning, using the same procedure as described above. In this phase, the hidden platform was located in a different quadrant in the pool, diagonal to its previous location. As before, measures were taken of latency to find the platform. On the day that the criterion for learning was met, the platform was removed from the pool, and the group was given a probe trial to evaluate reversal learning.
For the above behavioral profiling studies, subjects were 21 wild type (WT) mice (9 males and 12 females) and 18 GPR68 knockout (KO) mice (7 males and 11 females), on a C57BL/6 background. Sample sizes were not stistically predetermined. Testing began when animals were 6–7 weeks of age. For each procedure, measures were taken by an observer blind to mouse genotype (WT or KO) and no animals were excluded from analysis. Data were analyzed using one-way or repeated measures Analysis of Variance (ANOVA). Fisher’s protected least-significant difference (PLSD) tests were used for comparing group means only when a significant F value was determined. Within-group comparisons were conducted to determine side preference in the social behavior tests. For all comparisons, significance was pre-set at p < 0.05.
Effect of ogerin and its analogue ZINC32547799 on learning and memory
Contextual and cue-dependent learning and memory were evaluated using a Near-Infrared Video Fear Conditioning system (MED Associates, St. Albans, VT). Test chambers (29 × 25 × 25 cm) had transparent walls and metal rod floors, and were enclosed in sound-attenuating boxes. The conditioned fear procedure had 3 phases: training, a test for contextual learning, and a test for cue-dependent learning. Before each phase, mice were moved to a holding room adjacent to the test room and acclimated for at least 30 min. In the 8-min training phase, mice receive 3 pairings of a 30-sec, 90 dB, 5 kHz tone (the conditioned stimulus, CS) and a 2-sec, 0.6 mA foot shock (the unconditioned stimulus, US), in which the shock was presented during the last 2 sec of the tone. Context-dependent learning was evaluated 24 hours following the training phase. Mice were placed back into the original test chamber, and levels of freezing (immobility) were determined across a 5-min session, without the presence of the CS or US. 48 hours following the training phase, mice were evaluated for associative learning to the auditory cue (the CS) in a final 6-min session. The conditioning chambers were modified using a Plexiglas insert to change the wall and floor surface, and a novel odor (vanilla flavoring) was added to the sound-attenuating box. Baseline behavior was scored for 2 minutes, and then three 30-sec CS tones were presented across a 4 min period. Levels of freezing were automatically measured by the image tracking software (Med Associates, St Albans, VT). Freezing was defined as no movement (below the movement threshold) for 0.5 sec. To evaluate the effect of drug, strain-matched group of animals were given ogerin (10 mg/kg in 10% Tween 80 or saline) 30 min before the training.
For the learning and memory studies, sample sizes (number of animals) was not predetermined by a statistical method and minimum of 6 animals were used in each group (exact number of animals was specified in figure legends). Animals were assigned to groups randomly and experiments were not blinded to investigators. No animals were excluded from analysis. Statistical analyses were performed after first assessing the normality of distributions of data sets. Comparisons between groups were made using unpaired t-tests. Welch’s corrections were utilized when variances between groups were unequal. Comparisons between groups during conditioning, contextual and cued memory tests were assessed using two-way ANOVA with p-value < 0.05 being considered significant.
Extended Data
Extended Data Figure 1. Validation and confirmation of GPCR activation assays in yeast (a–k) and HEK293-T cells (l–o).
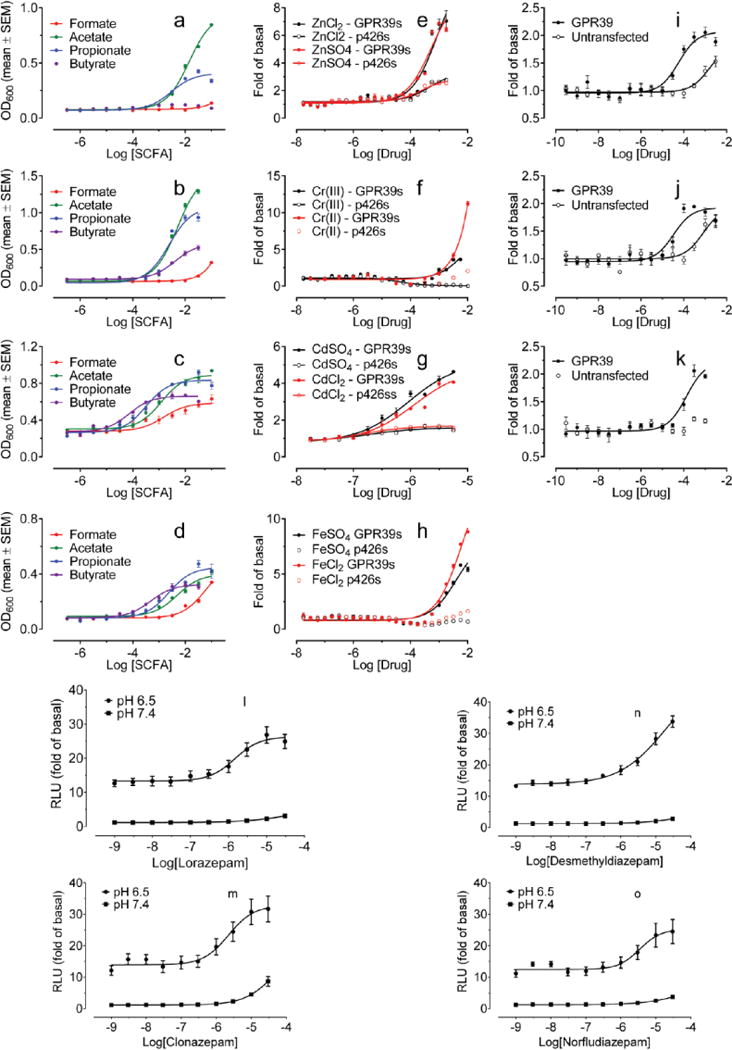
Concentration-dependent growth of GPR43-expressing Gs yeast (a), GPR43-expressing Gq yeast (b), GPR41-expressing Gs yeast (c), and GPR41-expressing Gq yeast (d) in response to various short-chain fatty acids (SCFAs). Concentration-dependent growth of GPR39-expressing Gs yeast (GPR39s) in response to zinc ions (e), chromium ions (f), cadmium ions (g), and iron ions (h). Concentration-dependent cAMP responses of GPR39-expressing HEK293-T cells to ZnCl2 (i), ZnSO4 (j), or CdSO4 (k) as measured by luciferase cAMP reporter assay. (l) N-unsubstituted benzodiazepines (lorazepam, clonazepam, desmethyldiazepam, norfludiazepam; 10 μM) stimulated cAMP production in a GPR68- and pH-dependent manner; data are mean ± SEM (n=3–66 measurements). Concentration-response curves of N-unsubstituted benzodiazepines lorazepam (m), desmethyldiazepam (n), clonazepam (o), and norfludiazepam (p) at pH 6.50 or 7.40 in GPR68-transfected HEK293-T cells (structures in Supplementary Table 1). Normalized results represent mean ± SEM (n = 3) and curves were analyzed in GraphPad Prism using the built-in 4 parameter logistic function.
Extended Data Figure 2. Lorazepam and ogerin have minimal GPR4 or GPR65 activity.
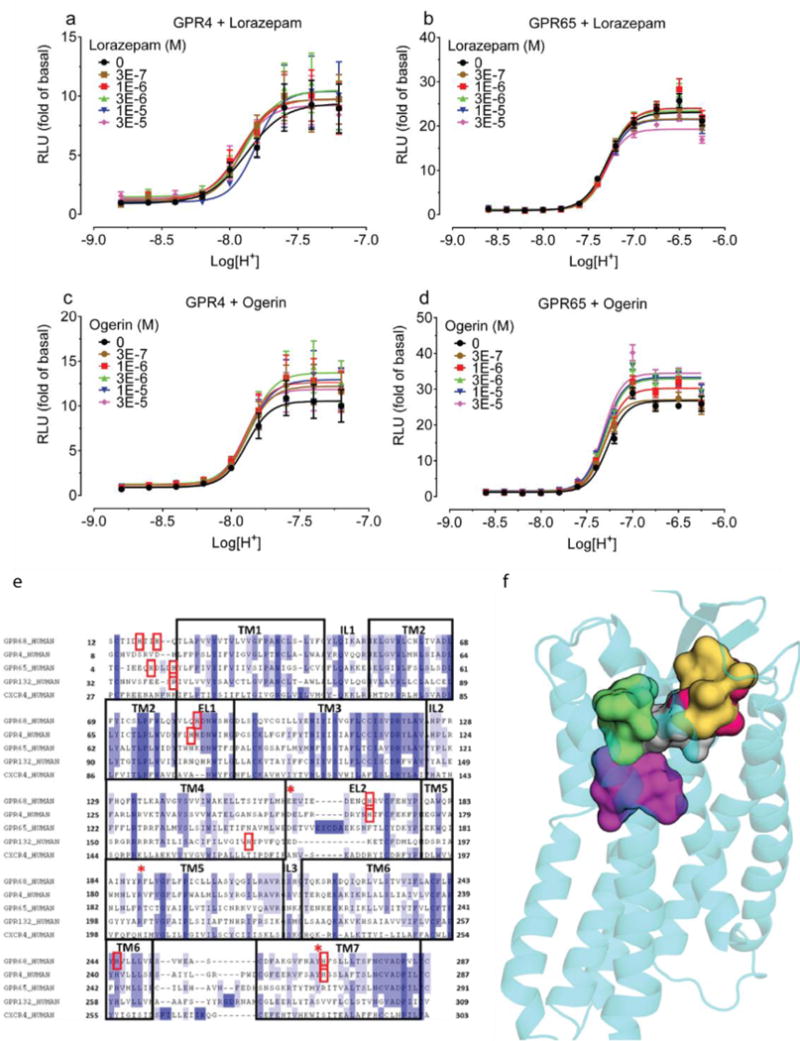
Effect of lorazepam (a, b) or ogerin (c, d) on GPR4 (a, c) or GPR65 (b, d); data represent normalized mean ± SEM (n = 3). Sequencing alignment proton-sensing receptor and docking poses for ogerin and its analogues. (e) GPR68 snake plot showing extracellular loops and transmembrane domains (upper portion); important residues are highlighted. Glu160, Arg189, and His269 were mutated in this study. (f) Sequence alignment of GPR4, GPR65, and GPR68 to CXCR4 (PDB 3ODU) (PROMALS-3D) was manually refined to reduce gaps and to position conserved residues (TM for transmembrane regions; IL for intracellular and EL for extracellular loop). Conserved residues highlighted in blue by degree of conservation while red boxes indicate residues important for receptor function. Red stars (*) indicate residues mutated in this study. (g) Sampling different regions for lorazepam binding modes in GPR68. Yellow and grey surfaces contour the binding site of 1T1t and CVX15 in CXCR4 crystal structures (PDB 3ODU, 3OE0, respectively) while green and red surfaces sample the entire binding pocket. The magenta surface represents the canonical orthosteric biogenic amine site. (h) ZINC32547799 in its predicted orientation and interactions with GPR68. (i) Optimization of ogerin (magenta, thin lines) to C2 (brown, structure in Fig 3a) by insertion of a single methylene is predicted to improve packing in the aryl pocket of the ogerin site. Adding a second methylene, thus creating a propyl linker in C3 (yellow, structure in Fig 3a), is predicted to disrupt the packing and thus to reduce the allosteric effect.
Extended Data Fig 3. Heat map of off-target activities of lead compounds at potential CNS drug targets.

Radioligand binding assays were carried out by the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH PDSP) as described previously56,57 (on-line protocols available at http://pdsp.med.unc.edu/pdspw/binding.php). Values represent mean binding affinities (pKi, n=2–4). Affinities lower than a pKi of 5, or less than 50% inhibition at 10 μM, are shown as a minimum of 5 on the pKi scale. The hERG inhibition activity was tested in a hERG functional assay as previously published59. ND for not determined; BZP for benzodiazepine receptor; DAT for dopamine transporter; NAT for norepinephrine transporter; SERT for serotonin transporter; DOR for delta (δ) opioid receptor; MOR for mu (μ) opioid receptor; KOR for kappa (κ) opioid receptor; PBR for peripheral benzodiazepine binding site; AMPA for aminomethylphosphonic acid receptor; KA for kainate acid receptor; NMDA for N-methyl-D-aspartate receptor; hERG for human Ether-a-go-go-related Gene (potassium channel Kv11.1).
Extended Data Figure 4. Confirmation of modeling results via mutagenesis.
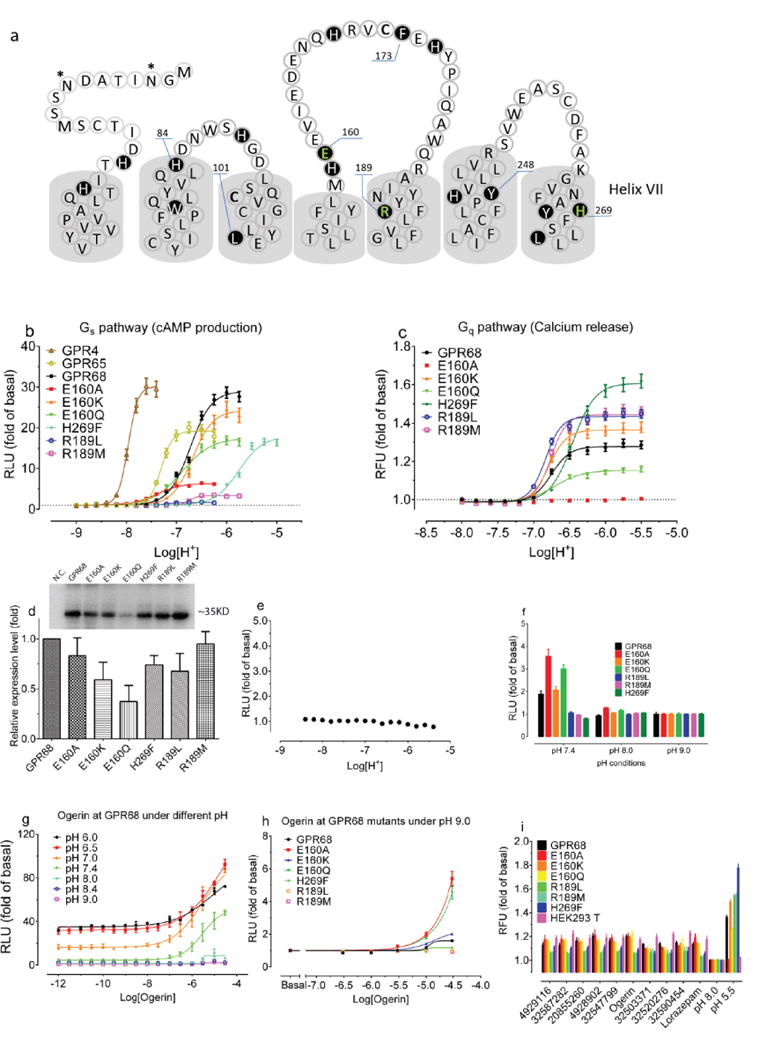
Protons showed agonist activity at GPR68 wild-type and mutant receptors in cAMP production (a) and calcium release (b); parameters are in Supplementary Table 4. (c) Relative GPR68 wild-type and mutant receptor expression levels determined by anti-Flag immunoblotting (n = 3). (d) Proton-mediated cAMP production in untransfected cells (n = 16). (e) Calcium release by lorazepam and selected ZINC compounds (10 μM at pH 8.00, n = 6 – 22 measurements). Effect of Ogerin and ZINC32547799 (10 μM) on proton-mediated cAMP production (f and g, n=4), calcium release (h and I, n=3), and PI hydrolysis (j, n=3) at GPR68 wild-type or mutant transfected HEK293 T cells. (k, n=3) Ogerin and ZINC32547799 mediated PI hydrolysis at pH 8.4 at GPR68 transfected GPR68 HEK293 T cells. Normalized results represent mean ± SEM and curves were analyzed using a 4-parameter logistic function.
Extended Data Figure 5. Control experiments for signaling and pharmacology.
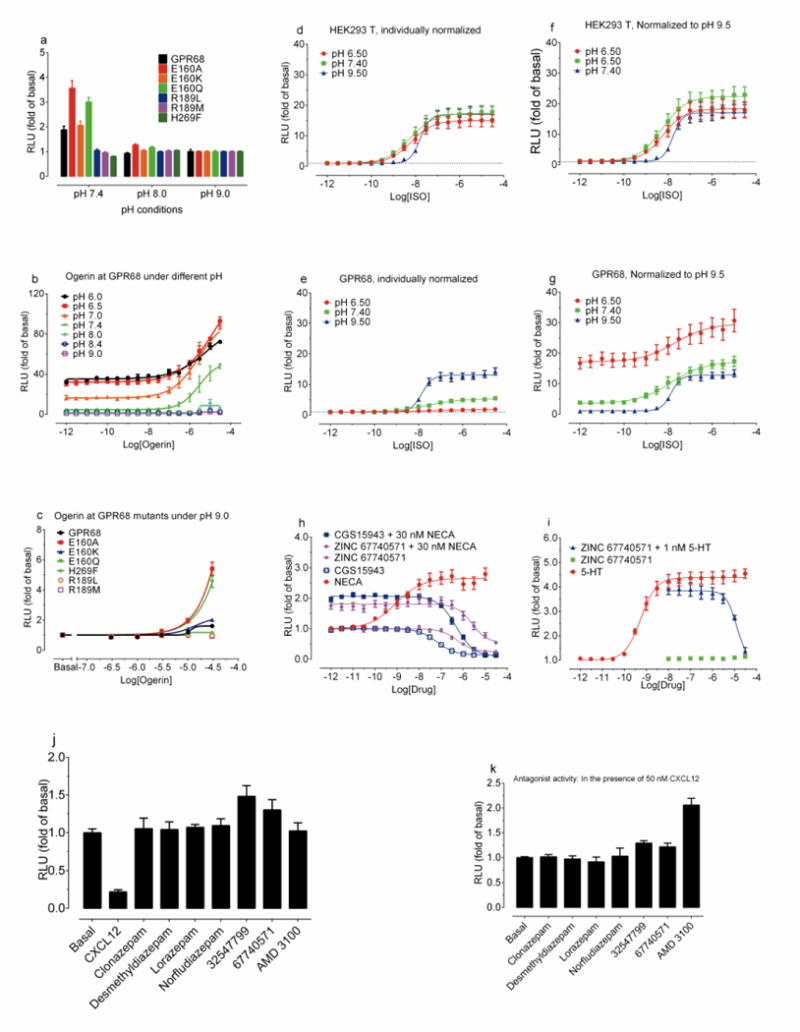
(a) Basal cAMP production of GPR68 wild-type and mutant receptors (mean ± SEM, n = 24 – 46 measurements). (b) pH-dependent activity of ogerin at GPR68 wild-type (mean ± SEM, n = 3). (c) Ogerin concentration-responses at GPR68 wild-type and mutant receptors at pH 9.0 (mean ± SEM, n = 3), under which cAMP reporter assay was not affected (d–f). Proton modulated ISO-mediated Gs-activation via β2-adrenergic receptors in untransfected (d, f) and GPR68-transfected cells (e, g). Normalized results (basal at pH 9.5 for d and e; or corresponding buffer control for f and g) represent mean ± SEM (n = 6). Inverse agonist and antagonist activity (Ki of 220 nM) of ogerin at A2A (cAMP production, h) and weak antagonist activity (Ki of 736 nM) at 5-HT2B receptors (calcium mobilization, i). 5′-N-Ethylcarboxamidoadenosine (NECA) and 2-Chloro-N6-cyclopentyladenosine (CCPA) served as agonist controls, while CGS15943 is an inverse agonist control for A2A receptors. Normalized results represent mean ± SEM (n=3). Curves were analyzed in GraphPad Prism with the built-in 4-parameter logistic function. Lead compounds (10 μM) showed no agonist (j) or antagonist (k) activity at CXCR4 receptors (cAMP production) with CXCL12 as an agonist control (1 or 3 μM) or AMD 3100 (10 μM) as an antagonist control. Results represent mean ± SD (n=2).
Extended Data Figure 6. Primary screening and comparison of allosteric parameters of 13 ogerin analogues at GPR68.
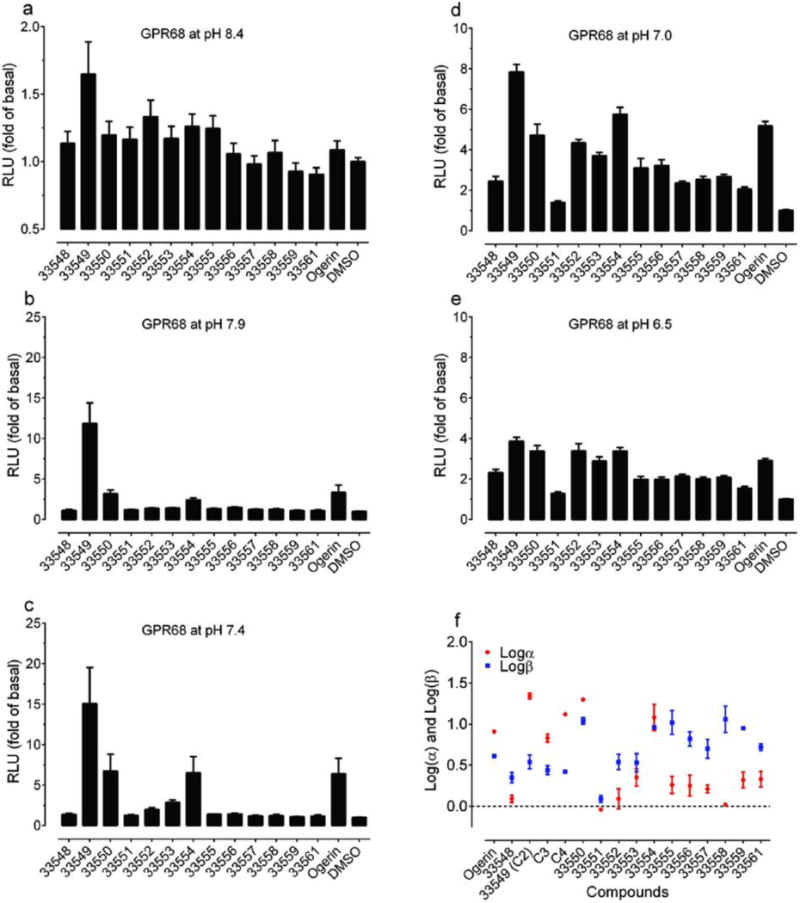
The 13 ogerin analogues (structures in Supplementary Table 9) identified from docking a virtual library of over 600 ogerin derivatives were synthesized (Supplementary Information). Production of cAMP was measured in transiently transfected HEK293-T cells at 10 μM and 5 different pH conditions, (a) pH 8.4; (b) pH 7.9; (c) pH 7.4; (d) pH 7.0; and (e) pH 6.5, to reveal any pH-dependent potentiation activity. Normalized results represent mean ± SEM (n = 8 – 16 measurements). (f) Graphic comparison of the allosteric parameters Logα and Logβ. Proton concentration-responses were carried out in the absence and presence of increasing concentrations of ogerin and its analogues, results were analyzed using a standard allosteric operational model to obtain allosteric parameters. Values represent mean ± SEM (n≥3; See details in Supplementary Table 8).
Extended Data Figure 7. Characterization of potent GPR68 PAMs.
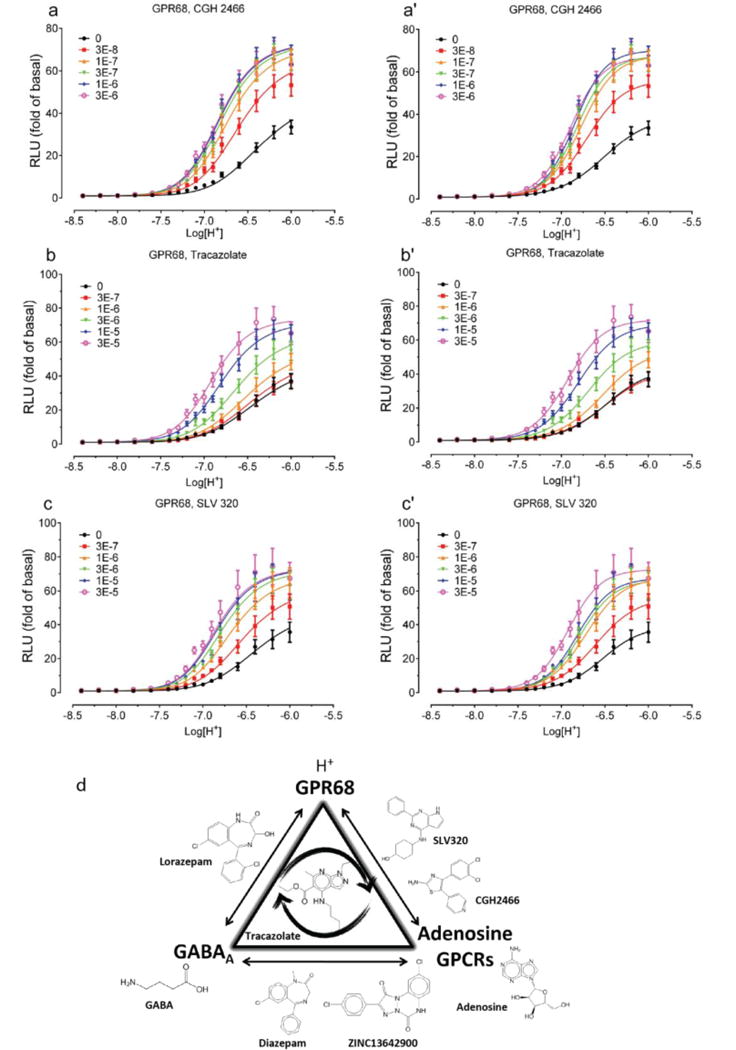
Concentration-response curves of H+ in the absence and presence of increasing concentrations of CGH2466 (a and a’), Tracazolate (b and b’), SLV320 (c and c’) at GPR68–expressing cells. Normalized results (mean ± SEM, n = 8 for CGH2466; n = 5 for Tracazolate; n = 5 for SLV320) were analyzed using a 4-parameter logistic function (right column, a’, b’, c’) and the standard allosteric operational model (left column, a, b, c). Allosteric parameters are summarized in Supplementary Table 8. For each pair of fittings, the proton potency value (pEC50) from the agonist concentration-response curve (right panel) in the absence of testing compound was used as the pKA for the allosteric operational model (left panel). (d) Schematic showing the shared pharmacology among GABAA, adenosine GPCRs, and GPR68 ligands. Molecules along each edge of the triangle have been shown to have activity at both targets, while tracazolate, in the middle, shows activity at all three.
Extended Data Figure 8. GPR68 mouse biology.
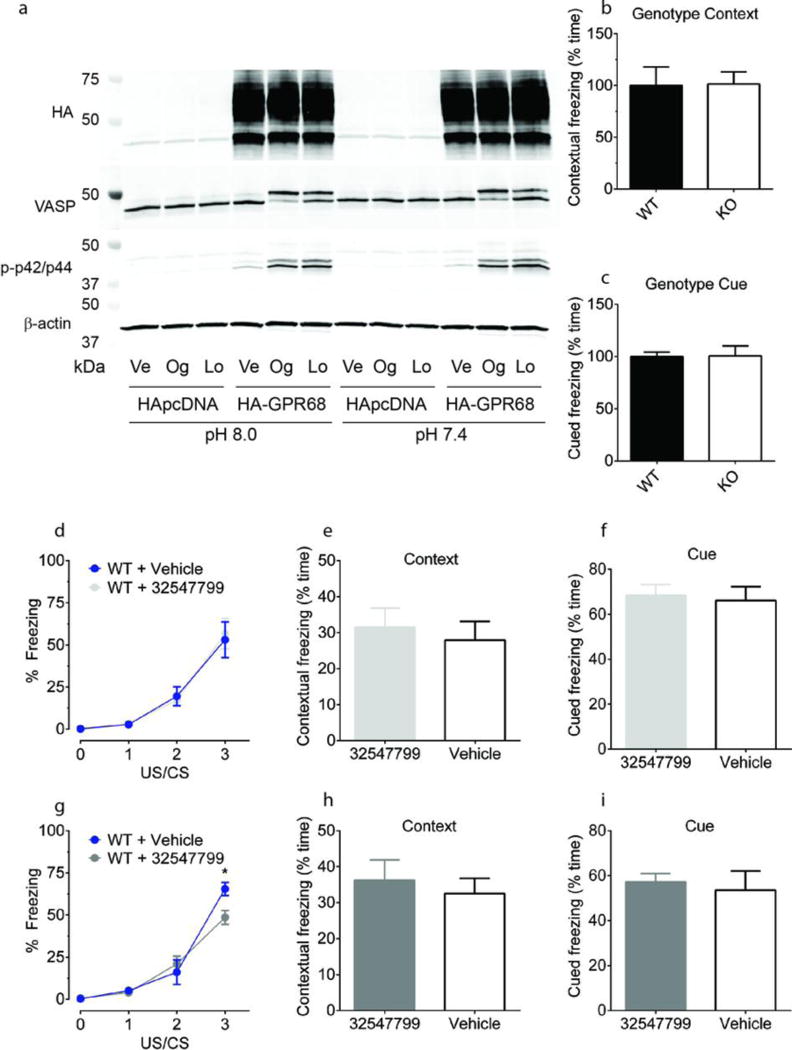
(a) Ogerin (Og) and lorazepam (Lo) activate PKA and p42/p44 MAP kinase in HEK293 cells stably expressing HA-GPR68 but not HApcDNA; vehicle (Ve). GPR68 KO (n = 7) mice exhibited no differences in contextual memory retrieval (b) or cued memory retrieval (c) as compared to wild-type mice (n = 8). At 10 mg/kg, the ogerin isomer ZINC32547799 had no effect on learning (d) or contextual and cue memory (e, f) in wild-type mice (vehicle, n = 6; drug, n = 7). At 30 mg/kg, ZINC32547799 enhanced wild-type learning (g, drug × time interaction, F(3.39) = 3.58, p = 0.022; drug alone F(1.39) = 1.19, p = 0.295; Bonferroni post-hoc test revealed a significant effect (p < 0.05) at the third US/CS training point, two-way ANOVA), but had no effect at contextual and cue memory (h, i) (vehicle, n = 7; drug n = 8). Normalized contextual memory retrieval (d) and cued memory retrieval (f) are presented in Fig 4c and 4d.
Extended Data Figure 9. Screening of ZINC compounds predicted to be active at GPR65 based on BTB09089 docking poses.
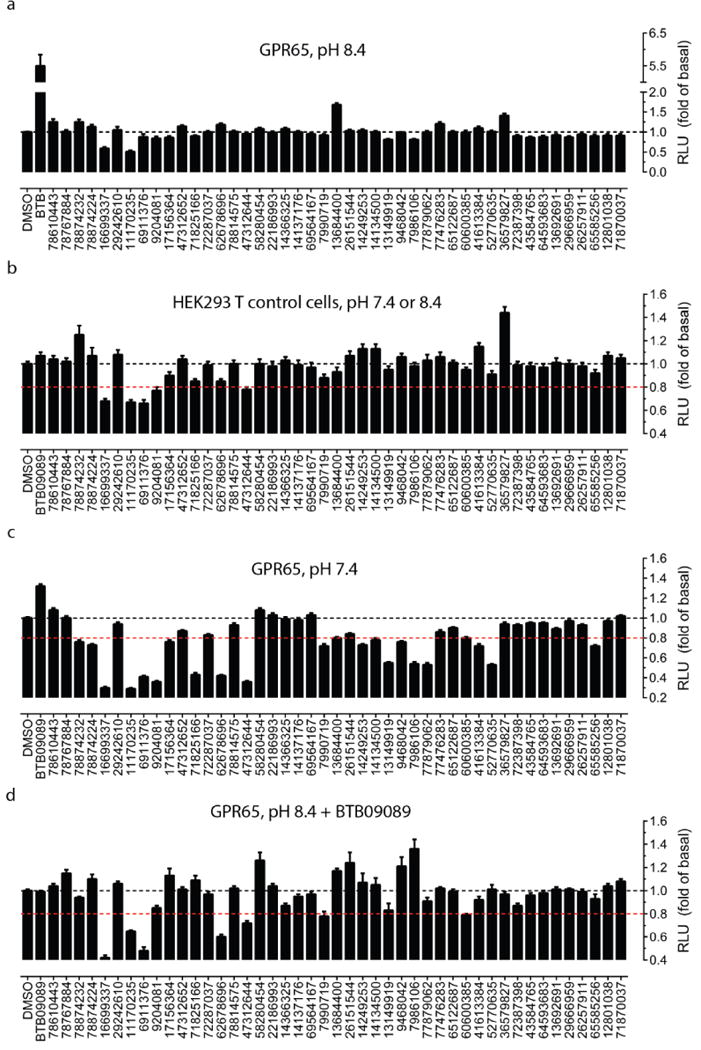
Primary screening with ZINC compounds (30 μM) for agonist activity at GPR65 when receptors were kept inactive at pH 8.40 (a); at control HEK293 T cells for nonspecific activity (b); at GPR65 when receptors were activated at pH 7.40 for modulator or antagonist activity (c); at GPR65 when receptors were activated by BTB09089 (30 μM) at pH 8.40 for modulator or antagonist activity (d). Normalized results represent mean ± SM from a minimum of three assays (each in minimum of triplicate and a total of ≥ 16 measurements). The red dashed line in panel b – d indicates the 20% inhibition line - an arbitrary cut-off line.
Extended Data Figure 10. Characterization of GPR65 allosteric modulators at wild-type and mutant receptors.
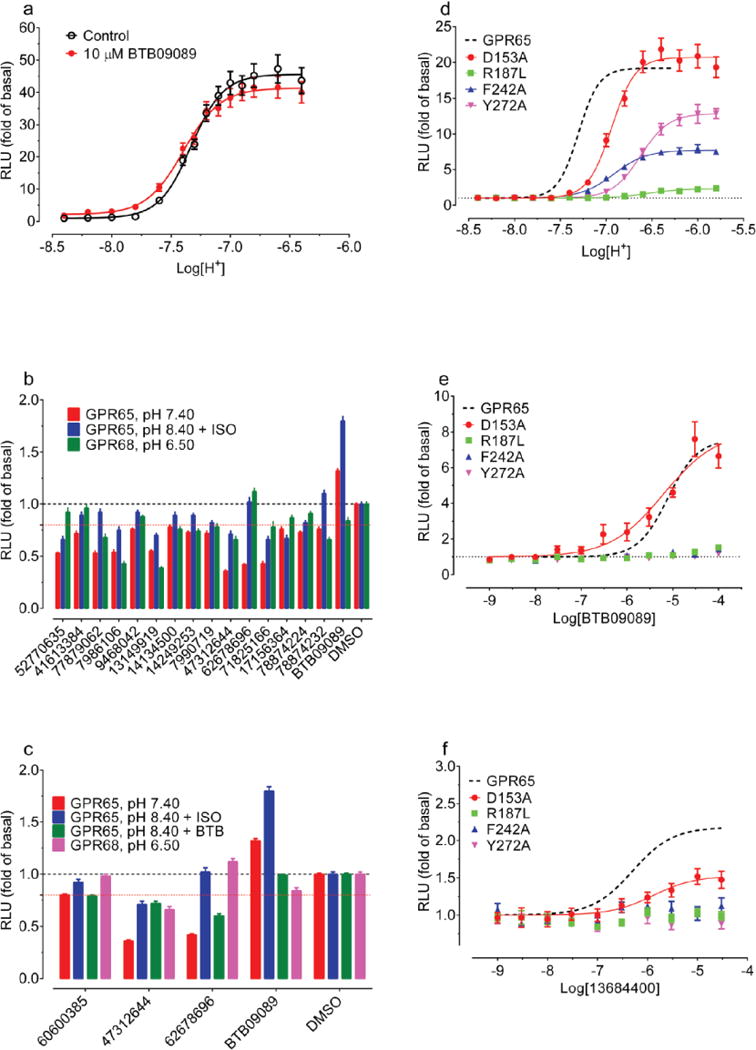
(a) BTB09089 showed weak agonist activity, but failed to potentiate proton activity at GPR65 (n = 8). (b) Selected compounds from Extended Data Figure 9b–c were tested for GPR65 specific inhibition (n = 16 – 56 measurements). Several compounds (such as ZINC41613384, ZINC9468042, and ZINC62678696) showed GPR65-specific inhibition. (c) Selected compounds from Extended Data Figure 9b and 9d were tested for antagonist activity against BTB09089-activated signal at GPR65 (n = 16 – 64 measurements). ZINC62678696 showed GPR65 specific inhibition when it was activated by either proton or BTB09089. (d) ZINC62678696 inhibited GPR65 activity. (e) Proton concentration-responses, (f) BTB09089 concentration-responses, and (g) ZINC13684400 concentration-responses at GPR65 mutant receptors. Normalized results represent mean ± SEM (n ≥3) and curves were analyzed in GraphPad Prism with a standard 4-parameter logistic function. Corresponding curves of proton at GPR65 wild-type receptors (from Extended Data Figure 4a) and BTB09089 and ZINC13684400 (from Figure 5e) are also included (dashed lines) for comparison. Pharmacological parameters are listed in Supplementary Table 13.
Extended Data Figure 11. In vivo behavioral profiling of GPR68 knock-out mice.
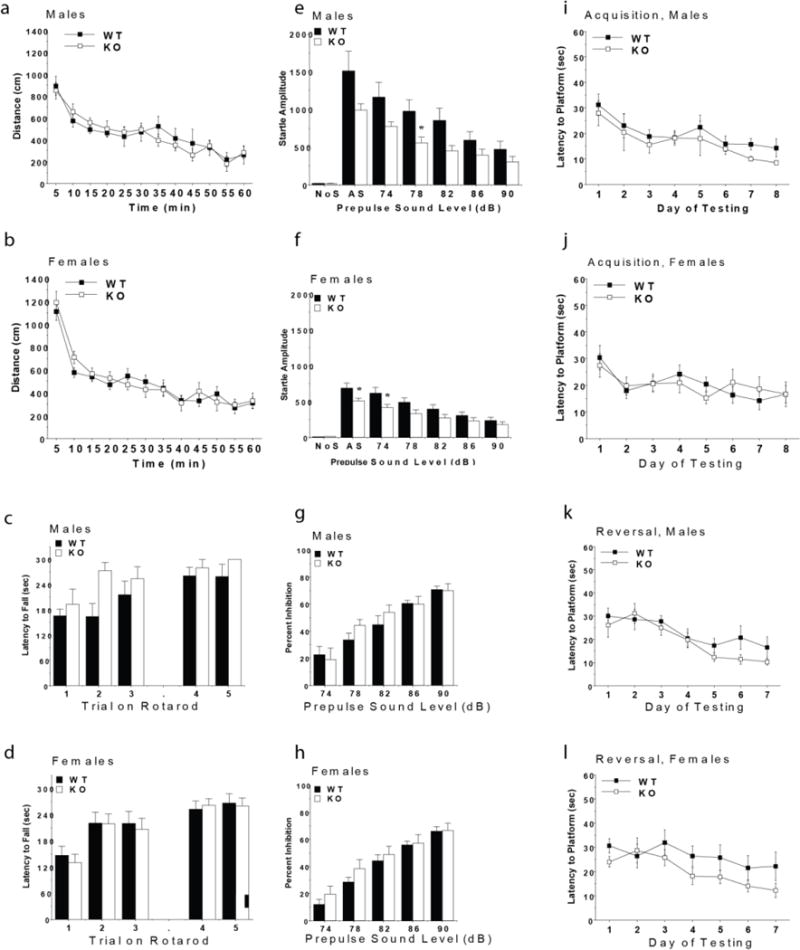
(a–b) No effects of GPR68 deletion on distance traveled in an open field. Data represent mean ± SEM for each group for a one-hour test session. (c–d) No difference on latency to fall from an accelerating rotarod. Data represent mean ± SEM for each group. (e–h) Decreased startle responses in GPR68 KO mice following presentation of acoustic stimuli (e, f). Data represent mean ± SEM for each group. No effects of genotype were found for levels of prepulse inhibition (g, h). Data represent means ± SEM for each group (*p<0.05). (i–l) No difference at acquisition and reversal learning in the Morris water maze. Data represent mean ± SEM of four trials per day. Subject numbers were 9 WT and 7 KO male mice, and 12 WT and 11 KO female mice.
Supplementary Material
Acknowledgments
This work was supported by NIH grants U01104974 (B.L.R, B.K.S and W.K.K.), R01 DA017204 (B.L.R. and W.K.K.) and the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH PDSP) (X.-P.H., H. Z., M. S. F., W. K. K., T. J. M., A. J., B. L. R.), the Michael Hooker Chair for Protein Therapeutics and Translational Proteomics to B. L. R.; Genentech Foundation Predoctoral Fellowship (J. K.); NIH grants GM59957 and GM71896 (B. K. S.) and the Structural Genomics Consortium (B. K. S.); grant P01 HL114471 (R. B. P. and D. A. D); NICHD grant U54 HD079124 (M. S. M., K. A. S., V. N.); NIH grant U19MH082441 (B.L.R., J. J. and X. C.). We thank Mark Pausch (Merck & Co.) for providing us Gs- and Gq-yeast strains for yeast screening assays.
Footnotes
Author contributions
X.-P.H. subcloned GPR68 for yeast screening, made GPR68 and GPR65 mutants, designed, carried out cell-based screening assays, analyzed results, and wrote the paper. J. K. designed and developed homology models, carried out docking screens, analyzed results, and wrote the paper. W. K. K. set up and carried out yeast screening assays, analyzed results, and wrote the paper. H. Z. and M. S. F. designed, carried out in vivo fear-conditioning studies, analyzed results, and wrote the paper. M. S. F. and B. L. R. dubbed ZINC67740571 “ogerin”. B. H. K. created the GPR68 KO mice. S. S. M., K. A. S., and V. N. carried out initial phenotypic characterization, analyzed results, and wrote the paper. X. C. and J. J. synthesized ZINC32547799, ZINC67740571 (ogerin), and ogerin analogues (compounds 33548– 33561, C3 and C4) for functional assays and in vivo studies, and wrote the paper. T. J. M. carried out radioligand binding assays. A. J. prepared drug plates and plasmids for initial screening. R. B. P. and D. A. D. designed and carried out anti-HA immunoblot assays, analyzed results, and wrote the paper. S. W. designed primers, prepared Flag-tagged GPR68 wild-type and mutant plasmids, performed anti-Flag Western blot assays, and analyzed results. T. K. analyzed results and wrote the paper. B. L. R. and B. K. S. coordinated and supervised the project, and with the other authors wrote the paper.
The authors state that there are no conflicting financial interests.
References
- 1.Roth BL, Kroeze WK. Integrated approaches for genome-wide interrogation of the druggable non-olfactory G protein coupled receptor superfamily. J Biol Chem jbc. 2015:R115.654764. doi: 10.1074/jbc.R115.654764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davenport AP, et al. International Union of Basic and Clinical Pharmacology. LXXXVIII G protein-coupled receptor list: recommendations for new pairings with cognate ligands. Pharmacol Rev. 2013;65:967–86. doi: 10.1124/pr.112.007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chung S, Funakoshi T, Civelli O. Orphan GPCR research. Br J Pharmacol. 2008;153(Suppl 1):S339–46. doi: 10.1038/sj.bjp.0707606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Knapp S, et al. A public-private partnership to unlock the untargeted kinome. Nat Chem Biol. 2013;9:3–6. doi: 10.1038/nchembio.1113. [DOI] [PubMed] [Google Scholar]
- 5.Ferguson FM, et al. Targeting Low-Druggability Bromodomains: Fragment Based Screening and Inhibitor Design against the BAZ2B Bromodomain. J Med Chem. 2013;56:10183–10187. doi: 10.1021/jm401582c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leung D, Hardouin C, Boger DL, Cravatt BF. Discovering potent and selective reversible inhibitors of enzymes in complex proteomes. Nat Biotechnol. 2003;21:687–691. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- 7.Ludwig MG, et al. Proton-sensing G-protein-coupled receptors. Nature. 2003;425:93–8. doi: 10.1038/nature01905. [DOI] [PubMed] [Google Scholar]
- 8.Mogi C, et al. Sphingosylphosphorylcholine antagonizes proton-sensing ovarian cancer G-protein-coupled receptor 1 (OGR1)-mediated inositol phosphate production and cAMP accumulation. J Pharmacol Sci. 2005;99:160–7. doi: 10.1254/jphs.fp0050599. [DOI] [PubMed] [Google Scholar]
- 9.Li J, et al. Ovarian cancer G protein coupled receptor 1 suppresses cell migration of MCF7 breast cancer cells via a Galpha12/13-Rho-Rac1 pathway. J Mol Signal. 2013;8:6. doi: 10.1186/1750-2187-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh LS, et al. Ovarian Cancer G Protein–Coupled Receptor 1, a New Metastasis Suppressor Gene in Prostate Cancer. J Natl Cancer Inst. 2007;99:1313–1327. doi: 10.1093/jnci/djm107. [DOI] [PubMed] [Google Scholar]
- 11.Schneider JW, et al. Coupling hippocampal neurogenesis to brain pH through proneurogenic small molecules that regulate proton sensing G protein-coupled receptors. ACS Chem Neurosci. 2012;3:557–68. doi: 10.1021/cn300025a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frick KK, Krieger NS, Nehrke K, Bushinsky DA. Metabolic acidosis increases intracellular calcium in bone cells through activation of the proton receptor OGR1. J Bone Min Res. 2009;24:305–13. doi: 10.1359/jbmr.081015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komarova SV, Pereverzev A, Shum JW, Sims SM, Dixon SJ. Convergent signaling by acidosis and receptor activator of NF-kappaB ligand (RANKL) on the calcium/calcineurin/NFAT pathway in osteoclasts. Proc Natl Acad Sci U A. 2005;102:2643–8. doi: 10.1073/pnas.0406874102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang M, et al. Expression of and role for ovarian cancer G-protein-coupled receptor 1 (OGR1) during osteoclastogenesis. J Biol Chem. 2006;281:23598–605. doi: 10.1074/jbc.M602191200. [DOI] [PubMed] [Google Scholar]
- 15.Russell JL, et al. Regulated expression of pH sensing G Protein-coupled receptor-68 identified through chemical biology defines a new drug target for ischemic heart disease. ACS Chem Biol. 2012;7:1077–83. doi: 10.1021/cb300001m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mohebbi N, et al. The proton-activated G protein coupled receptor OGR1 acutely regulates the activity of epithelial proton transport proteins. Cell Physiol Biochem. 2012;29:313–24. doi: 10.1159/000338486. [DOI] [PubMed] [Google Scholar]
- 17.Regard JB, Sato IT, Coughlin SR. Anatomical profiling of G protein-coupled receptor expression. Cell. 2008;135:561–71. doi: 10.1016/j.cell.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen YJ, Huang CW, Lin CS, Chang WH, Sun WH. Expression and function of proton-sensing G-protein-coupled receptors in inflammatory pain. Mol Pain. 2009;5:39. doi: 10.1186/1744-8069-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saxena H, et al. The GPCR OGR1 (GPR68) mediates diverse signalling and contraction of airway smooth muscle in response to small reductions in extracellular pH. Br J Pharmacol. 2012;166:981–90. doi: 10.1111/j.1476-5381.2011.01807.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Sun Y, Tomura H, Okajima F. Ovarian cancer G-protein-coupled receptor 1 induces the expression of the pain mediator prostaglandin E2 in response to an acidic extracellular environment in human osteoblast-like cells. Int J Biochem Cell Biol. 2012;44:1937–41. doi: 10.1016/j.biocel.2012.07.015. [DOI] [PubMed] [Google Scholar]
- 21.Li H, et al. Abnormalities in Osteoclastogenesis and Decreased Tumorigenesis in Mice Deficient for Ovarian Cancer G Protein-Coupled Receptor 1. PLoS ONE. 2009;4:e5705. doi: 10.1371/journal.pone.0005705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aoki H, et al. Proton-sensing ovarian cancer g protein-coupled receptor 1 on dendritic cells is required for airway responses in a murine asthma model. PLoS One. 2013;8:e79985. doi: 10.1371/journal.pone.0079985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mogi C, Nakakura T, Okajima F. Role of extracellular proton-sensing OGR1 in regulation of insulin secretion and pancreatic beta-cell functions [Review] Endocr J. 2013 doi: 10.1507/endocrj.ej13-0380. at < http://www.ncbi.nlm.nih.gov/pubmed/24088601>. [DOI] [PubMed]
- 24.Okajima F. Regulation of inflammation by extracellular acidification and proton-sensing GPCRs. Cell Signal. 2013;25:2263–71. doi: 10.1016/j.cellsig.2013.07.022. [DOI] [PubMed] [Google Scholar]
- 25.Dong S, Rogan SC, Roth BL. Directed molecular evolution of DREADDs: a generic approach to creating next-generation RASSLs. Nat Protoc. 2010;5:561–73. doi: 10.1038/nprot.2009.239. [DOI] [PubMed] [Google Scholar]
- 26.Mysinger MM, Shoichet BK. Rapid Context-Dependent Ligand Desolvation in Molecular Docking. J Chem Inf Model. 2010;50:1561–1573. doi: 10.1021/ci100214a. [DOI] [PubMed] [Google Scholar]
- 27.Evers A, Klebe G. Ligand-Supported Homology Modeling of G-Protein-Coupled Receptor Sites: Models Sufficient for Successful Virtual Screening. Angew Chem Int Ed. 2004;43:248–251. doi: 10.1002/anie.200352776. [DOI] [PubMed] [Google Scholar]
- 28.Cavasotto CN, et al. Discovery of Novel Chemotypes to a G-Protein-Coupled Receptor through Ligand-Steered Homology Modeling and Structure-Based Virtual Screening. J Med Chem. 2008;51:581–588. doi: 10.1021/jm070759m. [DOI] [PubMed] [Google Scholar]
- 29.Katritch V, Rueda M, Lam PCH, Yeager M, Abagyan R. GPCR 3D homology models for ligand screening: lessons learned from blind predictions of adenosine A2a receptor complex. Proteins. 2010;78:197–211. doi: 10.1002/prot.22507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leach K, Sexton PM, Christopoulos A. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28:382–9. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 31.Keiser MJ, et al. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007;25:197–206. doi: 10.1038/nbt1284. [DOI] [PubMed] [Google Scholar]
- 32.Kalk P, et al. The adenosine A1 receptor antagonist SLV320 reduces myocardial fibrosis in rats with 5/6 nephrectomy without affecting blood pressure. Br J Pharmacol. 2007;151:1025–1032. doi: 10.1038/sj.bjp.0707319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson SA, Wingrove PB, Connelly L, Whiting PJ, Wafford KA. Tracazolate Reveals a Novel Type of Allosteric Interaction with Recombinant γ-Aminobutyric AcidA Receptors. Mol Pharmacol. 2002;61:861–869. doi: 10.1124/mol.61.4.861. [DOI] [PubMed] [Google Scholar]
- 34.Tomura H, et al. Prostaglandin I(2) production and cAMP accumulation in response to acidic extracellular pH through OGR1 in human aortic smooth muscle cells. J Biol Chem. 2005;280:34458–64. doi: 10.1074/jbc.M505287200. [DOI] [PubMed] [Google Scholar]
- 35.Ichimonji I, et al. Extracellular acidification stimulates IL-6 production and Ca(2+) mobilization through proton-sensing OGR1 receptors in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2010;299:L567–77. doi: 10.1152/ajplung.00415.2009. [DOI] [PubMed] [Google Scholar]
- 36.Liu JP, et al. Ovarian cancer G protein-coupled receptor 1-dependent and -independent vascular actions to acidic pH in human aortic smooth muscle cells. Am J Physiol Heart Circ Physiol. 2010;299:H731–42. doi: 10.1152/ajpheart.00977.2009. [DOI] [PubMed] [Google Scholar]
- 37.Matsuzaki S, et al. Extracellular acidification induces connective tissue growth factor production through proton-sensing receptor OGR1 in human airway smooth muscle cells. Biochem Biophys Res Commun. 2011;413:499–503. doi: 10.1016/j.bbrc.2011.08.087. [DOI] [PubMed] [Google Scholar]
- 38.Gravius A, Barberi C, Schäfer D, Schmidt WJ, Danysz W. The role of group I metabotropic glutamate receptors in acquisition and expression of contextual and auditory fear conditioning in rats - a comparison. Neuropharmacology. 2006;51:1146–1155. doi: 10.1016/j.neuropharm.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 39.Daumas S, et al. Transient activation of the CA3 Kappa opioid system in the dorsal hippocampus modulates complex memory processing in mice. Neurobiol Learn Mem. 2007;88:94–103. doi: 10.1016/j.nlm.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 40.Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- 41.Onozawa Y, et al. Activation of T cell death-associated gene 8 regulates the cytokine production of T cells and macrophages in vitro. Eur J Pharmacol. 683:325–31. doi: 10.1016/j.ejphar.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 42.Pompeia S, Manzano GM, Tufik S, Bueno OF. What makes lorazepam different from other benzodiazepines? J Physiol. 2005;569:709. doi: 10.1113/jphysiol.2005.569005. author reply 710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greenblatt DJ, et al. Clinical pharmacokinetics of lorazepam. I. Absorption and disposition of oral 14C-lorazepam. Clin Pharmacol Ther. 1976;20:329–341. doi: 10.1002/cpt1976203329. [DOI] [PubMed] [Google Scholar]
- 44.Schoepp DD. Where will new neuroscience therapies come from? Nat Rev Drug Discov. 2011;10:715–6. doi: 10.1038/nrd3559. [DOI] [PubMed] [Google Scholar]
- 45.Eswar N, et al. Current Protocols in Protein Science. (John Wiley & Sons, Inc; 2001. at < http://onlinelibrary.wiley.com/doi/10.1002/0471140864.ps0209s50/abstract>. [Google Scholar]
- 46.Yang Q, Sharp KA. Building alternate protein structures using the elastic network model. Proteins Struct Funct Bioinforma. 2009;74:682–700. doi: 10.1002/prot.22184. [DOI] [PubMed] [Google Scholar]
- 47.Jacobson MP, Friesner RA, Xiang Z, Honig B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J Mol Biol. 2002;320:597–608. doi: 10.1016/s0022-2836(02)00470-9. [DOI] [PubMed] [Google Scholar]
- 48.Li J, Zhu T, Cramer CJ, Truhlar DG. New Class IV Charge Model for Extracting Accurate Partial Charges from Wave Functions. J Phys Chem A. 1998;102:1820–1831. [Google Scholar]
- 49.Chambers CC, Hawkins GD, Cramer CJ, Truhlar DG. Model for Aqueous Solvation Based on Class IV Atomic Charges and First Solvation Shell Effects. J Phys Chem. 1996;100:16385–16398. [Google Scholar]
- 50.Hert J, Keiser MJ, Irwin JJ, Oprea TI, Shoichet BK. Quantifying the Relationships among Drug Classes. J Chem Inf Model. 2008;48:755–765. doi: 10.1021/ci8000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gaulton A, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2011;40:D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mumberg D, Muller R, Funk M. Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene. 1995;156:119–22. doi: 10.1016/0378-1119(95)00037-7. [DOI] [PubMed] [Google Scholar]
- 53.Erlenbach I, et al. Functional expression of M(1), M(3) and M(5) muscarinic acetylcholine receptors in yeast. J Neurochem. 2001;77:1327–37. doi: 10.1046/j.1471-4159.2001.00344.x. [DOI] [PubMed] [Google Scholar]
- 54.Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U A. 2007;104:5163–8. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- 56.Besnard J, et al. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–20. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keiser MJ, et al. Predicting new molecular targets for known drugs. Nature. 2009;462:175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Horvat SJ, et al. A-kinase anchoring proteins regulate compartmentalized cAMP signaling in airway smooth muscle. FASEB J. 2012;26:3670–9. doi: 10.1096/fj.11-201020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang XP, Mangano T, Hufeisen S, Setola V, Roth BL. Identification of Human Ether-à-go-go Related Gene Modulators by Three Screening Platforms in an Academic Drug-Discovery Setting. ASSAY Drug Dev Technol. 2010;8:727–742. doi: 10.1089/adt.2010.0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.