

Abstract
Background
Bifidobacterium thermophilum RBL67 (RBL67), a human fecal isolate and health promoting candidate shows antagonistic and protective effects against Salmonella and Listeria spec. in vitro. However, the underlying mechanisms fostering these effects remain unknown. In this study, the interactions of RBL67 and Salmonella enterica subsp. enterica serovar Typhimurium N-15 (N-15) were explored by global transcriptional analysis.
Results
Growth experiments were performed in a complex nutritive medium with controlled pH of 6.0 and suitable for balanced growth of both RBL67 and N-15. RBL67 growth was slightly enhanced in presence of N-15. Conversely, N-15 showed reduced growth in the presence of RBL67. Transcriptional analyses revealed higher expression of stress genes and amino acid related function in RBL67 in co-culture with N-15 when compared to mono-culture. Repression of the PhoP regulator was observed in N-15 in presence of RBL67. Further, RBL67 activated virulence genes located on the Salmonella pathogenicity islands 1 and 2. Flagellar genes, however, were repressed by RBL67. Sequential expression of flagellar, SPI 1 and fimbrial genes is essential for Salmonella infection. Our data revealed that RBL67 triggers expression of SPI 1 and fimbrial determinants prematurely, potentially leading to redundant energy expenditure. In the competitive environment of the gut such energy expenditure could lead to enhanced clearing of Salmonella.
Conclusion
Our study provides first insights into probiotic-pathogen interactions on global transcriptional level and suggests that deregulation of virulence gene expression might be an additional protective mechanism of probiotica against infections of the host.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-016-0659-x) contains supplementary material, which is available to authorized users.
Keywords: Bifidobacterium thermophilum, Salmonella Typhimurium, Co-culture, RNA-seq, Probiotic, Anti-microbial, Virulence
Background
Probiotics are live organism that, when administered in adequate amounts, confer a health benefit on the host [1]. They exert their beneficial effect via a wide array of mechanisms including direct and indirect antagonism with enteropathogens, improvement of the intestinal barrier function and activation of the mucosal immune system [2, 3]. Direct antagonism with enteropathogens is mediated via production of antimicrobial compounds such as organic acids and bacteriocins, competition for nutrients and minerals, and occupation of adhesion sites [2]. Bifidobacteria and lactobacilli are important constituents of the human gut microbiota and have been associated with a good health status of the host [3, 4]. They are the two major genera used for probiotic applications and have a long history of safe use. Specific strains from bifidobacteria and lactobacilli have been shown to protect against pathogens, with strain specific effects [4].
Bifidobacterium thermophilum is a relatively oxygen tolerant Bifidobacterium species that has been isolated from bovine rumen, sewage, and from piglet, calf and baby feces [5, 6]. Peptidoglycans from B. thermophilum strain P2-91 protect mice against Escherichia coli infections and improve cytotoxic activity of mice lymphocytes [7, 8]. Furthermore, chicken were more resistant to E. coli infection after oral administration of B. thermophilum [9]. The infant feces isolate B. thermophilum RBL67 (RBL67) is a promising probiotic candidate which genome was sequenced [10]. The strain can grow under low oxygen, at pHs as low as 4.0 and at temperatures up to 47 °C. Further, it can reach high cell yield numbers in fermentation which makes it suitable to be applied in industrial fermentations [6, 11–14]. Furthermore, RBL67 decreases S. Typhimurium counts in an in vitro fermentation model of the gastrointestinal tract [14], reduces severity of rotavirus-associated diarrhea in suckling mice [15], and blocks invasion of S. Typhimurium and L. monocytogenes to human intestinal cell lines [13, 16]. However, the underlying mechanisms of RBL67-Salmonella interaction are not elucidated yet.
Salmonella species are a major cause of food-borne diseases with an estimated world-wide annual infection rate of 93.8 million cases and 155,000 deaths [17]. Salmonella usually infect humans after ingestion of contaminated food products [18, 19]. Salmonella enterica subsp. enterica serovar Typhimurium (S. Typhimurium) is a Salmonella serotype frequently encountered in clinical cases [18]. Its pathogenesis depends on multiple factors including motility and chemotaxis, adhesion, invasion and persistence. The majority of relevant virulence determinants are located on Salmonella pathogenicity islands (SPIs) and are regulated by a complex molecular network that transmits environmental signals of conditions prevailing in the host [18]. Salmonella invasion is dependent on the gut environment and is enhanced by low oxygen tension, high osmolarity, neutral pH and acetate, whereas cationic peptides, bile, propionate and butyrate suppress invasion [18, 20]. One of the key regulators for Salmonella invasion is HilA [21]. HilA expression is affected by environmental signals and enables Salmonella to express different invasive phenotypes under different conditions [18, 22, 23]. Modulation of the gut environment via pre- and/or probiotic treatments may alter the gene expression of pathogens like Salmonella, either indirectly via production of organic acids or directly via microbe-microbe interactions [2]. Indeed, probiotic strains were reported to modulate the transcriptional response of Salmonella. PhoP, a postulated repressor of hilA expression was activated and HilA was repressed during growth in the presence of supernatant of Lactobacillus rhamnosus GG [23]. However, information about modulation of gene expression in enteropathogens due to direct microbe-microbe interaction is still scarce and unraveling the transcriptomic response of these multifactorial interactions is challenging.
RNA-sequencing (RNA-seq) is a powerful tool to determine the transcriptional response of an organism in a complex culture because interference of signals from other organisms is limited [24]. In this study we investigated the potential of B. thermophilum RBL67 to modulate the transcriptome of S. Typhimurium N-15. The response of RBL67 and Salmonella Typhimurium N-15 in the co-culture was compared to mono-cultures using RNA-seq in attempt to provide insight in the protective mechanism of RBL67 against Salmonella infections.
Methods
Bacterial strains
Salmonella Typhimurium N-15 was isolated from a clinical case in Switzerland in 2007 and obtained from the National Reference Centre for Enteropathogenic Bacteria and Listeria (NENT; Zurich, Switzerland). Bifidobacterium thermophilum RBL67 (=LMG S-23614), originally isolated from infant feces [6], was obtained from our own culture collection.
Batch fermentation conditions
Two sets of fermentations were performed, each set consisting of six fermentations. The first set was composed of three RBL67 mono-cultures and three RBL67-N-15 co-cultures. The second set consisted of three N-15 mono-cultures and another set of three RBL67-N-15co-cultures. The first set of three co-cultures was used for sampling RBL67-RNA at t = 5 h and the second for sampling N15-RNA at t = 4 h. Bacteria were cultured in 350 mL scale Sixfors bioreactors (Infors AG, Bottmingen, Switzerland) using 310 mL YCFA medium [25] supplemented with 6 g/L glucose (Sigma-Aldrich Chemie GmbH, Buchs, Switzerland). Fermentations were performed at 38 °C with stirring at 200 rpm for 24 h. A constant pH of 6.0 was maintained by automated addition of 2.5 M NaOH. Anaerobic conditions were ensured by purging the headspace with CO2. Fermentations were inoculated with 4 % (v/v) of a 16 h grown pre-culture. Pre-cultures were prepared by propagating strains twice in 10 mL YCFA medium in Hungate tubes to adapt the strains to the medium and anaerobic conditions. The pre-cultures were centrifuged (6000 × g, 5 min), washed in 0.1 % peptone water reduced with 0.05 % L-cysteine hydrochloride (VWR International AG, Dietikon, Switzerland) and resuspended in 2 mL peptone water before inoculation to the fermenter.
Growth was monitored by optical density measurements at 600 nm (OD600) using a Biochrom WPA CO8000 cell density meter (Biochrom, Cambridge, United Kingdom). Samples were taken hourly until the stationary growth phase was reached, with a final sample taken after 24 h. Metabolite and sugar concentrations were determined by HPLC analysis (Thermo Fisher Scientific, Wohlen, Switzerland) as described previously [26]. Carbon balance as % of carbons recovered was calculated on the basis of consumed glucose and produced organic acids. Viable cell counts of RBL67 were determined by plating appropriate dilutions on MRS agar (Biolife, Milan, Italy), supplemented with 0.05 % L-cysteine hydrochloride (MRS-C). Viable cell counts of N-15 were determined on MacConkey Agar No. 2 (Oxoid AG, Pratteln, Switzerland). Co-culture effluent samples were plated on MRS-C agar supplemented with 5 g L−1 mupirocin (VWR International AG, Dietikon, Switzerland) to select for RBL67 [27], and on MacConkey Agar No. 2 to select for N-15. MRS plates were incubated anaerobically using anaerobic gas pack systems (AnaeroGen TM, Oxoid AG) at 37 °C for 48 h. MacConkey Agar plates were incubated aerobically at 37 °C for 24 h.
Maximum specific growth rates were calculated for each replication separately (N = 3) from the slope of the curve of the log cell counts versus time during the exponential growth phase.
Sampling for RNA extraction
RBL67 and N-15 mono- and co-culture samples were subjected to different procedures to allow optimal RNA extraction of both RBL67 and N-15.
Mono- and co-culture samples of N-15 cultures (20 mL each) were directly transferred to 20 mL 60 % glycerol (Sigma-Aldrich Chemie GmbH, Buchs, Switzerland) at −40 °C, kept on ice for 20 min and centrifuged for 15 min (3220 × g, 4 °C). The supernatant was discarded and the resulting pellets were immediately frozen at −80 °C until RNA extraction. Mono- and co-culture samples of RBL67 cultures were shortly centrifuged (10,000 × g, 20 s). The RBL67 mono-culture pellets were resuspended in 400 μl MRS-C and transferred to a pre-chilled screw cap tube, containing 500 mg glass beads (0.1 mm; Biospec Products Inc., Bartlesville, USA), 500 μl chloroform/phenol (1:1, v/v), 30 μl 3 M Na-acetate (pH 5.2) and 30 μl SDS 10 % [28]. The pellets of the RBL67 co-culture were resuspended in 12 mL of RNAprotect® Bacteria Reagent (Qiagen AG, Basel, Switzerland), incubated for 5 min at room temperature and centrifuged again (10,000 × g, 20 s). Both samples were then rapidly frozen in liquid nitrogen and stored at −80 °C until RNA extraction.
RNA-extraction and ribosomal RNA depletion
Frozen pellets from N-15 samples were resuspended in 200 μl 10 mM Tris-buffer (pH 8.0). Total RNA was extracted using the High Pure RNA isolation kit (Roche Diagnostics, Rotkreuz, Switzerland), according to the manufacturer’s instructions. Total RNA of RBL67 mono- and co-culture samples was extracted using a phenol/chloroform extraction method [28], followed by a purification using the High Pure RNA isolation kit (Roche Diagnostics). Prior to RNA extraction the sample from the RBL67 co-culture was resuspended in MRS-C medium and transferred to a pre-chilled mix of 500 mg glass beads (Biospec Products Inc.) and TRI Reagent® (Life Technologies Europe BV, Zug, Switzerland).
RNA quantity and purity was determined on a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific, Washington, USA) and RNA integrity was tested with an Agilent 2100 Bioanalyzer (Agilent, Basel, Switzerland). RBL67 samples with a RNA integrity number (RIN) ≥ 9.5 and a 16S/23S-rRNA ratio ≥1.6 were used for ribosomal RNA depletion and subsequent RNA-sequencing. Due to the aberrant nature of ribosomal RNA of S. Typhimurium [29], the RIN value and the 16S/23S-rRNA ratio could not be calculated for N-15. Hence we selected samples which were comparable to the profiles reported previously for Salmonella [30], i.e. a straight zero line (indicating no RNA degradation), absence of 23S RNA and appearance of two additional peaks neighboring the 16S peak.
Depletion of ribosomal RNA from 10 μg total RNA was performed using the MICROBExpress™ Bacterial mRNA Enrichment Kit (Life Technologies Europe BV, Zug, Switzerland) according to the manufacturer’s instructions. Additionally, EDTA (1 mM) was added to chelate divalent cations present in the RNA solution.
RNA-sequencing
RNA-sequencing was performed on an Illumina HiSeq 2000 sequencer (Illumina Inc., California, USA) at the Functional Genomics Center Zurich (FGCZ). Libraries were prepared using the TruSeq Stranded mRNA Sample Prep Kit (Illumina) according to the manufacturer’s protocol. The libraries were qualitatively and quantitatively checked using a Qubit® (1.0) Fluorometer (Life Technologies Europe BV, Zug, Switzerland) and a Bioanalyzer 2100 (Agilent, Basel, Switzerland) and were subsequently normalized at 10 nM in Tris-Cl (10 mM, pH 8.5) containing 0.1 % Tween20. Cluster generation was performed using the TruSeq SR Cluster Kit v3-cBot-HS (Illumina) using 8 pM of pooled normalized libraries on the cBOT and stranded sequencing of 100 bp was done using the TruSeq SBS Kit v3-HS (Illumina). Each set of samples (N = 6) was analyzed in a separate sequencing lane.
RNA-Seq data analysis
Illumina raw data reads (100 bp) were separated by barcode and mapped against the genome of RBL67 (GenBank accession no. CP004346) or Salmonella Typhimurium LT2 (GenBank accession no. AE006468) using the CLC Genomics Workbench 6.5.1 (CLCbio, Aarhus, Denmark) applying the default settings. Maximum allowance of mismatches was set at 2, minimum length fraction at 0.9 and minimum similarity fraction at 0.8.
Statistical analysis for differential gene expression of the mono- and co-cultures was done with the statistical software R (http://www.R-project.org) using the GLM method [31] included in the Bioconductor EdgeR software package [32–34] based on negative binomial distribution. Genes with low read numbers (sum of reads in all samples <3 counts per million (cpm)) or with high read numbers (number of reads >50,000 cpm in each sample) were filtered out before data normalization. A false discovery rate (FDR) value <0.05 and a differential expression of at least 2 fold (1 < log2 ratio < −1) was used as cut off for significant differentially expressed genes in mono-culture and co-culture [35]. Proteins of RBL67 and LT2 were assigned to gene ontology categories (GO) using Blast2GO at standard settings [36]. GO categories enrichment analyses were performed and visualized using the BiNGO plugin [37] in Cytoscape (v.3.0.1, [38]) applying the hypergeometric test with Benjamini and Hochberg false discovery rate correction option. The significance cutoff for overrepresented gene ontology categories was a corrected p-value of <0.05.
Virulence factors of Salmonella LT2 were identified by genome wide blast against the virulence factor database (VFDB) [39], using a cut off E-value of 1−20. Significant enrichment of virulence factors was calculated using the Fisher’s Exact Test Calculator for 2 × 2 Contingency at www.research.microsoft.com/en-us/um/redmond/projects/mscompbio/fisherexacttest/.
The RNAseq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus [40] and are accessible through GEO Series accession number GSE65716 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65716).
Statistical analysis
Statistical analysis for cell counts (log10 transformation) and growth rates were performed using JMP 10.0 (SAS Institute., Cary, NC). Cell counts and maximum specific growth rates of mono-and co-cultures were tested for significant differences using the non-parametric Kruskal-Wallis (P-value <0.05).
Results
Growth characteristics of RBL67 in mono- and co-culture with N-15
To analyze interactions between B. thermophilum RBL67 and Salmonella N-15, both strains were grown in pH controlled mono- and co-cultures (pH 6.0) and growth characteristics were compared. The maximum specific growth rate of RBL67 in mono-culture (μmax = 0.26 ± 0.05 h−1) was significantly lower compared to that in co-culture (μmax = 0.33 ± 0.01 h−1). The stationary growth phase was reached after approximately 8 h in both cultures, with final cell counts of 8.88 ± 0.10 log10 cfu mL−1 and 9.12 ± 0.14 log10 cfu mL−1 in the mono- and co-culture, respectively (Fig. 1a).
Fig. 1.
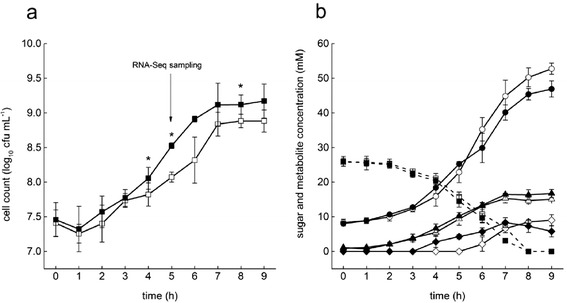
Cell counts and metabolic profiles of RBL67 in mono-culture and in co-cultures with N-15. a Cell counts in mono- (open symbols) and co-culture (closed symbols). b Metabolite concentration in mono- (open symbols) and co-culture (closed symbols). Means ± SD from three biological replicates are presented. *Cell counts significantly different between mono- and co-culture with the non-parametric Kruskal-Wallis Test (P < 0.05); square: glucose; circle: acetate; triangle: lactate and diamonds: formate
Glucose consumption and metabolite profiles were similar for RBL67 in mono- and co-culture (Fig. 1b). In both cultures, glucose was depleted after 8 h which corresponded to the onset of the stationary growth phase, indicating growth limitation by the carbon source. The main metabolites produced in mono-cultures were 50 ± 3 mM acetate, 15 ± 1 mM lactate, and 9 ± 0.3 mM formate after 8 h, corresponding to a calculated carbon recovery of 103 %. A slightly lower acetate concentration was observed in co-culture: 45 ± 2 mM acetate. Further, 16 ± 2 mM lactate and 7 ± 2 mM formate were produced after 8 h, corresponding to a carbon recovery of 100 %.
Taken together, RBL67 growth was slightly enhanced in the co-culture with Salmonella compared to mono-culture and only small differences in organic acid production were observed.
Global transcriptional response of RBL67 to co-culture with N-15
To elucidate the response of RBL67 to N-15 on a global level, the transcriptome profiles of B. thermophilum RBL67 grown in mono- and in co-culture were compared. Samples were taken after 5 h of growth, a time point at which RBL67 displayed exponential growth in both cultures (Fig. 1). Viable RBL67 cell counts at harvesting point were 8.07 ± 0.07 and 8.53 ± 0.04 log10 cfu mL−1, in the mono- and in co-culture, respectively. RNA sequencing of RBL67 cultures resulted in a mean read number of 37,365,651 and 31,752,403 for mono- and co-cultures, respectively. Thereof, 93 % of the reads deriving from the mono-cultures and 79 % of the reads from the co-cultures could be mapped onto the RBL67 genome. Differential gene expression analysis revealed 57 genes being significantly differentially expressed in mono- compared to co-cultures (Tables 1 and 2). An operon involved in lipid export (D805_0155-D805_0157), sugar transport (D805_1600-D805_1602), and an operon of undefined function (D805_1659-D805_1660), together with its putative regulator of the HxlR family (D805_1658) were higher expressed in co- culture (Table 1). Further, a stress response was triggered in co-cultures as revealed by higher expression of the heat shock protein regulator HspR (D805_1678), the SOS-response repressor and protease LexA (D805_0599) and the protease ClpB (D805_1594). The latter gene harbors a HspR-associated inverted repeat (HAIR) in its upstream region and is therefore likely activated by HspR. Additional functions of RBL67 genes higher expressed in co-cultures with Salmonella N-15 were related to metal transport (D805_1209) and amino acid metabolism (D805_1238 and D805_1530), including a glutamate-5-kinase (D805_1238) which catalyzes the first step for proline biosynthesis from glutamate.
Table 1.
Bifidobacterium thermophilum RBL67 genes higher expressed in co-culture with N-15 compared to mono-culture
| Locus tag | Function | logFCa | logCPMb | FDRc |
|---|---|---|---|---|
| D805_0058 | Oligopeptide transport ATP-binding protein OppF (TC 3.A.1.5.1) | −1.52 | 4.09 | 4E-06 |
| D805_0077 | hypothetical protein | −1.17 | 6.36 | 4.8E-07 |
| D805_0155 | Transcriptional regulator, MarR family | −1.95 | 6.80 | 1.2E-17 |
| D805_0156 | hypothetical protein | −1.50 | 3.64 | 0.00401 |
| D805_0157 | Lipid A export ATP-binding/permease protein MsbA | −1.17 | 8.10 | 6.2E-06 |
| D805_0382 | hypothetical protein | −1.13 | 7.94 | 1.4E-06 |
| D805_0466 | FIG 00672402: hypothetical protein | −1.00 | 7.21 | 9.5E-08 |
| D805_0503 | possible conserved integral membrane protein | −1.14 | 3.08 | 0.01355 |
| D805_0599 | SOS-response repressor and protease LexA (EC 3.4.21.88) | −1.24 | 6.59 | 7.9E-09 |
| D805_0600 | hypothetical protein | −2.14 | 6.18 | 2.6E-27 |
| D805_0707 | Inner membrane protein | −1.14 | 6.16 | 3.4E-06 |
| D805_1209 | Zinc ABC transporter, periplasmic-binding protein ZnuA | −1.15 | 4.69 | 1.3E-05 |
| D805_1238 | Cystathionine beta-synthase (EC 4.2.1.22) | −1.23 | 6.23 | 2.7E-09 |
| D805_1392 | putative aminotransferase | −1.23 | 4.66 | 1.2E-05 |
| D805_1393 | hypothetical protein | −1.03 | 4.30 | 0.00118 |
| D805_1530 | Glutamate 5-kinase (EC 2.7.2.11) | −1.08 | 8.00 | 3.9E-09 |
| D805_1531 | COG0536: GTP-binding protein Obg | −1.03 | 9.16 | 2.5E-06 |
| D805_1591 | DNA recombination protein RmuC | −1.27 | 7.58 | 3.6E-12 |
| D805_1594 | ClpB protein | −1.06 | 8.68 | 2.1E-06 |
| D805_1600 | Maltodextrin glucosidase (EC 3.2.1.20) | −1.17 | 6.23 | 5.6E-08 |
| D805_1601 | ABC-type sugar transport system, permease component | −1.85 | 4.02 | 3.9E-09 |
| D805_1602 | MSM (multiple sugar metabolism) operon regulatory protein | −1.68 | 3.28 | 1.1E-05 |
| D805_1621 | Sortase A, LPXTG specific | −1.09 | 3.52 | 0.00557 |
| D805_1622 | hypothetical protein | −1.44 | 4.42 | 5.6E-08 |
| D805_1637 | COG family: predicted phosphohydrolases | −1.63 | 6.91 | 1.8E-21 |
| D805_1658 | Transcriptional regulator, HxlR family | −1.07 | 4.37 | 0.00624 |
| D805_1659 | Rrf2-linked NADH-flavin reductase | −2.01 | 5.23 | 2.9E-14 |
| D805_1660 | COG2110, Macro domain, possibly ADP-ribose binding module | −1.83 | 4.90 | 2.4E-16 |
| D805_1678 | HspR, transcriptional repressor of DnaK operon | −1.08 | 5.37 | 4.9E-08 |
| D805_1702 | transport protein | −1.80 | 6.58 | 3.5E-23 |
a logFC log2 fold change, b logCPM log2 counts per million, c FDR false discovery rate
Table 2.
Bifidobacterium thermophilum RBL67 genes higher expressed in mono-culture compared to co-culture with N-15
| Locus tag | Function | logFCa | logCPMb | FDRc |
|---|---|---|---|---|
| D805_0063 | FIG 00519111: hypothetical protein | 1.76 | 5.67 | 1.8E-12 |
| D805_0064 | HTH domain protein | 2.18 | 3.98 | 4.1E-09 |
| D805_0075 | hypothetical protein | 1.04 | 5.15 | 0.00011 |
| D805_0178 | Ribonucleotide reductase of class Ib (aerobic), alpha subunit (EC 1.17.4.1) | 1.21 | 7.11 | 0.04089 |
| D805_0341 | Transcriptional regulator, GntR family domain/Aspartate aminotransferase (EC 2.6.1.1) | 1.93 | 3.72 | 0.02045 |
| D805_0345 | Manganese transport protein MntH | 1.84 | 3.71 | 0.04055 |
| D805_0351 | Glycosyl transferase, group 2 family protein | 1.49 | 4.48 | 0.00041 |
| D805_0352 | Glycosyltransferase (EC:2.4.1.-) | 1.74 | 4.96 | 1.3E-07 |
| D805_0354 | glycosyl transferase, group 1 family protein | 2.07 | 4.79 | 3.0E-10 |
| D805_0355 | hypothetical protein | 1.34 | 6.14 | 4.5E-06 |
| D805_0356 | Glycosyltransferase (EC 2.4.1.-) | 2.14 | 4.96 | 1.5E-15 |
| D805_0512 | hypothetical protein | 2.01 | 2.39 | 0.03026 |
| D805_0524 | D-lactate dehydrogenase (EC 1.1.1.28) | 2.85 | 3.15 | 0.00049 |
| D805_0525 | Aspartate aminotransferase (EC 2.6.1.1) | 1.91 | 3.11 | 0.01352 |
| D805_0652 | Oligopeptide transport system permease protein OppC (EC 3.A.1.5.1) | 2.25 | 2.47 | 0.00227 |
| D805_0656 | hypothetical protein | 1.57 | 3.01 | 0.01267 |
| D805_0665 | hypothetical protein | 1.18 | 5.85 | 2.0E-05 |
| D805_0693 | Acetyltransferase, GNAT family | 2.23 | 6.96 | 2.7E-31 |
| D805_0694 | hypothetical protein | 2.80 | 3.46 | 1.5E-14 |
| D805_0698 | hypothetical protein | 1.19 | 4.02 | 0.00369 |
| D805_0837 | putative TraA-like conjugal transfer protein | 2.75 | 3.13 | 0.00038 |
| D805_0885 | Ferric iron ABC transporter, iron-binding protein | 1.63 | 2.52 | 0.00624 |
| D805_0928 | hypothetical protein | 1.90 | 2.59 | 0.00374 |
| D805_0948 | hypothetical protein | 2.08 | 2.96 | 0.00118 |
| D805_1220 | hypothetical protein | 2.60 | 3.03 | 0.00015 |
| D805_1313 | Methionine ABC transporter permease protein | 1.52 | 2.03 | 0.04091 |
| D805_1771 | hypothetical protein | 1.01 | 4.56 | 0.01571 |
a logFC log2 fold change, b logCPM log2 counts per million, c FDR false discovery rate
Twenty-seven genes were down regulated in co-cultures compared to monoculture, of which 12 were classified as hypothetical proteins (Table 2). A putative operon encoding glycosyltransferases (ORF D805_0351-D805_0356), three genes involved in amino acid metabolism (D805_0341, D805_0525 and D805_1313), including the glutamate producing enzyme aspartate aminotransferase (EC 2.6.1.1) and two metal transporters (D805_0345 and D805_0885) were higher expressed.
Mapping the co-culture reads to the Salmonella Typhimurium LT2 genome resulted in less than 5 million reads mapped (data not shown), indicating that the majority of the RNA isolated form the co-culture after 5 h consisted of bifidobacterial RNA.
Growth characteristics of N-15 in mono- and co-culture with RBL67
In a next step Salmonella N-15 was grown in a mono- and co-culture with RBL67, the latter being a repetition of the co-culture presented above. Salmonella N-15 had similar maximum specific growth rates of μmax = 0.39 ± 0.02 h−1 and 0.38 ± 0.04 h−1 in mono- and co-culture, respectively. In the late exponential phase after approximately 5 h of fermentation, the growth rate in the co-culture was smaller compared to mono-culture (Fig. 2a). The difference in growth resulted in a higher Salmonella cell count of 9.10 ± 0.16 log10 cfu mL−1 in the mono-culture compared to 8.82 ± 0.08 log10 cfu mL−1 in the co-culture. Glucose was depleted after 10 and 8 h for mono- and co-cultures, respectively (Fig. 2b). The main metabolites produced by N-15 in mono-culture were 27 ± 0.4 mM acetate, 23 ± 2 mM lactate and 12 ± 2 mM formate, corresponding to a calculated carbon recovery of 93 %. The metabolite production in the co-culture was: 42 ± 4 mM acetate, 17 ± 2 mM lactate and 8 ± 3 mM formate. These values are virtually the same to those from the first co-culture experiments (Fig. 1b). The calculated carbon mass balance in the co-culture was 99 %.
Fig. 2.
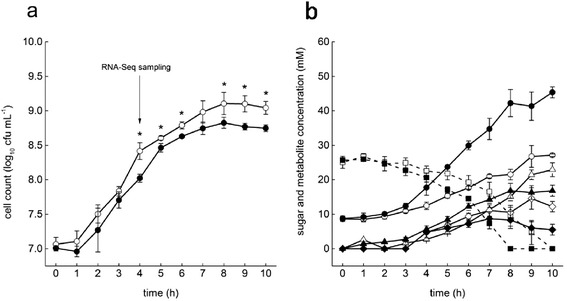
Cell counts and metabolic profiles of N-15 in mono-culture and in co-cultures with RBL67. a Cell counts in mono- (open symbols) and co-culture (closed symbols). b Metabolite concentration in mono- (open symbols) and co-culture (closed symbols). Means ± SD from three biological replicates are presented. *Cell counts significantly different between mono- and co-culture with the non-parametric Kruskal-Wallis Test (P < 0.05); square: glucose; circle: acetate; triangle: lactate and diamonds: formate
Salmonella reached slightly lower cell numbers in the co-culture with RBL67 compared to its mono-culture, but was further not affected by the presence of RBL67 concerning growth speed.
Global transcriptional response of N-15 to co-culture with RBL67
Because RNA-seq analyses of the co-culture after 5 h growth resulted in low read mapping the transcriptome of N-15 in mono-a-culture and in co-culture with RBL67 was analyzed after 4 h growth. This time-point corresponds to cell counts of 8.42 ± 0.12 and 8.02 ± 0.06 log10 cfu mL−1 for mono- and co-cultures, respectively. Acetate concentrations at sampling point was 17.8 ± 2.2 mM in the co-culture, slightly higher than the 12.5 ± 1.1 mM in the mono-culture Moreover, at this point Salmonella is growing exponentially and at comparable speed in both cultures (Fig. 2b). From the total mean read numbers of 38,838,013 (mono-culture) and 30,020,491 (co-culture), 91 and 52 % could be mapped onto the genome and plasmid of the sequenced strain Salmonella Typhimurium LT2, respectively. In total 701 genes were higher expressed in mono- culture and 1278 genes in the co-culture (Additional file 1: Tables S1 and S2).
GO category enrichment analysis revealed 88 categories significantly overrepresented in co-culture of which 47 belonged to the cluster “biological processes”, 29 to “molecular function” and 29 to “cellular component”. Within the cluster biological processes the categories “localization” (GO:051179), “establishment of localization” (GO:051234) and “transport” (GO:006810) were significantly overrepresented (Table 3). Detailed analysis of these categories revealed that they each contained the same 281 genes. At a lower hierarchical level, the category “protein secretion by the type III secretion system” was highly overrepresented (GO:030254, N = 49 genes). Further, “siderophore transport” and “carbohydrate transport systems”, including “PEP-dependent sugar phosphotransferase systems” (GO:009401, N = 33) were overrepresented in co-culture. Other categories overrepresented in biological processes included “multi-organism process” (GO:051704, N = 47), “pathogenesis” (GO:009405, N = 26) and “interspecies interaction between organisms” (GO:044419, N = 35). The majority of the genes (N = 26) in the latter category were also found in GO:052049: “interaction with host via protein secreted by type III secretion system”. The 26 genes assigned to this category were also present in already mentioned GO:030254: “type III secretion system” category.
Table 3.
Gene Ontology (GO) categories of the Salmonella Typhimurium N-15 transcriptome significantly overrepresented in the co-culture with RBL67 compared to mono-culture
| GO category | p-value | Ngenes in category | Description of category |
|---|---|---|---|
| Biological process | |||
| GO:051234 | 4.77E-28 | 281 | establishment of localization |
| GO:006810 | 4.77E-28 | 281 | transport |
| GO:051179 | 6.29E-25 | 281 | localization |
| GO:030254 | 8.34E-15 | 42 | protein secretion by the type III secretion system |
| GO:051704 | 1.80E-14 | 47 | multi-organism process |
| GO:051701 | 3.20E-12 | 35 | interaction with host |
| GO:044419 | 3.20E-12 | 35 | interspecies interaction between organisms |
| GO:044403 | 3.20E-12 | 35 | symbiosis, encompassing mutualism through parasitism |
| GO:008643 | 8.15E-11 | 52 | carbohydrate transport |
| GO:046903 | 4.51E-10 | 49 | secretion |
| GO:032940 | 4.51E-10 | 49 | secretion by cell |
| GO:009306 | 4.51E-10 | 49 | protein secretion |
| GO:052047 | 5.68E-10 | 26 | interaction with other organism via secreted substance involved in symbiotic interaction |
| GO:052049 | 5.68E-10 | 26 | interaction with host via protein secreted by type III secretion system |
| GO:052048 | 5.68E-10 | 26 | interaction with host via secreted substance involved in symbiotic interaction |
| GO:052210 | 5.68E-10 | 26 | interaction with other organism via protein secreted by type III secretion system involved in symbiotic interaction |
| GO:044046 | 5.68E-10 | 26 | interaction with host via substance released outside of symbiont |
| GO:051649 | 4.63E-09 | 49 | establishment of localization in cell |
| GO:051641 | 8.25E-09 | 49 | cellular localization |
| GO:009405 | 9.76E-08 | 26 | pathogenesis |
| GO:015031 | 2.55E-06 | 49 | protein transport |
| GO:045184 | 2.55E-06 | 49 | establishment of protein localization |
| GO:033036 | 2.96E-06 | 50 | macromolecule localization |
| GO:008104 | 3.67E-06 | 49 | protein localization |
| GO:009401 | 8.19E-06 | 33 | phosphoenolpyruvate-dependent sugar phosphotransferase system |
| GO:007047 | 2.10E-04 | 12 | cellular cell wall organization |
| GO:045229 | 2.10E-04 | 12 | external encapsulating structure organization |
| GO:071555 | 7.60E-04 | 12 | cell wall organization |
| GO:009242 | 2.19E-03 | 7 | colanic acid biosynthetic process |
| GO:052126 | 2.19E-03 | 7 | movement in host environment |
| GO:052192 | 2.19E-03 | 7 | movement in environment of other organism involved in symbiotic interaction |
| GO:044409 | 2.19E-03 | 7 | entry into host |
| GO:046377 | 2.19E-03 | 7 | colanic acid metabolic process |
| GO:051828 | 2.19E-03 | 7 | entry into other organism involved in symbiotic interaction |
| GO:022610 | 3.83E-03 | 17 | biological adhesion |
| GO:007155 | 3.83E-03 | 17 | cell adhesion |
| GO:030001 | 6.91E-03 | 33 | metal ion transport |
| GO:006814 | 8.26E-03 | 16 | sodium ion transport |
| GO:009235 | 1.23E-02 | 14 | cobalamin metabolic process |
| GO:009236 | 1.23E-02 | 14 | cobalamin biosynthetic process |
| GO:015891 | 2.34E-02 | 5 | siderophore transport |
| GO:019184 | 2.34E-02 | 5 | nonribosomal peptide biosynthetic process |
| GO:006811 | 2.34E-02 | 47 | ion transport |
| GO:015672 | 2.61E-02 | 27 | monovalent inorganic cation transport |
| GO:006812 | 3.45E-02 | 37 | cation transport |
| GO:006778 | 4.86E-02 | 16 | porphyrin metabolic process |
| GO:006779 | 4.86E-02 | 16 | porphyrin biosynthetic process |
| Molecular function | |||
| GO:005215 | 4.79E-20 | 215 | transporter activity |
| GO:015144 | 1.57E-09 | 39 | carbohydrate transmembrane transporter activity |
| GO:022892 | 1.31E-08 | 111 | substrate-specific transporter activity |
| GO:022891 | 9.42E-08 | 99 | substrate-specific transmembrane transporter activity |
| GO:051119 | 9.81E-08 | 34 | sugar transmembrane transporter activity |
| GO:022857 | 1.61E-07 | 107 | transmembrane transporter activity |
| GO:008324 | 1.82E-07 | 64 | cation transmembrane transporter activity |
| GO:005402 | 1.79E-06 | 29 | cation:sugar symporter activity |
| GO:015075 | 5.82E-06 | 70 | ion transmembrane transporter activity |
| GO:015291 | 6.89E-06 | 44 | secondary active transmembrane transporter activity |
| GO:015294 | 6.89E-06 | 34 | solute:cation symporter activity |
| GO:015293 | 6.89E-06 | 34 | symporter activity |
| GO:015295 | 6.89E-06 | 27 | solute:hydrogen symporter activity |
| GO:005351 | 6.89E-06 | 27 | sugar:hydrogen symporter activity |
| GO:022804 | 5.84E-05 | 71 | active transmembrane transporter activity |
| GO:015082 | 6.07E-03 | 13 | di-, tri-valent inorganic cation transmembrane transporter activity |
| GO:046873 | 7.20E-03 | 23 | metal ion transmembrane transporter activity |
| GO:022890 | 9.53E-03 | 28 | inorganic cation transmembrane transporter activity |
| GO:015149 | 1.06E-02 | 8 | hexose transmembrane transporter activity |
| GO:015145 | 1.06E-02 | 8 | monosaccharide transmembrane transporter activity |
| GO:015343 | 2.20E-02 | 5 | siderophore-iron transmembrane transporter activity |
| GO:042927 | 2.20E-02 | 5 | siderophore transporter activity |
| GO:005381 | 2.59E-02 | 8 | iron ion transmembrane transporter activity |
| GO:046915 | 2.59E-02 | 11 | transition metal ion transmembrane transporter activity |
| GO:042879 | 2.59E-02 | 6 | aldonate transmembrane transporter activity |
| GO:015128 | 2.59E-02 | 6 | gluconate transmembrane transporter activity |
| GO:005506 | 3.93E-02 | 16 | iron ion binding |
| GO:046943 | 4.82E-02 | 26 | carboxylic acid transmembrane transporter activity |
| GO:005342 | 4.82E-02 | 26 | organic acid transmembrane transporter activity |
| Cellular component | |||
| GO:016020 | 6.15E-12 | 381 | membrane |
| GO:030257 | 3.05E-10 | 26 | type III protein secretion system complex |
| GO:005886 | 2.84E-08 | 314 | plasma membrane |
| GO:044425 | 5.28E-06 | 129 | membrane part |
| GO:016021 | 5.28E-06 | 124 | integral to membrane |
| GO:031224 | 5.28E-06 | 124 | intrinsic to membrane |
| GO:009279 | 4.20E-05 | 47 | cell outer membrane |
| GO:019867 | 3.67E-04 | 47 | outer membrane |
| GO:043234 | 4.76E-04 | 50 | protein complex |
| GO:009289 | 9.12E-04 | 15 | pilus |
| GO:044462 | 2.45E-03 | 104 | external encapsulating structure part |
| GO:043190 | 3.32E-02 | 4 | ATP-binding cassette (ABC) transporter complex |
In the “molecular function” cluster, “transporter activity” was significantly overrepresented (GO:005215, N = 215), with transmembrane transporters being highly abundant (Table 3) The “cellular component” cluster included membrane-associated functions (GO:016020, N = 381) including again the overrepresented “type III protein secretion system complex” (GO:030257, N = 26) (Table 3). The GO enrichment in the “molecular function” and “cellular component” clusters was similar to that of the “biological processes” cluster.
Summarizing, the transcriptomic analyses of N-15 in co-culture compared to mono-culture revealed responses involved in carbohydrate and metal transport and in extracellular function, mainly secretion of proteins (secretion, cell wall organization, interaction with other organisms).
In the mono-culture, 133 GO categories were significantly enriched of which 101 were assigned to “biological processes”, 6 into “molecular functions” and 26 into “cellular components” (Table 4). Overrepresented categories included “cellular process” in the biological processes cluster (GO:009987; N = 276) and “structural molecule activity” in the molecular function cluster (GO:005198; N = 39). In the cellular components cluster the “intracellular parts” (GO:044424; N = 391), which includes the categories “ribosome” (GO:005840; N = 33) and “flagellum” (GO:019861; N = 19), were overrepresented. GO category 019861 (“flagellum”) includes the flagellar biosynthesis encoding operons flg and fli and cheZ. Overall, GO-categories related to flagella and to cell growth like “cellular processes” and “ribosomes” and were significantly overrepresented in the mono-culture transcriptome of Salmonella N-15.
Table 4.
Gene Ontology (GO) categories of Salmonella Typhimurium N-15 transcriptome significantly overrepresented in the mono-culture compared to co-culture with RBL67
| GO category | p-value | Ngenes in category | Description of category |
|---|---|---|---|
| Biological process | |||
| GO:044249 | 1.69E-20 | 168 | cellular biosynthetic process |
| GO:009058 | 2.27E-18 | 174 | biosynthetic process |
| GO:010467 | 1.16E-11 | 70 | gene expression |
| GO:009987 | 2.05E-11 | 276 | cellular process |
| GO:044237 | 1.12E-10 | 236 | cellular metabolic process |
| GO:006412 | 1.07E-09 | 49 | translation |
| GO:044238 | 2.26E-08 | 227 | primary metabolic process |
| GO:034645 | 5.65E-08 | 74 | cellular macromolecule biosynthetic process |
| GO:009059 | 1.43E-07 | 75 | macromolecule biosynthetic process |
| GO:044267 | 2.58E-07 | 66 | cellular protein metabolic process |
| GO:006633 | 1.86E-06 | 12 | fatty acid biosynthetic process |
| GO:019538 | 4.61E-06 | 82 | protein metabolic process |
| GO:044260 | 5.80E-06 | 118 | cellular macromolecule metabolic process |
| GO:008299 | 5.80E-06 | 12 | isoprenoid biosynthetic process |
| GO:006720 | 5.80E-06 | 12 | isoprenoid metabolic process |
| GO:008610 | 7.04E-06 | 32 | lipid biosynthetic process |
| GO:008152 | 1.73E-05 | 286 | metabolic process |
| GO:044255 | 1.80E-05 | 32 | cellular lipid metabolic process |
| GO:043170 | 1.93E-05 | 138 | macromolecule metabolic process |
| GO:006629 | 2.55E-05 | 34 | lipid metabolic process |
| GO:006631 | 4.06E-05 | 12 | fatty acid metabolic process |
| GO:048870 | 4.72E-04 | 13 | cell motility |
| GO:051674 | 4.72E-04 | 13 | localization of cell |
| GO:001539 | 4.72E-04 | 13 | ciliary or flagellar motility |
| GO:044283 | 6.74E-04 | 62 | small molecule biosynthetic process |
| GO:006928 | 8.07E-04 | 13 | cellular component movement |
| GO:006350 | 9.66E-04 | 9 | transcription |
| GO:043064 | 1.53E-03 | 8 | flagellum organization |
| GO:009141 | 2.48E-03 | 10 | nucleoside triphosphate metabolic process |
| GO:016070 | 2.77E-03 | 30 | RNA metabolic process |
| GO:009108 | 3.33E-03 | 17 | coenzyme biosynthetic process |
| GO:030030 | 3.43E-03 | 8 | cell projection organization |
| GO:009142 | 4.02E-03 | 9 | nucleoside triphosphate biosynthetic process |
| GO:006351 | 5.55E-03 | 7 | transcription, DNA-dependent |
| GO:040011 | 5.89E-03 | 17 | locomotion |
| GO:044281 | 8.06E-03 | 106 | small molecule metabolic process |
| GO:009296 | 8.41E-03 | 6 | flagellum assembly |
| GO:034641 | 9.65E-03 | 117 | cellular nitrogen compound metabolic process |
| GO:006139 | 9.87E-03 | 77 | nucleobase, nucleoside, nucleotide and nucleic acid metabolic process |
| GO:032774 | 9.87E-03 | 7 | RNA biosynthetic process |
| GO:019720 | 9.87E-03 | 7 | Mo-molybdopterin cofactor metabolic process |
| GO:032324 | 9.87E-03 | 7 | molybdopterin cofactor biosynthetic process |
| GO:043545 | 9.87E-03 | 7 | molybdopterin cofactor metabolic process |
| GO:051189 | 9.87E-03 | 7 | prosthetic group metabolic process |
| GO:006777 | 9.87E-03 | 7 | Mo-molybdopterin cofactor biosynthetic process |
| GO:016053 | 1.01E-02 | 31 | organic acid biosynthetic process |
| GO:046394 | 1.01E-02 | 31 | carboxylic acid biosynthetic process |
| GO:009219 | 1.10E-02 | 4 | pyrimidine deoxyribonucleotide metabolic process |
| GO:009394 | 1.10E-02 | 4 | 2′-deoxyribonucleotide metabolic process |
| GO:042180 | 1.26E-02 | 58 | cellular ketone metabolic process |
| GO:030031 | 1.55E-02 | 6 | cell projection assembly |
| GO:006732 | 1.55E-02 | 20 | coenzyme metabolic process |
| GO:042559 | 1.55E-02 | 7 | pteridine and derivative biosynthetic process |
| GO:042558 | 1.55E-02 | 7 | pteridine and derivative metabolic process |
| GO:046034 | 1.55E-02 | 7 | ATP metabolic process |
| GO:015985 | 1.55E-02 | 7 | energy coupled proton transport, down electrochemical gradient |
| GO:015986 | 1.55E-02 | 7 | ATP synthesis coupled proton transport |
| GO:006119 | 1.55E-02 | 7 | oxidative phosphorylation |
| GO:006754 | 1.55E-02 | 7 | ATP biosynthetic process |
| GO:044271 | 1.65E-02 | 52 | cellular nitrogen compound biosynthetic process |
| GO:006950 | 1.72E-02 | 26 | response to stress |
| GO:022607 | 1.93E-02 | 16 | cellular component assembly |
| GO:044085 | 2.28E-02 | 22 | cellular component biogenesis |
| GO:009152 | 2.52E-02 | 10 | purine ribonucleotide biosynthetic process |
| GO:009201 | 2.53E-02 | 7 | ribonucleoside triphosphate biosynthetic process |
| GO:009206 | 2.53E-02 | 7 | purine ribonucleoside triphosphate biosynthetic process |
| GO:009145 | 2.53E-02 | 7 | purine nucleoside triphosphate biosynthetic process |
| GO:006807 | 2.99E-02 | 121 | nitrogen compound metabolic process |
| GO:019748 | 3.10E-02 | 4 | secondary metabolic process |
| GO:009234 | 3.10E-02 | 4 | menaquinone biosynthetic process |
| GO:009233 | 3.10E-02 | 4 | menaquinone metabolic process |
| GO:042362 | 3.10E-02 | 4 | fat-soluble vitamin biosynthetic process |
| GO:042371 | 3.10E-02 | 4 | vitamin K biosynthetic process |
| GO:042373 | 3.10E-02 | 4 | vitamin K metabolic process |
| GO:006775 | 3.10E-02 | 4 | fat-soluble vitamin metabolic process |
| GO:009150 | 3.10E-02 | 10 | purine ribonucleotide metabolic process |
| GO:006164 | 3.35E-02 | 11 | purine nucleotide biosynthetic process |
| GO:009165 | 3.40E-02 | 15 | nucleotide biosynthetic process |
| GO:009205 | 3.58E-02 | 7 | purine ribonucleoside triphosphate metabolic process |
| GO:009199 | 3.58E-02 | 7 | ribonucleoside triphosphate metabolic process |
| GO:009144 | 3.58E-02 | 7 | purine nucleoside triphosphate metabolic process |
| GO:019752 | 3.59E-02 | 53 | carboxylic acid metabolic process |
| GO:043436 | 3.59E-02 | 53 | oxoacid metabolic process |
| GO:009211 | 3.59E-02 | 3 | pyrimidine deoxyribonucleoside triphosphate metabolic process |
| GO:009200 | 3.59E-02 | 3 | deoxyribonucleoside triphosphate metabolic process |
| GO:009120 | 3.59E-02 | 3 | deoxyribonucleoside metabolic process |
| GO:046125 | 3.59E-02 | 3 | pyrimidine deoxyribonucleoside metabolic process |
| GO:009221 | 3.59E-02 | 3 | pyrimidine deoxyribonucleotide biosynthetic process |
| GO:009263 | 3.59E-02 | 3 | deoxyribonucleotide biosynthetic process |
| GO:009265 | 3.59E-02 | 3 | 2′-deoxyribonucleotide biosynthetic process |
| GO:009260 | 3.68E-02 | 10 | ribonucleotide biosynthetic process |
| GO:006163 | 3.85E-02 | 11 | purine nucleotide metabolic process |
| GO:019438 | 3.88E-02 | 12 | aromatic compound biosynthetic process |
| GO:015992 | 3.97E-02 | 8 | proton transport |
| GO:006818 | 3.97E-02 | 8 | hydrogen transport |
| GO:006082 | 4.00E-02 | 54 | organic acid metabolic process |
| GO:090304 | 4.05E-02 | 52 | nucleic acid metabolic process |
| GO:043648 | 4.38E-02 | 9 | dicarboxylic acid metabolic process |
| GO:016043 | 4.38E-02 | 22 | cellular component organization |
| GO:009259 | 4.64E-02 | 10 | ribonucleotide metabolic process |
| GO:032787 | 4.92E-02 | 16 | monocarboxylic acid metabolic process |
| Molecular function | |||
| GO:005198 | 2.25E-10 | 39 | structural molecule activity |
| GO:003735 | 1.05E-08 | 32 | structural constituent of ribosome |
| GO:046983 | 1.50E-02 | 10 | protein dimerization activity |
| GO:003774 | 2.16E-02 | 10 | motor activity |
| GO:016810 | 2.24E-02 | 16 | hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds |
| GO:016814 | 3.39E-02 | 6 | hydrolase activity, acting on carbon-nitrogen (but not peptide) bonds, in cyclic amidines |
| Cellular component | |||
| GO:044424 | 7.14E-22 | 391 | intracellular part |
| GO:005622 | 8.72E-21 | 395 | intracellular |
| GO:005737 | 1.19E-16 | 371 | cytoplasm |
| GO:043228 | 7.58E-16 | 53 | non-membrane-bounded organelle |
| GO:043232 | 7.58E-16 | 53 | intracellular non-membrane-bounded organelle |
| GO:043229 | 7.58E-16 | 57 | intracellular organelle |
| GO:043226 | 7.58E-16 | 57 | organelle |
| GO:005840 | 1.95E-10 | 33 | ribosome |
| GO:030529 | 5.54E-10 | 33 | ribonucleoprotein complex |
| GO:044444 | 1.68E-09 | 41 | cytoplasmic part |
| GO:032991 | 2.06E-07 | 59 | macromolecular complex |
| GO:019861 | 1.39E-06 | 19 | flagellum |
| GO:009288 | 1.47E-05 | 15 | bacterial-type flagellum |
| GO:044422 | 3.09E-04 | 16 | organelle part |
| GO:033279 | 9.66E-04 | 9 | ribosomal subunit |
| GO:016469 | 3.31E-03 | 8 | proton-transporting two-sector ATPase complex |
| GO:042995 | 7.19E-03 | 19 | cell projection |
| GO:044446 | 7.85E-03 | 9 | intracellular organelle part |
| GO:015934 | 8.09E-03 | 5 | large ribosomal subunit |
| GO:009426 | 1.60E-02 | 3 | bacterial-type flagellum basal body, distal rod |
| GO:009424 | 1.60E-02 | 3 | bacterial-type flagellum hook |
| GO:009317 | 1.60E-02 | 3 | acetyl-CoA carboxylase complex |
| GO:044463 | 3.10E-02 | 7 | cell projection part |
| GO:044461 | 3.10E-02 | 7 | bacterial-type flagellum part |
| GO:044460 | 3.10E-02 | 7 | flagellum part |
| GO:030694 | 4.73E-02 | 3 | bacterial-type flagellum basal body, rod |
Effect of RBL67 to the virulence response of N-15
GO enrichment analysis revealed enriched differential expression of some virulence genes, such as the 42 genes belonging to “protein secretion by the type III secretion system” (GO:030254) in the co-culture. Therefore we analyzed the regulation of all putative virulence factors of Salmonella LT2. A comparison to the virulence database VFDB revealed 151 genes in LT2 putatively involved in virulence [39]. Of these 151 genes, one was higher expressed in mono-culture, i.e. phoP encoding the transcriptional regulator PhoP, a member of the two-component system PhoQ-PhoP. The PhoQ encoding gene was also overexpressed in the mono-culture, although not significant (FDR = 0.063, Additional file 1: Table S2).
In the co-culture, 122 virulence genes were higher expressed, a significant enrichment of expressed virulence genes (p = 7 × 10−39 in Fisher’s test). The large majority of genes were involved in secretion systems (N = 66) and fimbrial adherence determinants (N = 51). The pathogeny island 1 (SPI-1) encodes 39 genes [39] of which 30 were significantly higher expressed in the co-culture including the complete type III secretion system 1 (TTSS-1) consisting of sipB, sipD, prgIHK, invACBGH, spaSRQPO, and sicAP. Only avrAI, sprB, hilC, orgC and hilD were not higher expressed in co-culture. Additionally genes located on SPI-2 were higher expressed in co-culture, including the TTSS-2 genes ssrAB, ssaBCDE, ssaGHIJKLMVNOPQRSTU, sseAB, sseCDE, sseFG, sscA, and sscB. Further the main activation complex of type 1 fimbriae fimY, fimW and fimZ where higher expressed in co-culture, albeit the latter not significant.
Discussion
Antagonism and protective effects of selected B. thermophilum strains against enterobacteriaceae have been observed in several studies [7–9, 13, 14], but the underlying mechanisms of this antagonism are unknown. In this study we used RNA-sequencing to investigate the global transcriptional response of RBL67 and Salmonella N-15 in mono- and co-culture. To our knowledge we present the first study investigating the interaction of a probiotic Bifidobacterium strain with enteropathogenic S. Typhimurium using RNA-sequencing.
RNA-sequencing was previously shown to be a powerful method to investigate genome-wide transcript analysis in mixed-culture experiments [35]. In our study we could map at least 10 million reads specifically to one of the genomes, which is clearly above the 5 million reads needed for differential expression analyses in bacterial genomes [41]. The transcriptome of N-15 mapped to the genome of Salmonella Typhimurium LT2 had a similar efficiency as the mapping of the transcriptome of RBL67 to the RBL67 genome, suggesting that mapping reads to a closely related genome is possible. The pathogenicity islands of Salmonella Typhimurium strains are conserved and difference in virulence factors contents mainly occurs on plasmid [42]. We could map RNAseq reads against the plasmid of LT-2, indicating that N-15 has a virulence-genes-encoding plasmid similar to that of LT2. Hence, both strains seem highly similar and the RNAseq data presented resemble closely the transcriptome profile of Salmonella Typhimurium strain N-15. Sampling points were chosen when growth speed, cell number and metabolite concentrations were similar in both cultures to allow accurate transcriptomic profiling. Further, fermentations were performed under pH controlled condition to exclude low pH effects.
RBL67 growth was slightly but significantly enhanced in presence of Salmonella N-15. Also growth of other Bifidobacterium species (B. globosum, B. animalis, B. breve) was shown to be stimulated by S. Typhimurium and S. Enteriditis, albeit under pH uncontrolled conditions [43]. A glutamate producing enzyme was repressed and a glutamate consuming enzyme activated in RBL67 in co-culture, suggesting a change in glutamate availability in the presence of N-15. Interestingly, Salmonella accumulates glutamate under various conditions [44] and lysing Salmonella cells could provide B. thermophilum with additional glutamate resulting in the change in amino acid metabolism and possibly also in the observed growth rate. The elevated expression of 3 stress genes suggests that RBL67 is exposed to weak stress in the presence of Salmonella N-15, but the enhanced growth performance indicates that the microbe was able to cope with the stress in the co-culture.
The Salmonella N-15 transcriptome was clearly affected by presence of RBL67. Many virulence genes were higher expressed in Salmonella N-15 during co-culture with RBL67 and such increased expression may enhance infection rate. However, this would contradict with previous results showing reduced invasion capacity of Salmonella to HT29-MTX cells in presence of RBL67 [13]. Salmonella virulence is tightly controlled and the activity of virulence factors at the right time, correct place and in appropriate amounts is crucial for virulence [45]. Further, environmental factors such as acetate can trigger virulence gene expression in Salmonella [20]. A low concentration of 15 mM acetate at pH 6.7 induces the three invasion determinants hilA, invF and sipC in S. Typhimurium and the induction is dependent on acetate kinase (ackA) and phosphotransacetylase (pta) activity [46]. The genes hilA, and invF were higher expressed in co-culture but ackA and pta were down regulated in co-cultures (Additional file 1: Table S2) and therefore hilA was likely not activated by acetate. The two-component system PhoQ-PhoP was down-regulated in the co-culture. PhoQ-PhoP is a repressor of hilA, a key regulator for Salmonella invasion [18, 21] and the higher expression of hilA observed in the co-culture seems therefore due to a repressor release mediated by PhoQ-PhoP.
Invasion of Salmonella follows sequential expression of first flagellar genes, followed by genes encoded on SPI-1, and eventually type 1 fimbrial genes [47]. Flagellar genes were repressed while genes of SPI-1 and type 1 fimbriae genes were activated in co-culture compared to mono-culture. This shows that N-15 in co-culture is further progressed in the sequential expression for infection and the balance in virulence gene expression is disturbed by the presence of RBL67. In fact, the expression of SPI-1 and SPI-2 and repression of flagellar genes observed in the co-culture resembles the transcriptional profile of Salmonella cells in fibroblast after infection [48]. Further, an early activation of the type III secretion system-1 (TTSS-1) located on SPI-1 was observed in co-culture. A TTSS-1 expressing S. Typhimurium subpopulation is essential for infection, but this subpopulation is also vulnerable to overgrowth by the non-TTSS-1 expressing subpopulation [49]. An imbalance in the regulation of TTSS-1 results in an inappropriate fraction of TTSS-1 expressing cells and eventually to a decreased infection rate [50]. This results in situ in reduced invasion of human intestinal cells and ultimately eliminates Salmonella from the lumen [50]. In vitro, a reduced infection of human intestinal cells by Salmonella in presence of RBL67 and repression of Salmonella by RBL67 in a continuous intestinal fermentation model was reported [13, 14].
Our data provide a first clue on a possible mechanism that could contributes to the antagonistic effects of RBL67 against Salmonella spec and other pathogens [7–9, 13–15]. The expression of virulence gene at early stage is a burden for the pathogen and may result in lower infection rate and subsequent wash-out from the lumen. In addition, the repression of flagellar genes reduces motility thereby preventing colonization of other areas. Whether the imbalance in virulence gene expression observed in vitro also occurs in situ remains to be elucidated. The effect may be reinforced by simultaneous protection by other probiotic mechanisms such as competition for adhesion sites and nutrients, and acetate production.
Conclusion
Our study provides first insights into the transcriptome response of B. thermophilum RBL67 and S. Typhimurium grown in co-cultures under simplified conditions and reveals possible molecular mechanisms of probiotic-pathogen interaction. Our data show that RBL67 has a huge impact on the transcriptome of Salmonella and causes in an imbalanced virulence gene expression. This imbalance in the cascade pathway of virulence could represent a novel possible mechanism of how probiotic organisms can protect the host against infections.
Availability of data and materials
Data presented in this study are available under NCBI BioProject Record PRJNA274782 accessible through http://www.ncbi.nlm.nih.gov/bioproject/PRJNA274782. Gene expression data are directly accessible through GEO Series accession number GSE65716 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65716).
Acknowledgements
We thank Dr. Hubert Rehraurer and Dr. Lucy Poveda from the Functional Genomics Center Zurich for RNA-Sequencing and support in statistical analysis. This project was financed by the Commission for Technology and Innovation (CTI), Bern, Switzerland, under project no. 11962.1 PFLS-LS
Additional file
Salmonella Typhimurium N-15 genes higher expressed in mono-culture. Table S2: Salmonella Typhimurium N-15 genes higher expressed in co-culture with RBL 67. (DOC 2173 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ST, CC, CL and MS designed the research. ST and ER performed experiments. CC, CL, and MS supervised the research. ST and MS analyzed the data. ST, MS and CL wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Sabine A. Tanner, Email: sabine.tanner@hest.ethz.ch
Christophe Chassard, Email: Christophe.Chassard@hest.ethz.ch.
Eugenia Rigozzi, Email: rigozzi.e@gmail.com.
Christophe Lacroix, Email: Christophe.Lacroix@hest.ethz.ch.
Marc J. A. Stevens, Email: marc.stevens@hest.ethz.ch
References
- 1.Hill C, Guarner F, Reid G, Gibson GR, Merenstein DJ, Pot B, Morelli L, Canani RB, Flint HJ, Salminen S, et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat Rev Gastroenterol Hepatol. 2014;11(8):506–14. doi: 10.1038/nrgastro.2014.66. [DOI] [PubMed] [Google Scholar]
- 2.O’Toole PW, Cooney JC. Probiotic bacteria influence the composition and function of the intestinal microbiota. Interdiscip Perspect Infect Dis. 2008;2008:175285. doi: 10.1155/2008/175285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh CJ, Guinane CM, O’Toole PW, Cotter PD. Beneficial modulation of the gut microbiota. FEBS Lett. 2014;17;588(22):4120–30. [DOI] [PubMed]
- 4.Gaggia F, Mattarelli P, Biavati B. Probiotics and prebiotics in animal feeding for safe food production. Int J Food Microbiol. 2010;141(Suppl 1):15–28. doi: 10.1016/j.ijfoodmicro.2010.02.031. [DOI] [PubMed] [Google Scholar]
- 5.Biavati B, Mattarelli P. Genus I. Bifidobacterium. In: Whitman WB, Kämpfer P, Goodfellow M, Garrity GM, Ludwig W, editors. Bergey’s manual of systematic bacteriology The Actinobacteria, vol. 5. 2. New York, USA: Springer Verlag; 2009. pp. 171–206. [Google Scholar]
- 6.Toure R, Kheadr E, Lacroix C, Moroni O, Fliss I. Production of antibacterial substances by bifidobacterial isolates from infant stool active against Listeria monocytogenes. J Appl Microbiol. 2003;95(5):1058–69. doi: 10.1046/j.1365-2672.2003.02085.x. [DOI] [PubMed] [Google Scholar]
- 7.Sasaki T, Fukami S, Namioka S. Enhanced resistance of mice to Escherichia coli infection induced by administration of peptidoglycan derived from Bifidobacterium thermophilum. J Vet Med Sci. 1994;56(3):433–7. doi: 10.1292/jvms.56.433. [DOI] [PubMed] [Google Scholar]
- 8.Sasaki T, Fukami S, Namioka S. Enhancement of cytotoxic activity of lymphocytes in mice by oral administration of peptidoglycan (PG) derived from Bifidobacterium thermophilum. J Vet Med Sci. 1994;56(6):1129–33. doi: 10.1292/jvms.56.1129. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi C, Yokoyama H, Nguyen SV, Hashi T, Kuroki M, Kodama Y. Enhancement of chicken resistance against Escherichia coli infection by oral administration of Bifidobacterium thermophilum preparations. Avian Dis. 2002;46(3):542–6. doi: 10.1637/0005-2086(2002)046[0542:EOCRAE]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 10.Jans C, Lacroix C, Follador R, Stevens MJ. Complete genome sequence of the probiotic Bifidobacterium thermophilum strain RBL67. Genome Announc. 2013;1(3):e00191-13. doi: 10.1128/genomeA.00191-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Ah U. Identification of Bifidobacterium thermophilum RBL67 isolated from baby feces and partial purification of its bacteriocin. PhD thesis. Zurich, Switzerland: ETH Zurich, Switzerland; 2006.
- 12.von Ah U, Mozzetti V, Lacroix C, Kheadr EE, Fliss I, Meile L. Classification of a moderately oxygen-tolerant isolate from baby faeces as Bifidobacterium thermophilum. BMC Microbiol. 2007;7:79. doi: 10.1186/1471-2180-7-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zihler A, Gagnon M, Chassard C, Lacroix C. Protective effect of probiotics on Salmonella infectivity assessed with combined in vitro gut fermentation-cellular models. BMC Microbiol. 2011;11:264. doi: 10.1186/1471-2180-11-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zihler A, Le Blay G, Chassard C, Braegger C, Lacroix C. Bifidobacterium thermophilum RBL67 inhibits S. Typhimurium in an in vitro model of Salmonella infection in children. J Food Nutr Disord. 2014, in press.
- 15.Gagnon M. Rôle des probiotiques lors d’infections entériques d’origine bactérienne et virale: analyses in vitro et études in vivo chez des modèles murines. PhD thesis. Québec: Université de Laval; 2007.
- 16.Moroni O, Kheadr E, Boutin Y, Lacroix C, Fliss I. Inactivation of adhesion and invasion of food-borne Listeria monocytogenes by bacteriocin-producing Bifidobacterium strains of human origin. Appl Environ Microbiol. 2006;72(11):6894–901. doi: 10.1128/AEM.00928-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Majowicz SE, Musto J, Scallan E, Angulo FJ, Kirk M, O’Brien SJ, Jones TF, Fazil A, Hoekstra RM, International Collaboration on Enteric Disease ‘Burden of Illness’ Studies The global burden of nontyphoidal Salmonella gastroenteritis. Clin Infect Dis. 2010;50(6):882–9. doi: 10.1086/650733. [DOI] [PubMed] [Google Scholar]
- 18.Fabrega A, Vila J. Salmonella enterica serovar Typhimurium skills to succeed in the host: virulence and regulation. Clin Microbiol Rev. 2013;26(2):308–41. doi: 10.1128/CMR.00066-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.EFSA. ECDC The European Union summary report on trends and sources of zoonoses, zoonotic agents and food-borne outbreaks in 2012. EFSA Journal. 2014;12(2):3547–859. doi: 10.2903/j.efsa.2018.5500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altier C. Genetic and environmental control of Salmonella invasion. J Microbiol. 2005;43 Spec No:85–92. [PubMed] [Google Scholar]
- 21.Ellermeier JR, Slauch JM. Adaptation to the host environment: regulation of the SPI1 type III secretion system in Salmonella enterica serovar Typhimurium. Curr Opin Microbiol. 2007;10(1):24–9. doi: 10.1016/j.mib.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Lucas RL, Lee CA. Unravelling the mysteries of virulence gene regulation in Salmonella typhimurium. Mol Microbiol. 2000;36(5):1024–33. doi: 10.1046/j.1365-2958.2000.01961.x. [DOI] [PubMed] [Google Scholar]
- 23.de Keersmaecker SC, Marchal K, Verhoeven TL, Engelen K, Vanderleyden J, Detweiler CS. Microarray analysis and motif detection reveal new targets of the Salmonella enterica serovar Typhimurium HilA regulatory protein, including hilA itself. J Bacteriol. 2005;187(13):4381–91. doi: 10.1128/JB.187.13.4381-4391.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gong J, Yang CB. Advances in the methods for studying gut microbiota and their relevance to the research of dietary fiber functions. Food Res Int. 2012;48(2):916–29. doi: 10.1016/j.foodres.2011.12.027. [DOI] [Google Scholar]
- 25.Duncan SH, Hold GL, Barcenilla A, Stewart CS, Flint HJ. Roseburia intestinalis sp. nov., a novel saccharolytic, butyrate-producing bacterium from human faeces. Int J Syst Evol Microbiol. 2002;52(Pt 5):1615–20. doi: 10.1099/00207713-52-5-1615. [DOI] [PubMed] [Google Scholar]
- 26.Tanner SA, Zihler Berner A, Rigozzi E, Grattepanche F, Chassard C, Lacroix C. In vitro continuous fermentation model (PolyFermS) of the swine proximal colon for simultaneous testing on the same gut microbiota. PLoS One. 2014;9(4) doi: 10.1371/journal.pone.0094123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rada V, Sirotek K, Petr J. Evaluation of selective media for bifidobacteria in poultry and rabbit caecal samples. J Veterinary Med Ser B. 1999;46(6):369–73. doi: 10.1046/j.1439-0450.1999.00241.x. [DOI] [PubMed] [Google Scholar]
- 28.Stevens MJ, Wiersma A, de Vos WM, Kuipers OP, Smid EJ, Molenaar D, Kleerebezem M. Improvement of Lactobacillus plantarum aerobic growth as directed by comprehensive transcriptome analysis. Appl Environ Microbiol. 2008;74(15):4776–8. doi: 10.1128/AEM.00136-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Winkler ME. Ribosomal ribonucleic acid isolated from Salmonella typhimurium: absence of the intact 23S species. J Bacteriol. 1979;139(3):842–9. doi: 10.1128/jb.139.3.842-849.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith NH, Crichton PB, Old DC, Higgins CF. Ribosomal-RNA patterns of Escherichia coli, Salmonella typhimurium and related Enterobacteriaceae. J Med Microbiol. 1988;26(3):223–8. doi: 10.1099/00222615-26-3-223. [DOI] [PubMed] [Google Scholar]
- 31.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40(10):4288–97. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robinson MD, Smyth GK. Moderated statistical tests for assessing differences in tag abundance. Bioinformatics. 2007;23(21):2881–7. doi: 10.1093/bioinformatics/btm453. [DOI] [PubMed] [Google Scholar]
- 33.Robinson MD, Smyth GK. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics. 2008;9(2):321–32. doi: 10.1093/biostatistics/kxm030. [DOI] [PubMed] [Google Scholar]
- 34.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenthal AZ, Matson EG, Eldar A, Leadbetter JR. RNA-seq reveals cooperative metabolic interactions between two termite-gut spirochete species in co-culture. ISME J. 2011;5(7):1133–42. doi: 10.1038/ismej.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 37.Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21(16):3448–9. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- 38.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen L, Xiong Z, Sun L, Yang J, Jin Q. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 2012;40(Database issue):D641–5. doi: 10.1093/nar/gkr989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.next-generation-sequencing-guide (2015). Available at https://genohub.com/next-generation-sequencing-guide/. Accessed 15 Oct 2015.
- 42.Dhanani AS, Block G, Dewar K, Forgetta V, Topp E, Beiko RG, Diarra MS. Genomic comparison of non-typhoidal Salmonella enterica Serovars Typhimurium, Enteritidis, Heidelberg, Hadar and Kentucky isolates from broiler chickens. PLoS One. 2015;10(6) doi: 10.1371/journal.pone.0128773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bielecka M, Biedrzycka E, Smoragiewicz W, Smieszek M. Interaction of Bifidobacterium and Salmonella during associated growth. Int J Food Microbiol. 1998;45(2):151–5. doi: 10.1016/S0168-1605(98)00150-0. [DOI] [PubMed] [Google Scholar]
- 44.Yan D, Ikeda TP, Shauger AE, Kustu S. Glutamate is required to maintain the steady-state potassium pool in Salmonella typhimurium. Proc Natl Acad Sci U S A. 1996;93(13):6527–31. doi: 10.1073/pnas.93.13.6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kato A, Groisman EA. The PhoQ/PhoP regulatory network of Salmonella enterica. In: Utsumi R, editor. Bacterial signal transduction: networks and drug targets. New York, USA: Springer Science + Business Media, LLC Landes Bioscience; 2008. [Google Scholar]
- 46.Lawhon SD, Maurer R, Suyemoto M, Altier C. Intestinal short-chain fatty acids alter Salmonella typhimurium invasion gene expression and virulence through BarA/SirA. Mol Microbiol. 2002;46(5):1451–64. doi: 10.1046/j.1365-2958.2002.03268.x. [DOI] [PubMed] [Google Scholar]
- 47.Saini S, Slauch JM, Aldridge PD, Rao CV. Role of cross talk in regulating the dynamic expression of the flagellar Salmonella pathogenicity island 1 and type 1 fimbrial genes. J Bacteriol. 2010;192(21):5767–77. doi: 10.1128/JB.00624-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nunez-Hernandez C, Tierrez A, Ortega AD, Pucciarelli MG, Godoy M, Eisman B, Casadesus J, Garcia-del Portillo F. Genome expression analysis of nonproliferating intracellular Salmonella enterica serovar Typhimurium unravels an acid pH-dependent PhoP-PhoQ response essential for dormancy. Infect Immun. 2013;81(1):154–65. doi: 10.1128/IAI.01080-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sturm A, Heinemann M, Arnoldini M, Benecke A, Ackermann M, Benz M, Dormann J, Hardt WD. The cost of virulence: retarded growth of Salmonella Typhimurium cells expressing type III secretion system 1. PLoS Pathog. 2011;7(7) doi: 10.1371/journal.ppat.1002143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Diard M, Garcia V, Maier L, Remus-Emsermann MN, Regoes RR, Ackermann M, Hardt WD. Stabilization of cooperative virulence by the expression of an avirulent phenotype. Nature. 2013;494(7437):353–6. doi: 10.1038/nature11913. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data presented in this study are available under NCBI BioProject Record PRJNA274782 accessible through http://www.ncbi.nlm.nih.gov/bioproject/PRJNA274782. Gene expression data are directly accessible through GEO Series accession number GSE65716 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65716).