

Abstract
Telomerase consists of two essential components, the telomerase RNA template (TR) and telomerase reverse transcriptase (TERT). The haplo-insufficiency of TR was recently shown to cause one form of human dyskeratosis congenita, an inherited disease marked by abnormal telomere shortening. Consistent with this finding, we recently reported that mice heterozygous for inactivation of mouse TR exhibit a similar haplo-insufficiency and are deficient in the ability to elongate telomeres in vivo. To further assess the genetic regulation of telomerase activity, we have compared the abilities of TR-deficient and TERT-deficient mice to maintain or elongate telomeres in interspecies crosses. Homozygous TERT knockout mice had no telomerase activity and failed to maintain telomere length. In contrast, TERT+/− heterozygotes had no detectable defect in telomere elongation compared to wild-type controls, whereas TR+/− heterozygotes were deficient in telomere elongation. Levels of TERT mRNA in heterozygous mice were one-third to one-half the levels expressed in wild-type mice, similar to the reductions in telomerase RNA observed in TR heterozygotes. These findings indicate that both TR and TERT are essential for telomere maintenance and elongation but that gene copy number and transcriptional regulation of TR, but not TERT, are limiting for telomerase activity under the in vivo conditions analyzed.
The replicative capacity of eukaryotic cells is potentially limited by the fact that, in the absence of compensatory mechanisms, chromosomal synthesis is incomplete, resulting in a loss of terminal chromosomal nucleotides with each cell division. The termini of linear eukaryotic chromosomes consist of telomeres [structures made up of terminal (TTAGGG)n hexanucleotide repeats in mammalian chromosomes] as well as associated proteins (2, 3, 9). When, as a result of successive cell divisions, telomeres shorten to a critical length, telomere function is compromised, leading to replicative senescence or cell death through pathways that are incompletely understood (2, 15, 17, 22, 29). The best-characterized mechanism capable of compensating for telomere loss is the synthetic elongation of telomeric termini by the enzyme telomerase. Telomerase consists of two essential molecular components, the telomerase RNA (TR) component, which includes a template for telomeric DNA, and telomerase reverse transcriptase (TERT), the RT that mediates telomere synthesis (2, 3, 7, 15, 22). In vitro and in vivo studies have demonstrated that both TR and TERT are essential to telomerase enzymatic activity and that, in the absence of either of these components, progressive telomere shortening and dysfunction ensue, eventually compromising cell division and cell survival (2, 3, 15, 16). The critical in vivo role of telomerase has been most dramatically demonstrated in seminal studies of TR-deficient mice, in which the absence of TR expression and telomerase activity results in progressive telomere shortening over sequential generations of mice, leading to sterility as a result of germ cell abnormalities, and phenotypic consequences, including reduced life span and multiple organ- and tissue-specific changes (5, 21, 27). However, it is not clear how TR and TERT expression are regulated in vivo and how normal or altered expression of the genes encoding these telomerase components contributes to normal or pathological states.
It has been reported that TR is expressed in vivo and in vitro in a relatively ubiquitous and constitutive manner, although notable exceptions have been observed (4, 11). In contrast, it has been suggested that TERT is under more rigorous transcriptional regulation and is expressed by tumor cells and stem cell populations but not by most normal somatic cells, although again there have been significant exceptions to this generalization (11, 34). The hypothesis that TERT expression is limiting for telomerase enzymatic activity is supported by studies in which transfection with human TERT has led to the induction of measurable telomerase activity, stabilization of telomere length, and effective immortalization of fibroblasts, lymphocytes, and other cell types in vitro (33, 36). It remains unclear, however, whether the expression of TR or TERT limits telomerase function during in vivo development and function and whether genetic alterations in the expression of either or both of these telomerase genes will have in vivo consequences.
It was recently demonstrated that an autosomally inherited form of dyskeratosis congenita (DKC) is caused by mutation in the human TR (hTR) gene (30-32). DKC is a pleomorphic disease characterized by bone marrow failure as well as alterations in skin and other organ systems, and it was found that patients with this syndrome have abnormally short telomeres; these observations collectively provide the most direct evidence to date that an alteration in telomerase can cause human disease. A striking feature of this form of DKC is that it is inherited as an autosomal dominant, suggesting that mutations create a dominant negative hTR product or that haplo-insufficiency is responsible for the disease. The fact that at least some mutations characterized in DKC appear to involve deletions and the failure to express a product from the mutated allele suggests that haplo-insufficiency, caused by the inactivation of one copy of the hTR gene, might be responsible for the phenotype (31, 32). In another experimental approach, the function of TR was assessed in a mouse model in which crosses between two mouse species with markedly different telomere lengths result in elongation of the shorter telomeres during in vivo development (18). It was found that this telomere elongation is telomerase dependent and did not occur in mouse TR (mTR)-deficient knockout mice. Moreover, elongation was deficient in mice heterozygous for the mTR knockout, demonstrating a haplo-insufficient phenotype. Levels of TR in heterozygous mice were reduced to levels one-third to one-half of those in wild-type mice (18). Collectively, these results indicate that gene copy number and expression of TR may be functionally limiting in vivo, with consequences for normal biology and for pathogenic pathways.
To compare the consequences of reduced expression and copy number of the TERT gene with the observed consequences of TR deficiency, we have generated TERT-deficient mice and compared heterozygous and homozygous knockouts for each of the telomerase components. Homozygous knockouts for either mTR or mTERT were unable to elongate telomeres in interspecies crosses, in contrast to wild-type mice. Mice that were heterozygous for mTERT deficiency had reduced levels of mTERT mRNA, which were one-third to one-half of those measured in wild-type mice. However, in contrast to the haplo-insufficiency found in mTR heterozygotes, mice that were heterozygous for mTERT were fully competent for telomere elongation. Thus, in this in vivo model of telomerase function, TR gene copy number and transcription appear to be limiting, whereas TERT does not. These observations are relevant to potential pathways for telomerase dysfunction in human disease as well as in experimental model systems.
MATERIALS AND METHODS
Construction of the targeting vector.
mTERT genomic DNA used for these studies was a 6-kb fragment derived from strain 129 and containing exon 1 and part of exon 2 of the mTERT gene. The DNA fragment was inserted into the pBluescript-KS vector at the EcoRI site (KS-tert). A BamHI-XbaI fragment was cut from KS-tert and subcloned into the pBluescript-KS vector (KS-BX). In KS-BX, a unique enzyme restriction site (XhoI) was created by PCR proximally to an ATG that corresponds to the translation initiation code of mTERT mRNA. Green fluorescent protein (GFP) and neomycin resistance genes were then inserted into the vector at the XhoI site. The resulting BamHI-XbaI DNA fragment, containing GFP and neomycin resistance genes, was used to replace the BamHI-XbaI fragment of KS-tert. The thymidine kinase gene was inserted at a NotI site that was outside of the genomic DNA fragment. The final construct was used as an mTERT gene-targeting construct.
Gene targeting in ES cells and production of mTERT knockout mice.
The mTERT-targeting construct was linearized with SalI and electroporated into embryonic stem (ES) cells. G418-resistant colonies were selected and isolated as previously described (8). Colonies of ES cells with homologous recombination events were identified by Southern blot analysis with a BamHI-XbaI fragment from KS-tert as a probe. Mutant mice were generated by standard protocol with the selected ES clones. Southern blotting, reverse transcription-PCR, and telomerase activity assays were used to confirm the disruption of the mTERT gene, as described below.
Mice.
mTERT heterozygotes were maintained in a Mus musculus C57BL/6 (B6) background. Mice used in the present study had been backcrossed with B6 mice for at least three generations. Mus spretus (SPRET/Ei) mice were originally obtained from the Jackson Laboratory (Bar Harbor, Maine). All animals were housed at Bioqual (Rockville, Md.).
Identification of mTERT-deficient mice by PCR.
mTERT genotypes were identified by PCR. Tail DNA was purified with a DNeasy tissue kit (QIAGEN, Valencia, Calif.). PCR primers were mTERT-TR (5′-CCC CAG GCG CCG CAC AAA GG-3′), mTERT-TF (5′GGT CCT GGC TGT TTT CTA AG-3′), and NEON2 (5′-CTG GAT TCA TCG ACT GTG GC-3′). PCRs were carried out in 50 μl of a PCR mix containing 25 μl of PCR buffer (Supermix PCR kit; Life Technologies, Rockville, Md.), 1 to 100 ng of tail DNA, and 0.2 μM concentrations of the primers. PCRs were carried out at 95°C for 30 s, 58°C for 30 s, and 72°C for 1 min for 35 cycles. The wild-type genomic mTERT DNA (150-bp) PCR product was visualized by agarose gel electrophoresis with ethidium bromide staining when mTERT-TR and mTERT-TF primer pairs were used. The knockout mTERT DNA (420-bp) PCR product was visualized when mTERT-TR and NEON2 pairs were used.
Reverse transcription-PCR analysis for mTERT-deficient mice.
Spleen lymphocytes were stimulated with concanavalin A (ConA; 5 μg/ml), lipopolysaccharide (LPS; 15 μg/ml), and recombinant interleukin-2 (rIL-2; 150 IU/ml) for 48 h (18, 20). A NucleoSpin kit (BD Biosciences) was used to isolate total RNA from spleen cells for reverse transcription-PCR analysis. Two primer pairs were used to detect mTERT expression. For the detection of 5′ mTERT cDNA, mTERT658F (5′-AGA TCA AGA GCA GTA GTC GCC AG-3′), starting at nucleotide 658 of the mTERT cDNA sequence, and mTERT950R (5′-TTT ACA GCA CAC CGA CCC AGA G-3′), ending at nucleotide 950 of the mTERT cDNA sequence, were used. For the detection of 3′ mTERT cDNA, mTERT2646F (5′-GCA AAA ACC TTC TCA GCA CC-3′), starting at nucleotide 2646 of the mTERT cDNA sequence, and mTERT2952R (5′-ACT TCA ACC GCA AGA CCG AC-3′), ending at nucleotide 2952 of the mTERT cDNA sequence, were used. Reverse transcription-PCRs were carried out in 50 μl of a PCR mix containing 25 μl of PCR buffer (One-Step reverse transcription-PCR kit; Life Technologies), 1 μg of total RNA, and 0.2 nM concentrations of the primers. Reverse transcription-PCR was carried out at 42°C for 30 min and then with 35 cycles of 95°C for 30 s, 58°C for 30 s, and 72°C for 1 min. Reverse transcription-PCR products were visualized by agarose gel electrophoresis with ethidium bromide staining. The 5′-mTERT cDNA (292-bp) PCR product was visualized when the primer pair mTERT658F and mTERT950R was used. The 3′-mTERT cDNA (306-bp) PCR product was visualized when mTERT2646F and mTERT2952R were used.
Identification of mTERT-deficient mice by Southern blot analysis.
Southern blot analysis was performed by a standard protocol as described previously (10). Mouse tail genomic DNA from wild-type or mutant mTERT mice was digested with BamHI or HindIII, electrophoresed on 0.7% agarose gel, and transferred onto nylon filters. The filters were hybridized with 32P-labeled probe in Hyb buffer (Stratagene) at 65°C for 4 to 8 h. The probe was cut from KS-BX with BamHI and XbaI and labeled with [α-32P]dCTP with a Room Temperature random primer kit (Stratagene). Filters were exposed after three washes with 0.2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate)-0.2% sodium dodecyl sulfate for 10 min at 60°C.
Pulsed-field gel electrophoresis and in-gel hybridization for telomere length determination.
Mouse spleen cells were prepared and embedded in agarose plugs as described previously (18, 37). DNA plugs were digested with HinfI and RsaI and electrophoresed in a 1% agarose gel at 14°C with a pulsed-field apparatus. Gels were dried and hybridized with 32P-labeled (CCCTAA)4 oligonucleotides. Telomere repeats were revealed by exposing gels to X-ray film or by phosphorimaging.
Q-FISH.
Spleen lymphocytes were stimulated with ConA (5 μg/ml), LPS (15 μg/ml), and rIL-2 (150 IU/ml) for 48 h (18, 20). Activated cells were treated with Colcemid and fixed with 3:1 methanol-acetic acid. Metaphase cells were dropped on slides that were then hybridized with Cy3-labeled (CCCTAA)3 oligonucleotides, and chromosomes were stained with DAPI (4′,6′-diamidino-2-phenylindole). Fluorescent signals were collected and analyzed as previously described (18, 20). By using FluoreSphere beads (Molecular Probes) as standards, fluorescent-signal intensities were calculated with quantitative fluorescence in situ hybridization (Q-FISH) software from the Lansdorp laboratory (14). At least 10 metaphases and 800 telomeres from each mTR mutant genotype and at least 20 metaphases and 1,600 telomeres from each mTERT mutant genotype were used to collect telomere fluorescent-signal data.
Telomerase activity assay.
Telomerase activity was measured with the TRAPeze telomerase detection kit (Chemicon) (19). Testis cells or spleen lymphocytes stimulated with ConA (5 μg/ml), LPS (15 μg/ml), and rIL-2 (150 IU/ml) for 48 h were lysed with CHAPS {3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate} buffer. PCR products were separated by electrophoresis on 12.5% nondenaturing polyacrylamide gels and visualized by SYBR Green staining.
Real-time PCR.
Total RNA was isolated as described above. A standard curve for mTERT was generated in each experiment by titrating purified plasmid containing mTERT cDNA. A standard curve for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was similarly generated from total B6 mouse spleen cell RNA. Real-time PCR was performed with a Superscript One-Step reverse transcription-PCR kit (Invitrogen) and a 7900HT sequence detection system (Applied Biosystems, Foster City, Calif.). Primers used for real-time PCR of mTERT were mTERT 5′-TGG GTC TCC CCT GTA CCA AAT-3′ and 5′-GGC CTG TAA CTA GCG GAC ACA-3′. Primers for real-time PCR of GAPDH were GAPDH 5′-TTC ACC ACC ATG GAG AAG GC-3′ and 5′-GGC ATG GAC TGT GGT CAT GA-3′. The fluorogenic probe for mTERT was FAM-5′-TGC CAC CAC GGA TAT CTG GCC CT-3′-TAMRA (where FAM is 6-carboxyfluorescein and TAMRA is 6-carboxytetramethylrhodamine). The fluorogenic probe for mGAPDH was FAM-5′-TGC ATC CTG CAC CAC CAA CTG CTT AG-3′-TAMRA.
Generation and genotyping of transgenic mTERT mice.
The mTERT construct used to generate transgenic mice was isolated from a mouse testis cDNA library by PCR. Briefly, a 3.4-kb mTERT cDNA fragment was isolated by NotI digestion. The 5′ end was blunted and cloned 3′ downstream of 1.2 kb of the human ubiquitin C promoter (28). The 3′ end of this construct contains 150 bp from human c-jun and simian virus 40 splice and poly(A) sequences. The entire transgene construct (5.5 kb) was released by NdeI-EcoRI digestion and microinjected into C57BL/6 mouse oocytes. Four different founder lines were identified by Southern blotting using a KpnI-NotI fragment of mTERT as a probe, and the experiments described here were carried out using two of the founder lines. Mice expressing the transgene were subsequently identified with a PCR specific for the TERT transgene. Tail DNA was purified with a DNeasy tissue kit (QIAGEN) and analyzed by PCR using the following transgenic mTERT-specific primers: WH33 (5′-CCC CAG GCG CCG CAC AAA GG-3′) and WH2953 (5′-GGT CCT GGC TGT TTT CTA AG-3′). PCRs were carried out in 50 μl of a PCR mix containing 5 μl of 10× PCR buffer (Applied Biosystems), 0.1 mM concentrations of the four combined deoxynucleoside triphosphates (Roche), 0.4 μM concentrations of the primers, 0.5 U of AmpliTaq (Applied Biosystems), and 1 to 100 ng of tail DNA. The PCR was carried out at 95°C for 10 min, followed by 30 cycles of 94°C for 30 s, 60°C for 1 min, and 72°C for 1 min. The reaction was completed by incubation at 72°C for 10 min. This PCR amplifies a single band of 561 bp from transgenic mTERT-containing mice; DNA prepared from mice that lack the transgene yields no amplified band. The expression of a functional transgenic mTERT product was confirmed by the ability of the transgene to reconstitute in vitro telomerase activity in mTERT −/− mice (data not shown).
RESULTS
Generation of mTERT-deficient mice.
mTERT-deficient mice were constructed as shown in Fig. 1A. A gene-targeting vector was used to transfect mouse ES cells. Three of 148 G418-resistant ES cell clones were identified as homologous recombination positive, and two of these clones were used to generate mTERT knockout mice. In the targeted allele, GFP and neomycin genes were inserted into the first exon of the mTERT gene, disrupting mTERT expression (Fig. 1A). mTERT-deficient mice were identified by using Southern blot analysis, genomic PCR, reverse transcription-PCR, and assays of telomerase activity. For Southern blot analysis, genomic DNA was digested with BamHI, electrophoresed, and hybridized with probe A (Fig. 1A). Figure 1B shows the results of a representative Southern blot experiment in which wild-type mouse DNA generated a single 7-kb band, homozygous mTERT knockout mouse DNA generated a single 9.3-kb band, and heterozygous mTERT mouse DNA generated one band of each length. Genotypes were also confirmed by PCR (Fig. 1C). mTERT RNA expression in activated spleen lymphocytes from mTERT wild-type and putative mTERT knockout mice was assessed by reverse transcription-PCR using primer pairs that are specific for both the 5′ and 3′ ends of mTERT. As assessed by either primer set, mTERT knockout mice did not express detectable mTERT mRNA, while heterozygous and wild-type mice expressed easily detectable mRNA (Fig. 1D). Similar levels of telomerase activity in extracts from testis cells of mTERT heterozygous and wild-type mice were detected by a telomeric repeat amplification (TRAP) assay (Fig. 1E), while in contrast, no telomerase activity was detected in testis cell extracts of mTERT knockout mice (Fig. 1E) or in extracts of mTERT knockout spleen lymphocytes stimulated with LPS, ConA, and rIL-2 (data not shown).
FIG. 1.

Generation of mTERT−/− mice by gene targeting. +/+, mTERT+/+ mice; +/−, mTERT+/− mice; −/−, mTERT−/− mice. (A) Gene targeting strategy and restriction map of the mTERT gene. The black boxes represent the exons of the mTERT gene. GFP and neomycin genes were inserted into exon 1 5′ of the translation initiation codon ATG. E, EcoRI; X, XbaI; EX1, exon 1; EX2, exon 2; B, BamHI; H, HindIII; TK, thymidine kinase. (B) Southern blot analysis of mouse tail DNA. The 7- and 9.3-kb bands represent the germ line and target allele, respectively. (C) PCR genotype of mTERT knockout mice. The wild-type band is 150 bp; the knockout band of an mTERT knockout mouse is 420 bp. (D) Reverse transcription-PCR analysis of mTERT gene expression. 5′mTERT represents the reverse transcription-PCR product from the mTERT658F and mTERT950R primers. 3′mTERT represents the reverse transcription-PCR product from the mTERT2646F and mTERT2952R primers. (E) TRAP assay for telomerase activity. Cell lysates were prepared from testes of mTERT+/+ (wild-type [WT]), mTERT+/−, and mTERT−/− (knockout [KO]) mice. Telomerase activity is shown for each type of mouse for (from left to right) 90, 30, 10, and 3.3 ng of lysate. ▵, serial dilutions of cell lysates made before TRAP analysis; ○, protein samples heated to 85°C for 10 min before TRAP assay; IS, internal standard.
Telomere elongation is deficient in mTERT−/− and mTR−/− mice.
The role of telomerase function in in vivo telomere elongation was assessed in interspecies crosses between M. musculus (B6) and M. spretus (SPRET/Ei) mice. Analysis of telomere length by pulsed-field gel electrophoresis demonstrated that telomeres from SPRET/Ei mice were substantially shorter than those from B6 mice, and as previously described, telomeres from (B6 × SPRET/Ei)F1 mice, expressing wild-type mTR and mTERT, had a bimodal distribution of telomere length (Fig. 2). Notably, the short telomeres from these F1 mice were substantially longer than those from parental SPRET/Ei mice, indicating that telomere elongation had occurred during development of these mice, as previously reported (18).
FIG. 2.
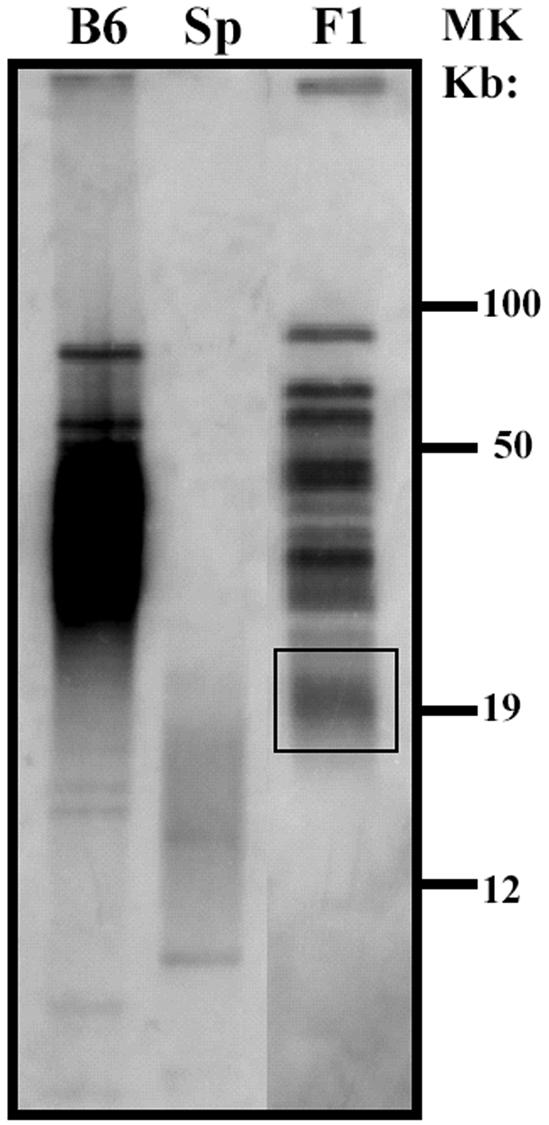
SPRET/Ei telomeres undergo elongation in (B6 × SPRET/Ei)F1 mice. Telomeric repeat fragment distribution was analyzed by pulsed-field gel electrophoresis and in-gel hybridization of telomeric DNA from B6, SPRET/Ei (Sp), and (B6 × SPRET/Ei)F1 (F1) mice. The boxed area represents elongated telomeres of parental SPRET/Ei origin. MK, molecular size marker.
To compare the functions of mTERT and mTR in telomere maintenance and elongation, mTERT+/− B6 mice werecrossed with wild-type SPRET/Ei mice, generating mTERT+/− and mTERT+/+ (B6 × SPRET/Ei)F1 offspring. Female mTERT+/− (B6 × SPRET/Ei)F1 mice were backcrossed with male mTERT+/− B6 mice to generate mTERT+/+, mTERT+/−, and mTERT−/− mice [(B6 × SPRET/Ei) × B6]. In parallel, mTR-deficient mice were bred to generate mTR+/− and mTR+/+ (B6 × SPRET/Ei)F1 offspring and mTR+/+, mTR+/−, and mTR−/− [(B6 × SPRET/Ei) × B6] offspring. In all experiments, comparisons were made between or among mice which differed in mTERT or mTR genotype but which were derived from the same breeding cohorts to control for effects of other genetic heterogeneities.
To first determine whether expression of both mTERT and mTR is essential for telomere elongation in interspecies mice, telomere lengths of spleen lymphocytes from mTERT+/+ and mTERT−/− [(B6 × SPRET/Ei) × B6] mice and from mTR+/+ and mTR−/− [(B6 × SPRET/Ei) × B6] mice were assayed. Q-FISH analysis demonstrated that, in mTERT+/+ [(B6 × SPRET/Ei) × B6] mice (Fig. 3B), the shortest telomeres were substantially longer than those from SPRET/Ei mice (Fig. 3A), indicating that telomere elongation had occurred during the development of these mice, as previously reported (18, 37). In contrast, short telomeres of putative SPRET/Ei origin were not elongated in mTERT−/− mice (Fig. 3C). As previously reported, short SPRET/Ei telomeres were similarly elongated in mTR+/+ mice (Fig. 3D) but not in mTR−/− mice (Fig. 3E) (18). Comparable results were observed from pulsed-field gel analysis of telomere length (data not shown). These findings indicate that expression of both mTERT and mTR is essential for telomere maintenance and elongation.
FIG. 3.

Telomere maintenance and elongation in [(B6 × SPRET/Ei) × B6] mice is dependent upon the expression of both mTR and mTERT. Q-FISH frequency distributions of telomere signals in SPRET/Ei mice (A), mTERT+/+ mice [(B6 × SPRET/Ei) × B6] (B), mTERT−/− mice [(B6 × SPRET/Ei) × B6] (C), mTR+/+ mice [(B6 × SPRET/Ei) × B6] (D), and mTR−/− mice [(B6 × SPRET/Ei) × B6] (E) are shown. All Q-FISH data represent the analysis of at least 10 metaphases and 800 telomeres for mTR mutant mice and 20 metaphases and 1,600 telomeres for mTERT mutant mice.
Telomere elongation is deficient in mTR+/− mice but not in mTERT+/− mice.
The measurement of telomere elongation in interspecies crosses provided an opportunity to further analyze whether quantitative levels of mTERT or mTR expression are limiting in this in vivo assay of telomerase function. To this end, telomere elongation was analyzed in mice that were heterozygous for inactivation of either mTR or mTERT. The shorter set of telomeres in mTR+/− (B6 × SPRET/Ei)F1 mice was substantially smaller than that of wild-type (B6 × SPRET/Ei)F1 mice, as measured by Q-FISH (Fig. 4A and B), consistent with results from a previous report and indicating that the expression of mTR is limiting for telomere elongation in vivo (18). In contrast, there was no detectable difference in the lengths of telomeres in mTERT+/+ and mTERT+/− (B6 × SPRET/Ei)F1 mice when they were assayed by Q-FISH (Fig. 4C and D), with mice of both genotypes exhibiting similar elongations of short, presumably SPRET/Ei, telomeres. Comparable results were again observed in pulsed-field gel analysis of telomere length (data not shown). Thus, the expression of mTR, but not mTERT, limits telomerase-dependent maintenance of telomere length under these in vivo conditions.
FIG. 4.

Telomere elongation is deficient in mTR+/− but not in mTERT+/− (B6 × SPRET/Ei)F1 mice. Q-FISH frequency distributions of telomere signals from mTR+/+ mice (B6 × SPRET/Ei)F1 (A), mTR+/− mice (B6 × SPRET/Ei)F1 (B), mTERT+/+ mice (B6 × SPRET/Ei)F1 (C), and mTERT+/− mice (B6 × SPRET/Ei)F1 (D) are shown. All Q-FISH data represent the analysis of at least 10 metaphases and 800 telomeres for mTR mutant mice and 20 metaphases and 1,600 telomeres for mTERT mutant mice.
TERT mRNA expression is reduced in mTERT+/− mice.
It has been reported that the defect in telomere elongation observed in mTR+/− mice is associated with decreased expression of mTR RNA, providing an explanation for the haplo-insufficiency in these mice (18). Because mTERT+/− mice did not exhibit a defect in telomere elongation, it was important to determine whether mTERT mRNA expression is altered in these heterozygous mice. When RNA from mTERT+/+ and mTERT+/− mouse testes was quantitated by real-time reverse transcription-PCR, it was found that mTERT+/+ and mTERT+/− mice had similar amounts of control GAPDH mRNA but that mTERT mRNA was reduced by two- to threefold in mTERT+/− mice (Fig. 5). Thus, a reduction in mTERT mRNA does not result in a functional deficit in telomere elongation, in contrast to the haplo-insufficiency that occurs in association with a similar decrease in mTR RNA in mTR+/− mice.
FIG. 5.

mTERT mRNA expression is reduced in mTERT+/− mice. Relative mRNA units were calculated from a standard curve. (A) Open circles represent mTERT mRNA from mTERT+/+ (B6 × SPRET/Ei)F1 mice, and filled circles represent mTERT mRNA from mTERT+/− (B6 × SPRET/Ei)F1 mice. (B) Open squares represent mGAPDH mRNA from mTERT+/+ (B6 × SPRET/Ei)F1 mice, and filled squares represent mGAPDH mRNA from mTERT+/− (B6 × SPRET/Ei)F1 mice.
Expression of transgenic mTERT does not affect telomere elongation.
Using a complementary strategy to further test the effect of altered mTERT expression, we generated transgenic lines that express mTERT under the control of a human ubiquitin promoter and have tested the effect of transgenic mTERT expression in interspecies crosses with M. spretus. Elongations of short M. spretus telomeres were equivalent in mice crossed with wild-type and mTERT-transgenic mice (Fig. 6). Thus, neither overexpression of mTERT in transgenic mice nor reduced mTERT expression in heterozygous mTERT+/− mice altered telomerase-dependent telomere elongation.
FIG. 6.

SPRET/Ei telomeres undergo the same elongation in wild-type and mTERT-transgenic B6 or (B6 × SPRET/Ei)F1 mice. Telomeric repeat fragment distribution was analyzed by pulsed-field gel electrophoresis and in-gel hybridization of telomeric DNA from SPRET/Ei (Sp), B6, and (B6 × SPRET/Ei)F1 mice with a wild-type genotype (WT) and/or the mTERT-transgene (+). The results shown are representative of those from six mTERT-transgenic (B6 × SPRET/Ei)F1 mice from two independent transgenic lines. The boxed area represents elongated telomeres of parental SPRET/Ei origin. MK, molecular size marker; Tg, transgene.
DISCUSSION
The regulation and maintenance of telomere length in mammalian cells is dependent upon the activity of telomerase as well as the modulating effects of additional telomere-associated proteins. Thus, mice that are rendered deficient in mTR by gene disruption are completely lacking in telomerase activity and undergo gradual erosion of telomere length over successive generations, resulting in insufficiency of germ line cells as well as multiple deficiencies in somatic cell lineages (4-6). Knockout mice that are completely deficient in mTERT also lack telomerase activity (19, 27), and telomere shortening has been reported to occur over successive generations of these mTERT-deficient mice (35; unpublished data).
Although these studies demonstrate the absolute requirement for both TR and TERT in vivo, they did not establish the role played by quantitative regulation of these two telomerase components under physiologic conditions or in disease pathogenesis. The recent mapping of genes responsible for the inherited human disease DKC has provided provocative evidence that altered telomere length or function can result in pathology (25). Autosomal dominant DKC is caused by mutation in TR, the RNA component of telomerase, and is marked by abnormal telomere shortening (31). The fact that DKC is caused by mutation in one copy of the hTR gene suggests that haplo-insufficiency might result from reduced overall hTR expression caused by the inactivation of one allelic copy. The possibility that haplo-insufficiency in TR expression can affect telomere length maintenance was supported by recent studies demonstrating that telomerase-dependent telomere elongation is deficient in interspecies mice which are heterozygous for mTR inactivation and which thus express reduced levels of mTR RNA, an effect that was shown to be independent of genetic mTR polymorphisms or species-specific differences (18). These results demonstrated that mTR expression can be limiting for telomerase-dependent telomere maintenance in vivo.
In the studies reported here, we generated mTERT-deficient mice and studied the effect of inactivating one or both copies of mTERT on telomere lengths of interspecies crosses. Fully deficient mTERT, homozygous knockout mice resembled mTR knockout mice in lacking detectable telomerase activity and in undergoing telomere shortening rather than the elongation observed in wild-type controls. Heterozygous mTERT+/− mice expressed two- to threefold-lower levels of mTERT mRNA than wild-type controls, similar to the two- to threefold reduction in telomerase RNA observed in mTR+/− heterozygotes. However, when telomere length was measured, it was found that telomere elongation occurred in mTERT+/− heterozygous mice to a degree equivalent to that seen in wild-type controls and in marked contrast to the deficiency observed in mTR+/− mice. Thus, the restriction of mTR expression, but not of mTERT expression, resulted in a reduced capacity for telomere elongation, indicating that it is mTR gene expression that is limiting for telomerase-dependent telomere maintenance in this in vivo model system.
While the studies presented here were under review, Erdmann et al. (12) reported an analysis of telomere length in mTERT heterozygous B6 mice. They found that, in successive generations of backcrossing mTERT+/− heterozygotes to wild-type B6 mice, mTERT+/− offspring demonstrated telomere shortening beyond that seen in wild-type littermates. These results are in contrast to the absence of any difference in telomere length observed in mTERT heterozygous or wild-type offspring of interspecies crosses between M. musculus (B6) and M. spretus (SPRET/Ei) mice. The difference in results between these two studies could reflect a difference in regulation of mTERT and mTR expression under the conditions studied. Thus, mTR but not mTERT expression may be limiting when telomerase is activated to achieve a new set point for telomere length in interspecies crosses, whereas limiting factors for telomere maintenance may differ under other circumstances. In this regard, it would be of interest to determine the effect of mTR heterozygosity under the conditions studied by Erdmann et al. It should be noted that differences in the strategies used for mTERT inactivation cannot be excluded as relevant to the findings of these studies.
It has been reported that overexpression of transgenic mTERT can increase cancer susceptibility (1, 13, 14) and the in vivo proliferation and function in normal tissues (14, 26). Transgenic mTERT overexpression in basal skin cells, driven by the K5 promoter, was reported to have no effect on telomere length in these cells (14, 26), whereas transgenic mTERT expression in cardiac myocytes, under the control of the α myosin heavy chain promoter, was found to “rescue” telomere shortening observed in nontransgenic control mice (26). In the studies reported here, we observed that transgenic overexpression of mTERT had no effect on telomere elongation in interspecies crosses, consistent with the conclusion that wild-type levels of mTERT expression are not limiting in this model system.
An important question, as yet unanswered, is whether the decrease in mTERT mRNA in mTERT+/− mice results in a reduction in the levels of mTERT protein produced. The absence of an effect of reduced levels of mTERT mRNA could be explained by a failure to produce a corresponding reduction in mTERT translation or steady-state protein levels. Alternatively, a reduction in mTERT protein may occur without an impact on telomere dynamics. We have to date been unable to quantitate protein levels with the available anti-mTERT antibodies and can therefore not yet distinguish between these possibilities.
Liu et al. (23, 24) analyzed telomere length in mTERT+/+, mTERT+/−, and mTERT−/− ES cells during progressive passage in vitro and observed that telomere length was maintained in wild-type cells but decreased progressively in mTERT−/− cells. In addition, they found that mTERT+/− ES cells failed to maintain telomere length and underwent shortening at a rate intermediate between the rates of shortening of wild-type and homozygous deficient cells. From these findings, they concluded that mTERT expression was limiting in ES cells, in contrast to the absence of any detectable defect in mTERT+/− interspecies mice reported in the present studies. In our own studies, we found that neither wild-type nor mTERT+/− ES cells underwent shortening during 5 months in continuous culture; in fact, ES cells of both genotypes exhibited modest and comparable degrees of telomere elongation during this period (data not shown). The basis for the difference in behavior of mTERT+/− ES cells in this study and in that of Liu et al. is unclear but may reflect differences in ES cell populations and/or culture conditions. Taken together, the results presented here suggest that the expression of mTR, but not mTERT, limits telomere maintenance during embryonic and early postnatal in vivo development in the interspecies model that we have analyzed.
Identifying the factors that limit and determine telomerase activity and telomere maintenance in vivo is significant both for studies of underlying telomere biology and for an understanding of potential roles of altered telomere expression in pathogenesis. The haplo-insufficiency observed in mice deficient in one copy of the TR gene appears to provide a model for the mechanism underlying the inherited disease autosomal dominant DKC. The absence of both haplo-insufficiency and functional defect in mice deficient in one copy of mTERT suggests that a recessively inherited deficiency in both copies of mTERT, or a distinct dominant negative mechanism, may be necessary to disrupt function and cause disease. The possibility that different mechanisms may limit telomerase function in different cell lineages and under different conditions remains an important area for further study.
Acknowledgments
We are grateful to Carol W. Greider for her important role in discussions of this research design and in the preparation of the manuscript. We thank Nanping Weng for his critical reading of the manuscript. We thank Mary Brooks for her help in assembling the mTERT cDNA and Genevieve Sanchez-Howard and the staff at Bioqual for expert animal care and breeding.
M.T.H. was supported by NIH grant CA16519. W.C.H. is supported in part by grant K01 CA94223 from the U.S. National Cancer Institute and a Kimmel Scholar Award.
REFERENCES
- 1.Artandi, S. E., S. Alson, M. K. Tietze, N. E. Sharpless, S. Ye, R. A. Greenberg, D. H. Castrillon, J. W. Horner, S. R. Weiler, R. D. Carrasco, and R. A. DePinho. 2002. Constitutive telomerase expression promotes mammary carcinomas in aging mice. Proc. Natl. Acad. Sci. USA 99:8191-8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blackburn, E. H. 2001. Switching and signaling at the telomere. Cell 106:661-673. [DOI] [PubMed] [Google Scholar]
- 3.Blackburn, E. H., C. W. Greider, E. Henderson, M. S. Lee, J. Shampay, and D. Shippen-Lentz. 1989. Recognition and elongation of telomeres by telomerase. Genome 31:553-560. [DOI] [PubMed] [Google Scholar]
- 4.Blasco, M. A., W. Funk, B. Villeponteau, and C. W. Greider. 1995. Functional characterization and developmental regulation of mouse telomerase RNA. Science 269:1267-1270. [DOI] [PubMed] [Google Scholar]
- 5.Blasco, M. A., H. W. Lee, M. P. Hande, E. Samper, P. M. Lansdorp, R. A. DePinho, and C. W. Greider. 1997. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell 91:25-34. [DOI] [PubMed] [Google Scholar]
- 6.Blasco, M. A., H. W. Lee, M. Rizen, D. Hanahan, R. DePinho, and C. W. Greider. 1997. Mouse models for the study of telomerase. Ciba Found. Symp. 211:160-170. [DOI] [PubMed] [Google Scholar]
- 7.Bryan, T. M., and T. R. Cech. 1999. Telomerase and the maintenance of chromosome ends. Curr. Opin. Cell Biol. 11:318-324. [DOI] [PubMed] [Google Scholar]
- 8.Capecchi, M. R. 1989. The new mouse genetics: altering the genome by gene targeting. Trends Genet. 5:70-76. [DOI] [PubMed] [Google Scholar]
- 9.Cech, T. R. 1994. Chromosome end games. Science 266:387-388. [DOI] [PubMed] [Google Scholar]
- 10.Chiang, Y. J., H. K. Kole, K. Brown, M. Naramura, S. Fukuhara, R. J. Hu, I. K. Jang, J. S. Gutkind, E. Shevach, and H. Gu. 2000. Cbl-b regulates the CD28 dependence of T-cell activation. Nature 403:216-220. [DOI] [PubMed] [Google Scholar]
- 11.Cong, Y.-S., W. E. Wright, and J. W. Shay. 2002. Human telomerase and its regulation. Microbiol. Mol. Biol. Rev. 66:407-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Erdmann, N., Y. Liu, and L. Harrington. 2004. Distinct dosage requirements for the maintenance of long and short telomeres in mTert heterozygous mice. Proc. Natl. Acad. Sci. USA 101:6080-6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.González-Suárez, E., J. M. Flores, and M. A. Blasco. 2002. Cooperation between p53 mutation and high telomerase transgenic expression in spontaneous cancer development. Mol. Cell. Biol. 22:7291-7301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez-Suarez, E., E. Samper, A. Ramirez, J. M. Flores, J. Martin-Caballero, J. L. Jorcano, and M. A. Blasco. 2001. Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes. EMBO J. 20:2619-2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greider, C. W. 1996. Telomere length regulation. Annu. Rev. Biochem. 65:337-365. [DOI] [PubMed] [Google Scholar]
- 16.Hahn, W. C., C. M. Counter, A. S. Lundberg, R. L. Beijersbergen, M. W. Brooks, and R. A. Weinberg. 1999. Creation of human tumour cells with defined genetic elements. Nature 400:464-468. [DOI] [PubMed] [Google Scholar]
- 17.Harley, C. B., A. B. Futcher, and C. W. Greider. 1990. Telomeres shorten during ageing of human fibroblasts. Nature 345:458-460. [DOI] [PubMed] [Google Scholar]
- 18.Hathcock, K. S., M. T. Hemann, K. K. Opperman, M. A. Strong, C. W. Greider, and R. J. Hodes. 2002. Haploinsufficiency of mTR results in defects in telomere elongation. Proc. Natl. Acad. Sci. USA 99:3591-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim, N. W., M. A. Piatyszek, K. R. Prowse, C. B. Harley, M. D. West, P. L. Ho, G. M. Coviello, W. E. Wright, S. L. Weinrich, and J. W. Shay. 1994. Specific association of human telomerase activity with immortal cells and cancer. Science 266:2011-2015. [DOI] [PubMed] [Google Scholar]
- 20.Lansdorp, P. M., N. P. Verwoerd, F. M. van de Rijke, V. Dragowska, M. T. Little, R. W. Dirks, A. K. Raap, and H. J. Tanke. 1996. Heterogeneity in telomere length of human chromosomes. Hum. Mol. Genet. 5:685-691. [DOI] [PubMed] [Google Scholar]
- 21.Lee, H. W., M. A. Blasco, G. J. Gottlieb, J. W. Horner II, C. W. Greider, and R. A. DePinho. 1998. Essential role of mouse telomerase in highly proliferative organs. Nature 392:569-574. [DOI] [PubMed] [Google Scholar]
- 22.Lingner, J., and T. R. Cech. 1998. Telomerase and chromosome end maintenance. Curr. Opin. Genet. Dev. 8:226-232. [DOI] [PubMed] [Google Scholar]
- 23.Liu, Y., H. Kha, M. Ungrin, M. O. Robinson, and L. Harrington. 2002. Preferential maintenance of critically short telomeres in mammalian cells heterozygous for mTert. Proc. Natl. Acad. Sci. USA 99:3597-3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu, Y., B. E. Snow, M. P. Hande, D. Yeung, N. J. Erdmann, A. Wakeham, A. Itie, D. P. Siderovski, P. M. Lansdorp, M. O. Robinson, and L. Harrington. 2000. The telomerase reverse transcriptase is limiting and necessary for telomerase function in vivo. Curr. Biol. 10:1459-1462. [DOI] [PubMed] [Google Scholar]
- 25.Mitchell, J. R., E. Wood, and K. Collins. 1999. A telomerase component is defective in the human disease dyskeratosis congenita. Nature 402:551-555. [DOI] [PubMed] [Google Scholar]
- 26.Oh, H., G. E. Taffet, K. A. Youker, M. L. Entman, P. A. Overbeek, L. H. Michael, and M. D. Schneider. 2001. Telomerase reverse transcriptase promotes cardiac muscle cell proliferation, hypertrophy, and survival. Proc. Natl. Acad. Sci. USA 98:10308-10313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rudolph, K. L., S. Chang, H. W. Lee, M. Blasco, G. J. Gottlieb, C. Greider, and R. A. DePinho. 1999. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell 96:701-712. [DOI] [PubMed] [Google Scholar]
- 28.Schorpp, M., R. Jager, K. Schellander, J. Schenkel, E. F. Wagner, H. Weiher, and P. Angel. 1996. The human ubiquitin C promoter directs high ubiquitous expression of transgenes in mice. Nucleic Acids Res. 24:1787-1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stewart, S. A., and R. A. Weinberg. 2002. Senescence: does it all happen at the ends? Oncogene 21:627-630. [DOI] [PubMed] [Google Scholar]
- 30.Theimer, C. A., L. D. Finger, L. Trantirek, and J. Feigon. 2003. Mutations linked to dyskeratosis congenita cause changes in the structural equilibrium in telomerase RNA. Proc. Natl. Acad. Sci. USA 100:449-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vulliamy, T., A. Marrone, F. Goldman, A. Dearlove, M. Bessler, P. J. Mason, and I. Dokal. 2001. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 413:432-435. [DOI] [PubMed] [Google Scholar]
- 32.Vulliamy, T. J., S. W. Knight, P. J. Mason, and I. Dokal. 2001. Very short telomeres in the peripheral blood of patients with X-linked and autosomal dyskeratosis congenita. Blood Cells Mol. Dis. 27:353-357. [DOI] [PubMed] [Google Scholar]
- 33.Wen, J., Y. S. Cong, and S. Bacchetti. 1998. Reconstitution of wild-type or mutant telomerase activity in telomerase-negative immortal human cells. Hum. Mol. Genet. 7:1137-1141. [DOI] [PubMed] [Google Scholar]
- 34.Weng, N. P., and R. J. Hodes. 2000. The role of telomerase expression and telomere length maintenance in human and mouse. J. Clin. Immunol. 20:257-267. [DOI] [PubMed] [Google Scholar]
- 35.Yuan, X., S. Ishibashi, S. Hatakeyama, M. Saito, J. Nakayama, R. Nikaido, T. Haruyama, Y. Watanabe, H. Iwata, M. Iida, H. Sugimura, N. Yamada, and F. Ishikawa. 1999. Presence of telomeric G-strand tails in the telomerase catalytic subunit TERT knockout mice. Genes Cells 4:563-572. [DOI] [PubMed] [Google Scholar]
- 36.Yudoh, K., H. Matsuno, F. Nakazawa, R. Katayama, and T. Kimura. 2001. Reconstituting telomerase activity using the telomerase catalytic subunit prevents the telomere shorting and replicative senescence in human osteoblasts. J. Bone Miner. Res. 16:1453-1464. [DOI] [PubMed] [Google Scholar]
- 37.Zhu, L., K. S. Hathcock, P. Hande, P. M. Lansdorp, M. F. Seldin, and R. J. Hodes. 1998. Telomere length regulation in mice is linked to a novel chromosome locus. Proc. Natl. Acad. Sci. USA 95:8648-8653. [DOI] [PMC free article] [PubMed] [Google Scholar]