


Abstract
While the cost of DNA sequencing has dropped by five orders of magnitude in the past decade, DNA synthesis remains expensive for many applications. Although DNA microarrays have decreased the cost of oligonucleotide synthesis, the use of array-synthesized oligos in practice is limited by short synthesis lengths, high synthesis error rates, low yield and the challenges of assembling long constructs from complex pools. Toward addressing these issues, we developed a protocol for multiplex pairwise assembly of oligos from array-synthesized oligonucleotide pools. To evaluate the method, we attempted to assemble up to 2271 targets ranging in length from 192–252 bases using pairs of array-synthesized oligos. Within sets of complexity ranging from 131–250 targets, we observed error-free assemblies for 90.5% of all targets. When all 2271 targets were assembled in one reaction, we observed error-free constructs for 70.6%. While the assembly method intrinsically increased accuracy to a small degree, we further increased accuracy by using a high throughput ‘Dial-Out PCR’ protocol, which combines Illumina sequencing with an in-house set of unique PCR tags to selectively amplify perfect assemblies from complex synthetic pools. This approach has broad applicability to DNA assembly and high-throughput functional screens.
INTRODUCTION
Traditionally, DNA has been synthesized by solid-phase phosphoramidite chemistry (1). Column-based synthesis generates up to 200-mers with error rates of about 1 in 200 nt (2) and yields of 10 to 100 nmol per product. Column-based DNA synthesis is limited in throughput to 384-well plates (2), and oligos cost from $0.05 to $1.00/bp depending on length and yield (2–4). The commercialization of inkjet-based printing of nucleotides with phosphoramidite chemistries (5–7) (Agilent) and semiconductor-based electrochemical acid production (8) arrays (CustomArray) have increased throughput and decreased the cost of oligo synthesis. These oligos range from $0.00001–0.001/bp in cost, depending on length, scale and platform (2). However, these platforms are limited by short synthesis lengths, high synthesis error rates, low yield and the challenges of assembling long constructs from complex pools.
Many methods have recently addressed the high error rates of array-synthesized oligos, with a trade-off between cost and fidelity. Low-cost methods include proteins such as MutS (9,10), polymerases (11–15) and other proteins that bind and cut heteroduplexes (3,16). However, as these methods rely on identifying mismatches and require the majority of sequences to be identical, they are not always compatible with complex libraries (17,18) and therefore must be performed after individual gene assemblies. Furthermore, as these methods retain error rates as high as 1 per 1000 bases, further screening is required to confirm the correct sequence. More recent methods such as Dial-Out PCR rely on DNA sequencing followed by retrieval of sequence-verified constructs, achieving error rates as low as 10−7 (17–19). While these methods can work on complex oligo pools and yield very low error rates, they are costly, time-intensive and do not always recover targeted molecules.
Despite their high error rates, inexpensive oligo pools cleaved from microarrays have recently enabled high-throughput analysis of promoter (20–22) and enhancer (23,24) function, providing novel insight into the vocabulary of these regulatory elements. They have also been used in deciphering the role of genetic variants in protein function (25). However, these studies were all limited by short synthesis lengths – about 160 bp for CustomArray and 230 bp for Agilent.
To our knowledge, Tian et al. was first to perform gene synthesis from pools of array-derived oligos. Since array synthesis only provides yields of 1–10 fmol per sequence (26), Tian et al. amplified all oligos with a common set of primers. However, the study limited synthesis to 21 genes in order to circumvent high synthesis error rates and the challenges of assembling constructs from complex pools (27–30). To address this, Kosuri et al. demonstrated pre-amplifying subsets of the oligo-pool involved in specific assemblies, to reduce the spurious cross-hybridization observed in large-scale assemblies (3). The study relied on amplifying fragments for each gene separately, which was successful but limited throughput to the assembly of 47 genes. In 2012, Kim et al. described shotgun synthesis on 228 array-derived oligos spanning the penicillin biosynthetic gene cluster (19). Similar to Tian et al., Kim amplified oligos with universal primers, and removed adaptor sequences with two restriction enzymes. After assembling all oligos via PCR, they selected for fragments between 300–500 bp. While successful, only 3% of sequenced products were error-free. In order to retrieve error-free constructs, they barcoded and sequenced their pool, identifying accurate fragments covering 88% of their targets. They then ordered primers corresponding to the barcodes to retrieve the fragments of interest.
Short synthesis lengths and high error rates present bottlenecks to the use of array-derived oligos for both functional assays and gene assembly. Here, we describe a method to assemble thousands of array-derived oligos into targets approaching length estimates of cis-regulatory elements (31,32) and protein domains (33). Compared to existing methods, our method does not limit sequence space by using restriction enzymes, it is high throughput, and it offers an efficient way to retrieve error-free assemblies.
MATERIALS AND METHODS
Target designs
Target sequences range from 156–216 bases of unique sequence and were split into 10 sets. Each target was fragmented into two pieces (A and B) using a custom python script that determines overlaps with the least chance of cross-hybridization (see Supplementary Material). Briefly, we automated the following procedure using python: bases for the overlap region were dynamically added starting from the midpoint-7 position until the melting temperature was >56°C (34). The overlap fragment was then checked against all sequences in the set and accepted if <15 consecutive bases aligned to any other sequence. To quickly evaluate alignments against all sequences in a given set, we utilized a simple sliding algorithm, which scores the longest consecutive alignments (35). If the overlap sequence failed these conditions, we swapped out up to 6 codons at random within this sequence region, and if the melting temperature was still >56°C, we repeated the alignment step. If conditions still were not met, the starting position for the overlap region was shifted and the procedure was repeated. A window of 6 bases around the starting position was explored. A common 18 bp adapter was appended to the 5′ end of A fragments and 3′ end of B fragments. Two adenines were appended to the 3′ end of A fragments, and two thymines were appended to the 5′ end of B fragments. Finally, depending on length, either one or two pool-specific primers site(s) were added to all oligo designs, and random bases were added on the 3′ side to reach 160 bases for each oligo design (Figure 1). The pools of oligos were then synthesized by CustomArray in duplicate to decrease oligo dropout and increase uniformity.
Figure 1.

Multiplex pairwise assembly. A total of 2271 targets were separated into 10 sets of 131–250 genes. Each gene was split into A and B fragments with overlapping sequences providing >56°C melting temperature (Tm) for PCR-mediated assembly. All oligos were cleaved off the array into one tube. We then amplified each sub-pool with one common and one uracil-containing pool-specific primer. The pool-specific primer was then removed with Uracil Specific Excision Reagent (USER) followed by New England BioLabs End Repair kit. During PCR assembly, corresponding sub-pools were allowed to anneal and extend through 5 cycles of PCR, before adding a set of common, outer primers for amplification. During PCR assembly, M13F and M13R sequences can be introduced to the constructs in order to allow for Dial-Out Tagging and retrieval of sequence-verified constructs. In this study, we assembled up to 252-mers from 160-mer CustomArray oligos.
Pairwise oligonucleotide assembly
Targets were separated into sets of complexity ranging from 131–250. Each pool of A and B fragments was amplified off of the array using one common primer and one pool-specific uracil-containing primer with the Kapa HiFi HotStart Uracil+ Readymix. Quantitative PCR (qPCR) was performed in 25 μl reactions with SYBR Green on a MiniOpticon Real-Time PCR system (Bio-Rad) with 2.5 ng template. Each pool was pulled from the thermocycler one cycle before plateauing, purified with 1.8x AMPure XP beads and eluted in 20 µl. Two microliters of NEB USER enzyme was mixed with the purified PCR pools, and incubated at 37°C for 15 min, followed by 15 min at room temperature. The pools were then treated with NEBNext End Repair Module per manufacturer's protocol to remove adapter sequences. The pools were purified and concentrated in 10 µl using Zymo DNA Clean and Concentrator.
Corresponding A and B fragment libraries were assembled with Kapa Hifi Hotstart Readymix (Kapa Biosystems) using qPCR with a total of 1.5 ng of the purified, corresponding input DNA pools. After 5 cycles of annealing and extension, 7.5 × 10−12 moles of each outer primer (YF-pu1L and YR-pu1R) were added, and the reaction was continued for additional cycles. Reactions were monitored on a real-time qPCR instrument, and terminated one or several cycles before plateauing. Typically, this required 20–25 cycles in addition to the first 5 cycles. For both phases, the following protocol was used: (i) 95°C for 2 min, (ii) 98°C for 20 s, (iii) 65°C for 15 s, (iv) 72°C for 45 s, (v) repeat steps ii–iv. Reactions were then purified with 1.8x AMPure XP beads and eluted in 20 µl.
Two nanograms of the purified reaction was used in another real-time PCR with Kapa HiFi Hotstart Readymix with Pu1L_Flowcell and Pu1R_Flowcell primers. Reactions were pulled from the cycler one cycle before plateauing, purified with 1.8x AMPure XP beads, and sequenced on an Illumina MiSeq with paired end 155 bp reads with Pu1_Sequencing_F, Pu1_Sequencing_R and Pu1_Sequencing_I (Supplementary Table S1). For complex sets of up to 2271 targets, input DNA from the corresponding sub pools were mixed together, maintaining the same total amount of 1.5 ng input DNA.
In silico design of static tag library
We generated random 13-mer sequences and screened them for several properties: no homoguanine or homocytosine stretches >5 bp, no homoadenine or homothymine stretches >8 bp and GC content between 45% and 65%. The 13-mers passing this filter were added to a potential set if the last 10 bases had <90% nucleotide identity with any other forward, reverse, complement or reverse complement already in the list. This pipeline was repeated several times, ultimately with 1.2 million iterations, to generate a library of 7411 13-mers.
The Gibbs free energy of every possible primer pair was calculated using Unafold (36) with the following settings: –NA = DNA, –run-type = html, –Ct = 0.000001, –sodium = 0.050, –magnesium = 0.002. All 13-mer pairs with dG > −9 kcal/mol were indexed and added to a MatrixMarket Matrix. The maximum library of 13-mers with all pairwise dG > −9 kcal/mol was then identified using the Parallel Maximum Clique Library (arXiv:1302.6256). The indexed 13-mers were converted back to their corresponding sequences, and an additional step was applied to remove any primers with potential homodimers. This left a set of 4637 13-mers, which was split into a forward library of 2318 tags and a reverse library of 2319 tags, with a total tag complexity of 5 444 982 (Figure 2).
Figure 2.

Pipeline for generation of static tag library. We generated 1.2 million random 13-mers, and screened them for no homoguanine or homocytosine stretches >5 bp, no homoadenine or homothymine stretches >8 bp and GC content between 45% and 65%. We also screened for <90% nucleotide identity in the last 10 bp, which generated a set of 7411 13-mers. From this set of 7411 sequences, we calculated every pairwise Gibbs free energy, and identified the maximum number of sequences such that no two members had a dG ≤ −9 kcal/mol. This left a set of 4637 sequences, which were split into a set of 2318 forward tags and 2319 reverse tags.
To the forward 13-mers, 5′-CGACAGTAACTACACGGCGA-3′ was added to the 5′ end as a bridge for the flow cell adapter, and M13 (5′-GTTTTCCCAGTCACGAC-3′) was added to the 3′ end as the Dial-Out seed sequence. To the reverse 13-mers, 5′-GTAGCAATTGGCAGGTCCAT-3′ was used as the bridge and M13R (5′-CAGGAAACAGCTATGAC-3′) was used as the seed sequence.
Design and synthesis of dial-out retrieval primers
For each 13-mer, the Tm was calculated using 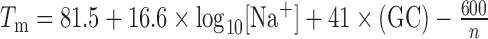 (37). Primer sequences were determined by recursively adding 2 bp from the bridge sequence to the 5′ end of the primer until the Tm was between 58°C and 61°C. After this procedure, all primers were 17 nt or 19 nt long, with Tm between 58.2°C and 60.6°C. Primers were ordered from IDT in 96-well plate format with standard desalting.
(37). Primer sequences were determined by recursively adding 2 bp from the bridge sequence to the 5′ end of the primer until the Tm was between 58°C and 61°C. After this procedure, all primers were 17 nt or 19 nt long, with Tm between 58.2°C and 60.6°C. Primers were ordered from IDT in 96-well plate format with standard desalting.
Static tag library synthesis and preparation
The 4637 tags were synthesized using CustomArray's semiconductor electrochemical process in duplicate. Forward and reverse tag sets were amplified in 24 parallel 50 µl reactions from 1.25 × 10−14 moles template/reaction using FP: 5′- CGACAGTAACTACACGGCGA -3′ and RP: 5′- GTCGTGACTGGGAAAAC -3′ with Kapa Hifi Hotstart Readymix for 17 cycles. Ten nanomolar PCR products were digested with NEB lambda exonuclease following manufacturer's protocol. A 113 ng sample was mixed with equivolume Novex TBE Urea Sample Buffer and heated at 70°C for 3 min, then chilled on ice. Samples and ladder were run on a Novex TBE Urea Gel, and the corresponding 50 bp band was cut. The bands were diced and spun through a 600 ml Eppendorf with a hole from a 22 gauge needle. The slurries were incubated with TE buffer at 65°C for 2 h and purified on a Spin-X column (Corning). Purified DNA was treated with the Qiagen nucleotide removal kit per manufacturer's protocol.
Tagging of assembled targets
Several concentrations of tags and input were tested for optimal tagging with several different polymerases (Supplementary Table S2). We identified that 8.5 × 10−14 moles of tags with 3 ng input (a 10:1 tag:input molecular ratio) with Kapa HiFi HotStart Readymix, yielded optimal performance. During the assembly process, we amplified targets with primers containing M13F and M13R, following the assembly protocol above. Libraries were purified with 1.8x AMPure XP beads and eluted in 20 µl. Three nanograms of purified assembly library was tagged with 8.5 × 10−14 moles of dial-out tags (Dial-Out Tags F and Dial-Out Tags R) using Kapa HiFi HotStart Readymix using qPCR and the following cycling conditions: (i) 95°C for 2 min, (ii) 98°C for 20 s, (iii) 65°C for 15 s, (iv) 72°C for 45 s, (v) repeat steps ii–iv 30 times and (vi) 72°C for 5 min and (vi) 72°C for 5 min. After the first 5 cycles, the reaction was paused, and 1.5 × 10−11 moles of barcoded forward and reverse flow-cell primers (Dial-Out_Flow_Cell_F and Dial-Out_Flow_Cell_R) were added. The tagged libraries were removed from the cycler one cycle before plateauing, and purified using 1.8x AMPure XP beads.
Sequence-verification of dial-out tagged targets
The tagged library was sequenced on an Illumina MiSeq with PE 155 bp reads using Dial-Out_Sequencing_F, Dial-Out_Sequencing_R and Dial-Out_Sequencing_I primers. Reads were merged with PEAR using default settings and tag pairs for all reads were identified (38). Using a custom python script (Supplementary file), we identified all reads containing sequence-verified constructs, and their corresponding tag pairs. One correctly-assembled molecule per target meeting the following criteria was randomly selected for retrieval: (i) containing a unique tag set not identified on any other molecule and (ii) represented in at least 5 sequencing reads.
Dial-out retrieval
Selected oligonucleotides were retrieved via PCR with Kapa HiFi Hotstart Readymix using real-time PCR with 0.135 ng template and 1.5 × 10−11 moles each of the corresponding forward and reverse dial-out retrieval primer with the following conditions: (i) 95°C for 3 min, (ii) 98°C for 20 s, (iii) 65°C for 15 s, (iv) 72°C for 40 s, (v) repeat steps ii–iv 34 times and (vi) 72°C for 5 min. Reactions were removed from the cycler just before plateauing, purified with 1.8x Ampure and quantified using a Qubit (Invitrogen). Equal concentrations of each retrieval reaction were mixed for sequencing.
Analysis of average nucleotide accuracy
All sequencing reads were aligned to a reference of intended target sequences using BWA v.0.7.3. The average nucleotide accuracy was calculated from bases with aligned reads with base and quality mapping score >20. To compare accuracy rates between experiments, we analyzed error rates for set 5 before and after assembly. We performed Exact Poisson Tests on the 15 935 028 bases of the assembled set and 9 325 493 bases of the corresponding oligo pools passing our quality cutoffs. We also performed the test on the 1 546 665 bases of the overlapping region in the assembled set and 1 617 760 in the oligo pools.
RESULTS
Assembling targets in sets of 131–250
We designed in silico 2271 targets ranging from 192–252 bases (156–216 of unique sequence) to assemble from array-derived oligos. All targets consisted of a unique sequence flanked by the same 18 bp 5′ and 3′ common adapters. Each target sequence was split into two fragments, A and B, containing an overlap region with a Tm >56°C. The 2271 target sequences were split into 10 sets of 131–250 targets, and each set received unique adapters flanking the 3′ end of the A fragments and the 5′ end of the B fragments designed for uracil incorporation (Figure 1). The corresponding oligos (160-mers with buffer sequence) were synthesized by CustomArray in duplicate to reduce oligo dropout and increase uniformity.
We first amplified each pool of oligos off the array with a sub pool specific primer (A fragment uniqueF or B fragment uniqueR) on one end and a common primer (YF/YR) on the other (Supplementary Table S1). Sequencing of the oligo library showed good uniformity, with an interquartile range of 5.5 (Figure 3A).
Figure 3.

Assembling targets in sets of 131–250. (A) Uniformity plot of error-free array-derived oligos by rank-ordered percentile for all 2271 targets. (B) Number and size of targets, and error-free yield for each target set. (C) Each target is placed into a bin based on the limiting oligo count, which is the number of error-free reads out of 1.2 million that are limiting for its corresponding target. The %Yield of assemblies is the percentage of targets in that bin with at least one perfect assembly. (D) The percentage of perfect, mismatch only, small indel (<5 bp), large indel (≥5 bp), truncations and unmapped reads for all oligos. (E) The percentage of perfect, mismatch only, small indel (<5 bp), large indel (≥5 bp), chimeras, truncations and unmapped reads for each assembled library. (F) Uniformity of each set of targets. Note that set 10 only has 131 targets.
The oligo pools provided by CustomArray were then amplified using either Uracil-containing A fragment primer and YF or Uracil-containing B fragment primer and YR (Supplementary Table S1), and the corresponding specific adapters were removed with Uracil Specific Excision Reagent (USER). For two pools, we tested amplifying oligos with either one or two unique primer sites and observed no difference in assembly composition or uniformity (Supplementary Figure S1). The corresponding A and B fragments were mixed for each set of targets and assembled through 5 cycles of annealing with extension and approximately 25 cycles of amplification with Kapa HiFi. In all cases, the correct size band was observed. Each assembled set was barcoded and sequenced.
For each set, we identified error-free assembled constructs for 72.7–96.4% of targets at a sequencing depth of 90 000 reads (Figure 3B). For each target, we examined the number of error-free reads for the corresponding A and B oligos (out of 1.2 million reads). Of the 223 targets with no error-free assemblies identified, 55 (24.7%) fell in the bottom 10th percentile of limiting oligo concentration (<6 error-free reads out of 1.2 million) and 97 (43.5%) fell in the bottom 20th percentile of limiting oligo counts (<11 error-free reads out of 1.2 million). Figure 3C shows higher yield (% of targets with at least one perfect assembled sequence) for targets assembled from better-represented limiting oligos in the array pool, suggesting that increasing oligo uniformity would likely improve the yield of full-length designs. We next looked at the composition of the raw oligo pools and the assembled target libraries (Figure 3D and E). A total of 23.8% of molecules represented error-free assemblies, 36.2% contained indel-free assemblies and 53.4% contained small indels (<5 bp). An additional 2.3% contained large indels (>5 bp), 4.8% contained chimeras, 2.1% contained truncated constructs and 0.6% unmapped reads. Within each set, 6 of 10 sets had <15-fold difference in the interquartile range. While this may be an issue for some applications, the uniformity is tight enough to use the sets directly for some downstream screening applications, such as functional protein screens. Uniformity plots are shown in Figure 3F.
Of the 2271 targets synthesized in individual sets, we assembled error-free constructs for 2055 (90.5%). Much of the drop-out appears to be due to poor representation of the corresponding oligos in the array pool (Figure 3C). Additionally, the majority of errors identified in the assembled sets is likely from the array-synthesis, since similar error profiles are identified in the oligo pools (Figure 3D and E). Chimeric assembly (assembly of the wrong A and B fragments) is rare.
Multiplex assembly of 2271 pairs of fragments
To test the limitations of our assembly protocol, we increased complexity by adding one additional set at a time, up to a complexity of 2271 designs. At a complexity of 2271 we assembled error-free constructs for 70.6% of targets at a sequencing depth of 300 000 reads (Figure 4A and B). We observed an even greater correlation between yield and representation of the limiting oligo in the array pool compared to the smaller sets (Figure 4C).
Figure 4.

Effect of complexity on assembly performance. (A) Percentage of targets with at least one error-free assembly for each level of complexity. (B) Yield (number of targets with at least one perfect read) versus complexity. Red bars show the total number of targets with error free assemblies at each level of complexity. Black bars show the number of targets from the corresponding sets with error-free assemblies, which were individually assembled in sets of complexity ranging from 131–250. (C) Each target is placed into a bin based on the limiting oligo count, which is the number of error-free reads (out of 1.2 million), that are limiting for its corresponding target.The %Yield of assemblies is the percentage of targets in that bin with at least one perfect assembly. (D) Percentage of perfect, mismatch only, small indels (<5 bp), large indels (≥5 bp), chimeras, truncations and unmapped reads in sets of increasing complexity. (E) Uniformity of each set of targets.
We were also interested in whether increasing complexity would affect the composition of assembled libraries. While the two lowest complexity sets (250 and 462 targets) show the highest percentage of perfect and indel-free reads, it is likely due to the fact that these two sets are composed of sets 2 and 3, which individually showed high percentages of perfect and indel-free reads (Figure 3E). The remaining libraries all share similar compositions. For all complexity levels, 11.8–31.3% of reads represented perfect constructs, 10.0–18.7% represented constructs with mismatches only, 41.4–48.5% represented small indels, 2.6–3.5% represented large indels, 3.7–21.5% represented chimeras, 2.5–4.9% represented truncations and 0.1–0.7% unmapped reads (Figure 4D). Within each set, there was a 10- to 34-fold difference in the interquartile range. Uniformity plots are shown in Figure 4E.
Error correction of assembled targets
Oligo pools were sequenced and aligned to a reference of intended target sequences. For error analysis, we chose to examine one set of 250 targets, each 237 bases long (set 5). We calculated average nucleotide accuracy from bases with aligned reads having quality mapping score >20. We identified a 98.68% average nucleotide accuracy of oligos after amplification off the array. Since our assembly process relies on two priming sites and an overlap region, we hypothesized that assembly might intrinsically increase accuracy in these regions. Indeed, we found that the average nucleotide accuracy of all aligning molecules in the 250-plex reaction was 99.02% (Poisson rate ratio 95% CI 1.36–1.38), showing the highest accuracy around the two priming sites and overlap region (Figure 5A). In particular, the average nucleotide accuracy for the overlapping region increased from 98.53 to 99.44% (Poisson rate ratio 95% CI 2.64–2.77).
Figure 5.

Error correction of assembled constructs. (A) The per base accuracy of assembled constructs in black and their corresponding oligos in red and blue. Increased accuracy is seen at both priming sites and the overlap region. (B) Bar graphs for the percentage of tags identified on only one, two, three, four or at least 5 different molecules in the sequenced library. Orange and purple bars are two different assembly sets, each with 250 targets. (C) The percentage of aligning reads that contain no errors for each of the 25 retrieved assemblies.
While we see a significant increase in accuracy at the nucleotide level (P∼4.9e-324), we were still limited to a maximum of 37% perfect reads in an assembled set. For downstream applications relying on accurate molecules, such as gene assembly, we were interested in retrieving perfect assemblies from our assembled sets. To do so, we modified the Dial-Out PCR protocol (17) to incorporate a set of in-house static Dial-Out tags to allow for cost-efficient PCR retrieval of sequence-verified constructs.
We designed primers that append M13F and M13R during the assembly reaction for targets from sets 2 and 6 (each 250 targets). The assembled libraries were then tagged with the static Dial-Out tags, and sequenced for verification. We first analyzed the distribution of tag pairs, and found that 84.0% and 85.6% of all molecules in assembled and tagged sets 2 and 6 contained a unique, retrievable tag pair (out of 1.3 million reads for set 2 and 1.6 million reads for set 6) (Figure 5B). 98.4% and 95.6% of targets had a sequence-verified assembly with a unique tag pair.
From set 2, we chose 25 targets to retrieve, each of which was represented in at least 5 out of 1.3 million reads. All 25 targets amplified, and we evaluated retrieval accuracy by pooling all 25 retrieval reactions together and sequencing them with 1 million reads. All 25 targets were sequenced between 8600 and 62 000 times, revealing error-free reads to the detection limit of Illumina sequencing chemistry, which is more quantitative than Sanger sequencing (Figure 5C). A total of 78% of all sequencing reads aligned to one of the 25 targets. When aligned to all 2271 potential targets, >99% of reads aligned, suggesting some background amplification of low abundance assemblies that we did not observe in our sequencing but that happen to share the same dial-out primer combinations. Consistent with this, Sanger sequencing revealed clean traces for 22 of the 25 targets, but high levels of noise for three traces (Supplementary Figure S2).
DISCUSSION
We sought to develop a protocol to overcome the limitations of array-derived oligonucleotides for library generation and gene assembly. Our method relies on multiplex pairwise assembly and Dial-Out molecular tagging. This method produced sequence-verified, individually retrievable 192–252-mers from CustomArray oligonucleotides.
We tested multiplex pairwise assembly in sets of 131–2271. In addition to perfect sequences, the composition of the assembled sets consisted of mostly small indels and mismatches, similar to the raw array-derived oligo pools. The composition of sets did not change noticeably with complexity beyond 712 targets, suggesting that increasing the number of targets per reaction does not strongly alter the amount of resulting chimeras or error-containing assemblies. While we observed reduced yield with increasing complexity, we were still able to assemble 70.6% of all targets in a 2271-plex reaction. By parallelizing similar complexity reactions in a 96-well plate, we could theoretically assemble a set of 200 000 constructs with 70% yield. If the experiment relies on representation of all targets, our data suggest that uniformity can be improved by performing assembly in sets of 250, to achieve >90% yield.
The main limitations in our protocol currently are the relatively high DNA synthesis error rate (e.g. mismatches and indels), moderate DNA assembly error rate (e.g. chimeras) and low uniformity. Low uniformity of input oligos impairs target uniformity in assembled sets. This is apparent in Figure 4C, as well as a separate array in which oligos were not duplicated (Supplementary Figure S3). We therefore suggest that for increased yield and uniformity, all oligos be duplicated during synthesis.
High-throughput functional screens would benefit from highly accurate and uniform assemblies. However, in many applications, error-containing molecules can be filtered in the analysis stage, or may provide additional diversity for directed evolution. The spread in uniformity may also be accounted for with a post-hoc analysis by normalizing a post-selection sample to a pre-selection sample. For gene assembly requiring very high accuracy, we implemented Dial-Out PCR to isolate perfect gene sequences. However, for hierarchical assembly, yield is a concern, as every fragment must be represented in order to assemble larger constructs. In applications for hierarchical gene assembly, constructs should be assembled in smaller sets, as we are able to achieve yields up to 96% in sets of 250.
We believe that with the exception of chimeras, both the high error rate and lack of uniformity are due to our input reagents, and not the multiplex pairwise assembly protocol. The error profiles of the assembled sets match closely with the profile of the raw oligos (Figure 3). In fact, we saw an increase in accuracy at priming and assembly sites from our assembly protocol. Moreover, we assembled at least one error-free sequence for each target with high representation of both oligos, suggesting that much of the dropout and uniformity issues are due to poor uniformity in array synthesis. Therefore, using a higher-fidelity and more uniform array should also reduce these limitations.
Our protocol inherently is prone to producing chimeras. While these can be filtered out in most downstream applications, they may cause issues in more complex reactions by diluting the designed library. We were able to minimize chimeras, to a maximum of 21.5%, by utilizing a custom script that examines all possible cross-hybridizations. In a separate experiment without the script, we identified chimera rates as high as 42% (Supplementary Figure S3). However, since the designs were different, we cannot make a direct comparison of chimera rates.
Through Dial-Out PCR, we were able to retrieve error-free assemblies for 25/25 targets. However, we did notice some background amplification, accounting for up to 22% of the sequenced pool. To reduce this noise in future experiments, we suggest either increasing the sequencing depth of the tagged pool or applying a more stringent filter for the number of times a construct was observed in the tagged pool.
We were limited to synthesizing 252-mers by the maximum length of oligonucleotides that we were able to synthesize in our input oligo pool (CustomArray, 160-mers). However, as we did not observe a decrease in yield with increasing target sizes from 191–252 bp (Supplementary Figure S4), we believe that target size can be increased by simply using longer oligo pools. For example, Agilent's 230-mers would allow the assembly of 392-mers using our current technique. As array technologies develop and longer oligos become available, our protocol will scale proportionately. Moreover, it is possible that our pairwise pools could be used for hierarchical assembly. This could occur directly after assembly, or after a round of multiplex Dial-Out PCR retrieval to reduce complexity and increase uniformity. Finally, it is possible that the protocol could be modified to assemble sets of three or more oligos instead of pairs, in a refined version of the shotgun synthesis technique described by Kim et al.
Our protocol for multiplex pairwise assembly of array-derived DNA oligonucleotides provides a method for inexpensive, sequence-verified, oligonucleotide assembly from array synthesis. To our knowledge, this is the first study to assemble thousands of array-derived oligos in multiplex, and to use a static set of PCR tags to retrieve sequence-verified molecules. We suggest the applicability of this protocol for both complex library generation and gene synthesis. Creating a library of 3118 such 200-mers would be ∼38-fold less expensive than column-based synthesis methods (∼0.84 USD/target). Retrieving individual sequence-verified assemblies for each of the 3118 would still be 17-fold less expensive with in-house Dial-Out tags and retrieval primers, and 4-fold less expensive including the one-time costs of the Dial-Out tag and retrieval primer libraries (Supplementary Table S3). While column-based synthesis is limited to 200 bases, our protocol synthesized 252-mers at 0.84 USD/target (0.0042 USD/base) with the similar efficiency as 200-mers (Supplementary Figure S4). With the advent of next-generation sequencing, high-throughput functional screens of DNA have shed light on the mechanisms of gene regulation (20–24) and the classification of variants of uncertain significance (25). The ability to synthesize defined libraries at an unprecedented cost will allow researchers to address these questions using precisely designed sequences rather than relying on biased mutagenesis methods. Moreover, gene synthesis has contributed to novel pharmaceuticals and a better understanding of genome organization, and we expect that increasing the length of DNA assemblies that can be produced with low-cost, high complexity DNA synthesis will provide new opportunities for protein design and synthetic biology.
Supplementary Material
Acknowledgments
We thank members of the Shendure and Baker laboratories (particularly E. Boyle, M. Snyder, M. Kircher, M. Gasperini and C. Lee), as well as S. Brothers for helpful discussions. D.B. and J.S. are investigators of the Howard Hughes Medical Institute.
Author contributions: The project was conceived and designed by J.C.K., M.J.L., J.J.S., J.N., D.B. and J.S. J.C.K. and J.J.S. designed in silico Dial-Out tags. E.M.S. designed script to fragment targets. J.C.K. performed experiments and data analysis. J.C.K. and J.S. wrote the manuscript. All authors read and approved the final version of the manuscript.
Footnotes
Present Address: Jerrod Schwartz, Google Life Sciences, Mountain View, CA 94043, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Department of Energy-Lawrence Berkeley National Laboratory-Joint Genome Institute [DE-AC02–05CH11231 to J.S.]; National Institutes of Health (NIH) [1R21CA160080 to J.S.]; Bill & Melinda Gates Foundation [OPP1120319 to D.B.]; Washington Research Foundation [Washington Research Foundation Innovation Postdoctoral Fellowship to M.J.L.]. Funding for open access charge: Dr. Shendure's HHMI funds.
Conflict of interest statement. None declared.
REFERENCES
- 1.Beaucage S.L., Caruthers M.H. Deoxynucleoside phosphoramidites-a new class of key intermediates for deoxynucleotide synthesis. Tetrahedron Lett. 1981;22:1859–1862. [Google Scholar]
- 2.Kosuri S., Church G.M. Large-scale de novo DNA synthesis: technologies and applications. Nat. Methods. 2014;11:499–507. doi: 10.1038/nmeth.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kosuri S., Eroshenko N., LeProst E.M., Super M., Way J., Li J.B., Church G.M. Scalable gene synthesis by selective amplification of DNA pools from high-fidelity microchips. Nat. Biotechnol. 2010;28:1295–1299. doi: 10.1038/nbt.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kong D.S., Carr P.A., Chen L., Zhang S., Jacobson J.M. Parallel gene synthesis in a microfluidic device. Nucleic Acids Res. 2007;25:e61. doi: 10.1093/nar/gkm121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blanchard A.P., Kaiser R.J., Hood L.E. High-density oligonucleotide arrays. Biosens. Bioelectron. 1996;11:687–690. [Google Scholar]
- 6.Hughes T.R., Mao M., Jones A.R., Burchard J., Marton M.J., Shannon K.W., Lefkowitz S.M., Ziman M., Schelter J.M., Meyer M.R., et al. Expression profiling using microarrays fabricated by an ink-jet oligonucleotide synthesizer. Nat. Biotechnol. 2001;19:342–347. doi: 10.1038/86730. [DOI] [PubMed] [Google Scholar]
- 7.Saaem I., Ma K.S., Marchi A.N., LaBean T.H., Tian J. In situ synthesis of DNA microarray on functionalized cyclic olefin copolymer substrate. ACS Appl. Mater. Interfaces. 2010;2:491–497. doi: 10.1021/am900884b. [DOI] [PubMed] [Google Scholar]
- 8.Ghindilis A.L., Smith M.W., Schwarzkopf K.R., Roth K.M., Peyvan K., Munro S.B., Lodes M.J., Stover A.G., Bernards K., Dill K, et al. Combimatrix oligonucleotide arrays: genotyping and gene expression assays employing electrochemical detection. Biosens. Bioelectron. 2007;22:1853–1860. doi: 10.1016/j.bios.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 9.Carr P.A., Park J.S., Lee Y.J., Zhang S., Jacobson J.M. Protein-mediated error correction for de novo DNA synthesis. Nucleic Acids Res. 2004;32:e162. doi: 10.1093/nar/gnh160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wan W., Ll L., Xu Q., Wang Z., Yao Y., Wang R., Zhang J., Liu H., Gao X., Hong J. Error removal in microchip-synthesized DNA using immobilized MutS. Nucleic Acids Res. 2014;42:e102. doi: 10.1093/nar/gku405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Binkowski B.F., Richmond K.E., Kaysen J., Sussman M.R., Belshaw P.J. Correcting errors in synthetic DNA through consensus shuffling. Nucleic Acids Res. 2005;33:e55. doi: 10.1093/nar/gni053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bang D., Church G.M. Gene synthesis by circular assembly amplification. Nat. Methods. 2008;5:37–39. doi: 10.1038/nmeth1136. [DOI] [PubMed] [Google Scholar]
- 13.Smith J., Modrich P. Removal of polymerase-produced mutant sequences from PCR products. Proc. Natl. Acad. Sci. U.S.A. 1997;94:6847–6850. doi: 10.1073/pnas.94.13.6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Young L., Dong Q. Two-step total gene synthesis method. Nucleic Acids Res. 2004;32:e59. doi: 10.1093/nar/gnh058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuhrmann M., Oertel W., Berthold P., Hegemann P. Removal of mismatched bases from synthetic genes by enzymatic mismatch cleavage. Nucleic Acids Res. 2005;33:e58. doi: 10.1093/nar/gni058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dormitzer P.R., Suphaphiphat P., Gibson D.G., Wentworth D.E., Stockwell T.B., Algire M.A., Alperovich N., Barro M., Brown D.M., Craig S., et al. Synthetic generation of influenza vaccine viruses for rapid response to pandemics. Sci. Transl. Med. 2013;5:185ra68. doi: 10.1126/scitranslmed.3006368. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz J.J., Lee C., Shendure J. Accurate gene synthesis with tag-directed retrieval of sequence-verified DNA molecules. Nat. Methods. 2012;9:913–915. doi: 10.1038/nmeth.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matzas M., Stahler P.F., Kefer N., Siebelt N., Boisguerin V., Leonard J.T., Keller A., Stahler C.F., Haberle P., Gharizadeh B., et al. High-fidelity gene synthesis by retrieval of sequence-verified DNA identified using high-throughput pyrosequencing. Nat. Biotechnol. 2010;28:1291–1294. doi: 10.1038/nbt.1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim H., Han H., Ahn J., Lee J., Cho N., Jang H., Kim H., Kwon S., Bang D. ‘Shotgun DNA synthesis’ for the high-throughput construction of large DNA molecules. Nucleic Acids Res. 2012;40:e140. doi: 10.1093/nar/gks546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patwardhan R.P., Lee C., Litvin O., Young D., Pe'er D., Shendure J. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat. Biotechnol. 2009;27:1173–1175. doi: 10.1038/nbt.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharon E., Kalma Y., Sharp A., Raveh-Sadka T., Levo M., Zeevi D., Keren L., Yakhini Z., Weinberger A., Segal E. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat. Biotechnol. 2012;30:521–530. doi: 10.1038/nbt.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schlabach M.R., Hu J.K., Li M., Elledge S.J. Synthetic design of strong promoters. Proc. Natl. Acad. Sci. U.S.A. 2010;107:2538–2543. doi: 10.1073/pnas.0914803107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melnikov A., Murugan A., Zhang X., Tesileanu T., Wang L., Rogov P., Feizi S., Gnirke A., Callan C.G., Kinney J.B., et al. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat. Biotechnol. 2012;30:271–277. doi: 10.1038/nbt.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith R.P., Taher L., Patwardhan R.P., Kim M.J., Inoue F., Shendure J., Ovcharenko I., Ahituv N. Massively parallel decoding of mammalian regulatory sequences supports a flexible organizational model. Nat. Genet. 2013;45:1021–1028. doi: 10.1038/ng.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Findlay G.M., Boyle E.A., Hause R.J., Klein J.C., Shendure J. Saturation editing of genomic regions by multiplex homology-directed repair. Nature. 2014;513:120–123. doi: 10.1038/nature13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Quan J.Y., Saaem I., Tang N., Ma S.M., Negre N., Gong H., White K.P., Tian J.D. Parallel on-chip gene synthesis and application to optimization of protein expression. Nat. Biotechnol. 2011;29:449–452. doi: 10.1038/nbt.1847. [DOI] [PubMed] [Google Scholar]
- 27.Tian J., Gong H., Sheng N., Zhou X., Gulari E., Gao X., Church G. Accurate multiplex gene synthesis from programmable DNA microarrays. Nature. 2004;432:1050–1054. doi: 10.1038/nature03151. [DOI] [PubMed] [Google Scholar]
- 28.Zhou X., Cai S., Hong A., You Q., Yu P., Sheng N., Srivannavit O., Muranjan S., Rouillard J.M., Xia Y., et al. Microfluidic PicoArray synthesis of oligodeoxynucleotides and simulataneous assembling of multiple DNA sequences. Nucleic Acids Res. 2004;32:5409–5417. doi: 10.1093/nar/gkh879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Borovkov A.Y., Loskutov A.V., Robida M.D., Day K.M., Cano J.A., Olson T.L., Patel H., Brown K., Hunter P.D., Sykes K.F. High-quality gene assembly directly from unpurified mixtures of microarray-synthesized oligonucleotides. Nucleic Acids Res. 2010;38:e180. doi: 10.1093/nar/gkq677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Linshiz G., Yehezkel T., Kaplan S., Gronau I., Ravid S., Adar R., Shapiro E. Recursive construction of perfect DNA molecules from imperfect oligonucleotides. Mol. Syst. Biol. 2008;4 doi: 10.1038/msb.2008.26. 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kristiansson E., Thorsen M., Tamas M.J., Nerman O. Evolutionary forces act on promoter length: identification of enriched cis-regulatory elements. Mol. Biol. Evol. 2009;26:1299–1307. doi: 10.1093/molbev/msp040. [DOI] [PubMed] [Google Scholar]
- 32.Levine M., Tjian R. Transcription regulation and animal diversity. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- 33.Xu D., Nussinov R. Favorable domain size in proteins. Fold. Des. 1998;3:11–17. doi: 10.1016/S1359-0278(98)00004-2. [DOI] [PubMed] [Google Scholar]
- 34.Allawi H.T., SantaLucia J., Jr Thermodynamics and NMR of internal G.T mismatches in DNA. Biochemistry. 1997;36:10581–10594. doi: 10.1021/bi962590c. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen-Dumont T., Pope B.J., Hammet F., Southey M.C., Park D.J. A high-plex PCR approach for massively parallel sequencing. Biotechniques. 2013;55:69–74. doi: 10.2144/000114052. [DOI] [PubMed] [Google Scholar]
- 36.Markham N., Zuker M. UNAFold: software for nucleic acid folding and hybridization. Methods Mol. Biol. 2008;453:3–31. doi: 10.1007/978-1-60327-429-6_1. [DOI] [PubMed] [Google Scholar]
- 37.Sambrook J., Fritsch E.F., Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 38.Zhang J., Kobert K., Flouri T., Stamatakis A. PEAR: a fast and accurate Illumina Paired-End read merger. Bioinformatics. 2014;30:614–620. doi: 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.