


Abstract
To study the mechanisms involved in the maintenance of a linear mitochondrial genome we investigated the biochemical properties of the recombination protein Mgm101 from Candida parapsilosis. We show that CpMgm101 complements defects associated with the Saccharomyces cerevisiae mgm101–1ts mutation and that it is present in both the nucleus and mitochondrial nucleoids of C. parapsilosis. Unlike its S. cerevisiae counterpart, CpMgm101 is associated with the entire nucleoid population and is able to bind to a broad range of DNA substrates in a non-sequence specific manner. CpMgm101 is also able to catalyze strand annealing and D-loop formation. CpMgm101 forms a roughly C-shaped trimer in solution according to SAXS. Electron microscopy of a complex of CpMgm101 with a model mitochondrial telomere revealed homogeneous, ring-shaped structures at the telomeric single-stranded overhangs. The DNA-binding properties of CpMgm101, together with its DNA recombination properties, suggest that it can play a number of possible roles in the replication of the mitochondrial genome and the maintenance of its telomeres.
INTRODUCTION
Homologous recombination plays an important role in the telomerase-independent telomere maintenance pathway which operates in the mitochondria of the yeast Candida parapsilosis (1). The mitochondria of this species contain two topologically distinct types of DNA replicons: linear, 30.9-kbp-long DNA molecules which terminate in arrays of 738-bp-long, tandemly repeating telomeric motifs (t-arrays) (2–4) and circular molecules (telomeric circles or t-circles) consisting of integral repetitions of the telomeric repeat (5). These t-circles appear to be required for the maintenance of mitochondrial telomeres. The likely mechanism is as follows. The t-circles are generated by intramolecular recombination between two of the telomeric repeats of a t-array. They then replicate via a rolling-circle mechanism, producing longer t-arrays, which are then used as substrates for recombination-dependent elongation of the termini of the linear mitochondrial genome (5,6).
To better understand this mechanism, we have begun to systematically examine the proteins involved. We have identified several C. parapsilosis genes coding for homologs of proteins involved in Saccharomyces cerevisiae mitochondrial DNA (mtDNA) recombination, including Abf2, Cce1, Rim1, Mhr1, Pif1 and Mgm101. In addition to the roles they share with their S. cerevisiae counterparts, these genes may also actively participate in the recombination-mediated maintenance of the linear mitochondrial genome or mitochondrial telomeres of C. parapsilosis. One such example has already been described: Mtp1/mtTBP, a homolog of S. cerevisiae Rim1, serves both as a mitochondrial single-stranded DNA-binding protein (SSB), its S. cerevisiae function, and as a specific, mitochondrial telomere-binding factor (7–9).
Another key player involved in S. cerevisiae mtDNA recombination is Mgm101 (ScMgm101). It is involved in the recombination-dependent repair of oxidatively damaged mtDNA molecules and is essential for the maintenance of the wild-type (ρ+) mitochondrial genome (10–12). The 247-amino-acid mature form of ScMgm101 has been shown to be a component of actively replicating mitochondrial nucleoids and to be important for mtDNA recombination in S. cerevisiae (10,13–15). In addition, ScMgm101 is also involved in the repair of interstrand crosslinks in nuclear DNA (16). Biochemical analyses have shown that this protein shares structural and functional features with the bacteriophage members of the Rad52 protein family, which catalyze the annealing of single-stranded DNA to the complementary sequence of a donor strand during homologous recombination. Concomitantly, the conserved ‘core’ region of ScMgm101 exhibits sequence similarity to the single-stranded DNA annealing domains of Rad52 and Rad59. On the other hand, its N-terminal region displays only limited homology to proteins from closely related species. ScMgm101 molecules form large oligomeric ring structures, and loss of this ability leads to loss of its activity (15,17,18).
In a recent study, we demonstrated that recombination structures and forks are the predominant DNA replication intermediates detected in C. parapsilosis mitochondria, suggesting that mtDNA replication proceeds through a recombination-initiated mechanism (19). Since ScMgm101 is important for mtDNA recombination in S. cerevisiae, it seems likely that its C. parapsilosis homolog is a key player in the dynamics of the C. parapsilosis linear mitochondrial genome. To investigate this possibility, we subjected C. parapsilosis Mgm101 (CpMgm101) to a detailed biochemical analysis and compared it with its S. cerevisiae counterpart. Our experiments revealed that CpMgm101 possesses a number of characteristics that support the hypothesis that it has a role in the recombination-dependent replication of the C. parapsilosis linear mitochondrial genome or the maintenance of its telomeres.
MATERIALS AND METHODS
Growth media and yeast strains
Candida parapsilosis strain CDU1 (CLIB214 ura3Δ::FRT/ura3Δ::FRT) (20) and S. cerevisiae strain M2915–7C (MATα, ade2, his4, leu2, ura3, mgm101–1ts) (11) were used in the experiments described in this study. Yeasts were cultivated in YPD (1% (w/v) yeast extract, 2% (w/v) peptone, 2% (w/v) glucose) or in synthetic media (0.67% (w/v) yeast nitrogen base (Difco)) supplemented with appropriate amino acids and nucleotide bases with 2% (w/v) glucose (SD), 2% galactose (w/v) (SGal) or 3% (v/v) glycerol (SGly) as the carbon sources.
Cloning and expression of CpMgm101 in S. cerevisiae mgm101–1ts mutant cells
The C. parapsilosis MGM101 gene was amplified from the genomic DNA of the CBS604 strain using primers CpMGM101_Up and CpMGM101_down2 (Supplementary Tables S1 and S2). The polymerase chain reaction (PCR) product was phosphorylated with T4 polynucleotide kinase and inserted into the SmaI site of the pUC19 vector (New England Biolabs). The resulting pUC19-CpMgm101 construct was digested with EcoRI and BamHI and the 843-bp fragment was inserted into the corresponding sites of the pYES2/CT vector (Life Technologies). Both the pYES2/CT-CpMgm101 construct and the pYES2/CT vector were transformed into S. cerevisiae M2915–7C cells (mgm101–1ts) using the lithium acetate protocol (21) and the transformants were selected for uracil prototrophy in SD medium. The GAL1 promoter-driven expression of CpMgm101 was induced by cultivation in SGal medium for 1 day followed by cultivation for another 2 days at either permissive (25°C) or restrictive (37°C) temperatures. Subsequently, serial dilutions of the cell suspensions were spotted onto solid SGly plates, which were incubated at the same temperature as the original cultures for 7 days. A control experiment was also performed with the ScMGM101 gene cloned into the pYES2 vector.
Fluorescence microscopy
The pBP8-CpMgm101 plasmid expressing full-length CpMgm101 tagged with yEGFP3 at the C-terminus was constructed as follows. The CpMGM101 coding sequence was amplified using the primers CpMGM101_BamHI_up and CpMGM101_BamHI_dn (Supplementary Tables S1 and S2), digested with BamHI and ligated into the BamHI site of the pBP8 vector, a multi-copy vector under the control of the medium-strength MET25 promoter from S. cerevisiae (22). The plasmid was transformed into C. parapsilosis cells by electroporation (23).
Intracellular localization of the CpMgm101-yEGFP3 fusion protein was determined by fluorescence microscopy using an Olympus FV1000 confocal microscope. C. parapsilosis CDU1 cells transformed with pBP8-CpMgm101 were grown for 36 h in SD media with 20 μg/ml methionine and 2 μg/ml adenine at 28°C. The cells were centrifuged 10 min at 1500 × g and washed 2× with sterile distilled water and resuspended in SD media without methionine. After 12 h, the cells were centrifuged 10 min at 1500 × g, washed 2× with sterile water and incubated for 30 min on ice. Then the cells were treated with 25 mM H2O2 for 1 h at 30°C. After the treatment, the cells were pelleted, washed with sterile water, resuspended in fresh SD media without methionine and stained with 57 μM DAPI for 2 h, then washed with phosphate buffered saline and visualized by fluorescence microscopy.
Expression and purification of recombinant proteins
The recombinant version of CpMgm101-Δ84 (core domain) from C. parapsilosis and ScMgm101 from S. cerevisiae lacking the mitochondrial import signal (Δ1–21 aa) was prepared using the Champion pET100 Directional TOPO Expression Kit (Life Technologies). The CpMGM101-Δ84 and ScMgm101 PCR products were amplified using the CpMGM101core_up_topo and CpMGM101_down2 primers and ScMgm_up2_topo and ScMgm_down, respectively (Supplementary Tables S1 and S2) and inserted into a linearized pET100/D-TOPO expression vector (Life Technologies). CpMgm101, chimera1 and chimera2 with an N-terminal 6× His-tag followed by a linker region from the pProEx-1 vector (Merck) lacking the mitochondrial import signal were prepared using in-fusion cloning (Clontech) and the primers CpMGM101_pProEx_up and CpMGM101_pProEx_down, chimera1_FW and chimera1_RV and chimera2_FW and chimera2_RV (Supplementary Tables S1 and S2). The CpMgm101-NoTag version was prepared by in-fusion cloning into a pOPINJ vector (24) as a 6× His-GST tagged protein. The 6× His-GST tag was removed by overnight incubation with PreScission protease at 6°C during purification of the protein according to the GE Healthcare protocol.
The recombinant proteins were expressed in Escherichia coli strain Rosetta 2 (DE3) (Merck). Bacteria were grown at 28°C in 1% (w/v) bactotryptone, 0.5% (w/v) yeast extract and 1% (w/v) NaCl, with 100 μg/ml carbenicillin and 34 μg/ml chloramphenicol and induced with 0.5 mM isopropylthio-β-D-galactoside for 3 h at 28°C. The cells were harvested and suspended in buffer A (50 mM Tris–HCl, pH 7.4, 1 M NaCl, 20% (v/v) glycerol, 5 mM CHAPS, 5 mM betaine, 10 mM imidazole) and sonicated on ice. The cell lysate was centrifuged 20 min at 100 000 × g and the supernatant was loaded onto a Ni Sepharose™ 6 Fast Flow column (GE Healthcare). The column was washed with 5 column volumes of buffer A containing 50 mM imidazole and then with buffer B (50 mM Tris–HCl, pH 7.4, 400 mM NaCl, 5 mM CHAPS, 5% (v/v) glycerol) with 50 mM imidazole. The protein bound to the column was then eluted in three steps with buffer B or buffer C (50 mM Tris–HCl, pH 7.4, 400 mM NaCl, 5% (v/v) glycerol) containing 0.1, 0.3 and 0.5 M imidazole. Purified proteins were analyzed by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie Brilliant Blue staining.
Gel filtration analysis
Analytical gel filtration was performed with a Superose 6 10/300 GL (GE Healthcare) column using buffer B or buffer C. The flow rate was 0.4 ml/min. The peak fractions were analyzed by SDS-PAGE, concentrated on a Microsep Advance 30K column (Pall) and stored at −25°C. The concentration of protein was determined using a BCA kit (Thermo Scientific).
Electrophoretic mobility shift assay (EMSA)
The sequences and structures of the oligonucleotides used for the electrophoretic mobility shift assays (EMSAs) are shown in Supplementary Table S3. The oligonucleotides were end labeled with FITC at the 5′ end. All substrates were prepared as described in (25).
The given amounts of both forms of CpMgm101 were incubated with 3 nM of fluorescently‐labeled DNA substrates at 30°C in 10 μl of buffer D (25 mM Tris–HCl, pH 7.4, 100 mM NaCl and 0.1 mg/ml bovine serum albumin (BSA)) for 10 min. After adding the gel loading buffer, the reaction mixtures were resolved on 0.6% (w/v) agarose gel in 0.5×TBE buffer (45 mM Tris-borate, 1 mM EDTA) at room temperature. The gels were scanned using an FLA-9000 Starion (Fujifilm) and quantified using ImageJ (http://imagej.nih.gov/ij/) (26). Dissociation constants (Kd) were calculated by fitting a 3-parameter Hill equation to the quantified intensities using OriginPro 8.5.1 (OriginLab, USA). Rabbit polyclonal antibodies against CpMgm101-Δ84 were prepared (Biogenes, Germany). The IgG fraction of the rabbit serum against CpMgm101 was purified on a Protein A-Sepharose column (GE Healthcare) and used for supershift assay.
Single-strand annealing assay
This assay was performed similarly as in Altmannova et al. (27). A total of 8 nM of Oligo2 and 3 nM of fluorescently labeled Oligo1 (Supplementary Table S3) were incubated at 30°C in 30 μl of buffer E (25 mM Tris–HCl, pH 7.5, 50 mM KCl, 1 mM DTT and 100 μg/ml BSA) with increasing concentrations of yRad52 or CpMgm101. A total of 10 μl aliquots were withdrawn after 4 and 10 min and treated with 0.15% SDS, 500 μg/ml proteinase K and 20 nM unlabeled Oligo1 for 3 min at 30°C. The samples were resolved by electrophoresis on 12% native polyacrylamide gels in 0.5×TBE buffer. The gels were then scanned using an FLA-9000 Starion (Fujifilm) and quantified using MultiGauge software (Fujifilm).
Strand invasion assay
The D-loop assay was performed as described by Sebesta et al. (28). Briefly, 40 nM of fluorescently labeled Oligo8 (a 90-mer) (Supplementary Table S3) was incubated for 10 min with increasing concentrations of either CpMgm101 (at 30°C) or hRad52 (at 37°C) in 10 μl of buffer F (10 mM Tris–HCl pH 7.5, 50 mM KCl, 1 mM DTT). The reaction was initiated by adding pBluescript replicative form I (90 ng/μl) and the mixture was incubated for 15 min. The reaction was stopped by the addition of 0.15% SDS and 500 μg/ml proteinase K and incubated for 5 min. The samples were resolved in a 0.9% w/v agarose gel in 1xTAE buffer (40 mM Tris-acetate, 1 mM EDTA). The gels were scanned using an FLA-9000 Starion (Fujifilm) and quantified using MultiGauge software (Fujifilm).
SEC-MALLS
MALLS measurements were performed with an Agilent Technologies 1260 Infinity HPLC system equipped with a MiniDAWN TREOS light-scattering detector (Wyatt Technology Corporation) and a SHODEX refractive index detector. A Superdex 200 10/300 GL column (GE Healthcare) was equilibrated in buffer C and purified CpMgm101 at a concentration of 8 mg/ml and Mgm101 chimera1 at a concentration 5 mg/ml were loaded onto the column in the same buffer. The data were analyzed using ASTRA software (Wyatt Technology Corporation).
Electron microscopy
The DNA template for CpMgm101 binding was a 738-bp EcoRI fragment derived from the mitochondrial telomere of the C. parapsilosis linear mitochondrial DNA. The fragment was prepared by digestion of the pTZ19R-E1 plasmid (2) with EcoRI followed by a 1 min incubation with exonuclease III to generate single-stranded overhangs at the 5′ termini. CpMgm101 and this DNA were mixed in a 10:1 ratio (w/w) and incubated for 10 min at 30°C in a buffer B. Immediately after incubation they were diluted in spraying buffer (100 mM ammonium acetate, 30% (v/v) glycerol, pH adjusted to 8.0 with NaOH) to a final concentration of 225 μg/ml CpMgm101 and 20 μg/ml DNA. The diluted samples were sprayed onto freshly cleaved mica chips and transferred into a BalTec MED020 high vacuum evaporator equipped with electron guns. The rotating samples were coated with 0.7 nm platinum at an angle of 7°, followed by 7 nm carbon at 45°. The produced replicas were floated off from the mica chips and picked up with 400 mesh Cu/Pd grids. The grids were inspected in an FEI Morgagni 268D TEM operated at 80 kV. Images were acquired using an 11 megapixel Olympus-SIS Morada CCD camera.
SAXS data collection and analysis
SAXS experiments were conducted at the P12 beamline at the PETRA III storage ring (EMBL, Hamburg, Germany). A Pilatus 2M pixel X-ray detector (Dectris) with a wavelength of 0.124 nm, a sample-to-detector distance of 3 m and a collection temperature of 20°C were used (29). CpMgm101 data were acquired by taking 20 successive 0.05 s frames of each 20 μl sample in buffer C; for buffer subtraction of the raw data, the buffer was analyzed under the same conditions. Guinier plots of data at 5, 4 and 2 mg/ml were linear at all but the smallest angles (30) and showed very similar scattering patterns. No significant time-dependent protein aggregation or radiation damage were observed under these conditions. The CpMgm101–DNA complex was prepared by mixing the protein with 100 μM of 34-bp-long ssDNA (5′-GCAGAATCTGGCCATGGTTGGCGAAGTCTCCCTC-3′) in a 3:1 ratio to a final protein concentration of 5 mg/ml. This mixture was incubated for 20 min at 30°C, then diluted with buffer C to concentrations appropriate for measurement.
CpMgm101-FPLC data (FPLC) were acquired at the P12 beamline, which possesses an on-line FPLC system, allowing an eluting sample to be continuously scanned. A 100 μl sample at a concentration of 11.39 mg/ml was separated by SEC (Size Exclusion Chromatography) on a Superose 6 10/300 GL column (GE Healthcare) using a Malvern FPLC system in buffer C. As the sample eluted, 3600 successive 0.995 s frames were collected. A contiguous set of 74 frames with the same radius of gyration (Rg) was selected from the top of the elution peak. These 74 were then averaged and the buffer was subtracted to give the dataset used for subsequent analysis.
After initial data integration and background subtraction, the SAXS data were analyzed using the ATSAS package (31). The Rg, the pair distance distribution function (P(r)) and the maximum intermolecular distance (Dmax) of the protein and protein–DNA complex were calculated using GNOM (32). All plots were visualized using R 3.2.1 (http://www.R-project.org/) (33). The web version of DAMMIF (34) was used to generate twenty ab initio low-resolution dummy-atom models, assuming no (that is, P1) symmetry. These models were then filtered and averaged using DAMAVER (35). The averaged structure as then refined against the experimental data using DAMMIN (36). The models were visualized in PyMol (37) and pictures were produced using either PyMol or Chimera (38).
RESULTS
CpMgm101 stabilizes the mitochondrial genome of the S. cerevisiae mgm101–1ts mutant
The CPAR2_106590 (CPAG_04901) open reading frame of the C. parapsilosis CDC317 genome encodes a putative 30 kDa protein (266 amino acids), which exhibits a high degree of homology with the S. cerevisiae Mgm101 protein (Supplementary Figure S1). The core region (Ile-99–Lys-236 in ScMgm101) predicted to bind mtDNA is highly conserved. The pI of the CpMgm101 protein lacking the mitochondrial presequence is 8.48 and molecular weight is 26.58 kDa.
To verify that CpMgm101 is in fact a functional homolog of ScMgm101 we tested its ability to suppress a mtDNA maintenance defect in a thermosensitive mutant of S. cerevisiae (mgm101–1ts). At restrictive temperature (37°C), the mtDNA synthesis of this mutant is blocked and it progressively loses mtDNA during subsequent cell divisions, resulting in respiratory deficiency (11,12). To test the ability of CpMgm101 to complement this mutant, we transformed S. cerevisiae mgm101–1ts cells with plasmids containing either CpMGM101 or ScMGM101 under the control of a GAL1 promoter. Following an initial growth period in galactose-containing medium (SGal), these cells were grown at either permissive (25°C) or restrictive (37°C) temperatures for 2 days in SGal medium to allow a certain level of Mgm101 to accumulate. Because galactose is primarily a fermentation substrate, the culture was then plated onto glycerol-containing media (SGly) to select for respiratory-competent cells maintaining wild-type mtDNA and incubated for an additional 7 days. Such long incubation times are needed because the cells grow slowly in synthetic media and the loss of mtDNA in mutant cells can only become apparent after several generations have passed. Figure 1 shows these plates at the end of the incubation period. Although this particular strain has a fairly high background at 37°C, there is still a visible difference in the number of respiratory-competent cells between the control strains and those carrying the ScMGM101 and CpMGM101 genes. It can also be seen that CpMgm101 is able to functionally replace the defect in S. cerevisiae mgm101–1ts nearly as well as ScMgm101, indicating that both proteins may play similar roles in the maintenance of the mitochondrial genome.
Figure 1.

Functional complementation of the mgm101–1ts mutation in Saccharomyces cerevisiae with CpMgm101. S. cerevisiae M2915–7C cells transformed with the indicated plasmids were cultivated for 2 days in SGal medium at permissive (25°C) or restrictive (37°C) temperatures. The cultures were spotted in serial dilutions onto SGly plates and cultivated at the indicated temperatures for 7 days. pYES2 and pYES2/CT are empty vectors while pYES2-ScMgm101 and pYES2-CpMgm101 contain ScMgm101 and CpMgm101, respectively.
CpMgm101 co-localizes with mtDNA in C. parapsilosis cells
Mitochondrial nucleoids can be easily identified as discrete structures within the mitochondria using the dsDNA-specific fluorescent dye 4′,6-diamidino-2-phenylindole (DAPI) (39,40). An analysis of 528 C. parapsilosis cells stained with DAPI indicates that each cell contains on average 10.7 ± 4.3 nucleoids (Supplementary Figure S2). To confirm the subcellular localization of CpMgm101, it was tagged at the C-terminus with yEGFP3. Expressing CpMgm101-yEGFP3 in C. parapsilosis gave rise to discrete dots within the mitochondria, which co-localized with the DAPI-stained mitochondrial nucleoids (Figure 2). In contrast to ScMgm101, which does not extensively co-localize with the nucleoids (10), CpMgm101 associates with the entire visualized cellular nucleoid population and also localizes to the cell nucleus (Figure 2). Because ScMgm101 is required for the repair of mtDNA that has been oxidatively damaged by H2O2 (10), we tested the influence of H2O2 on the intracellular localization of CpMgm101-yEGFP3 (Figure 2A). No change in CpMgm101 localization was detected by this treatment. CpMgm101 therefore has a dual cellular localization, reflecting its function in both the nucleus and mitochondria of C. parapsilosis.
Figure 2.

Intracellular localization of CpMgm101-yEGFP3. The expression of CpMgm101-yEGFP3 from pBP8-CpMgm101 was induced by transferring the Candida parapsilosis cells into methionine-free SD media for 12 h. The cells were then stained with 57 μM DAPI for 2 h in the same medium. The blue staining color of DAPI was changed to red in the microscope operating software. (A) Cells treated with 25 mM H2O2 for 1 h. (B) Control cells without such treatment. The cells were visualized using an Olympus FV1000 confocal microscope. The bar represents 5 μm. No obvious change in CpMgm101 localization is apparent.
CpMgm101 forms homo-oligomers in vitro
To biochemically characterize CpMgm101, two different recombinant forms were designed and expressed in E. coli: a full-length, mature form (CpMgm101) and a truncated form lacking the first 84 amino acids (CpMgm101-Δ84). Both proteins possessed an N-terminal 6× His-tag. An untagged version of the full-length protein (CpMgm101-NoTag) was also prepared from a 6× His-GST tagged form by cleaving off the tag during purification as described in ‘Materials and Methods’ section above. It should be noted that both full-length forms lack the first 29 amino acids; since this region is predicted to be a mitochondrial import presequence by MitoProt II (41), it was designed out of the full-length constructs. The purity of the His-tagged CpMgm101 protein after SEC was assessed by SDS-PAGE (Supplementary Figure S3).
To determine the molecular size of CpMgm101, we performed size-exclusion chromatography using a Superdex 200 10/300 GL (GE Healthcare). As shown in Supplementary Figure S4A, CpMgm101 eluted as a single peak with a molecular weight of ∼100 kDa. No protein fraction corresponding to the monomeric form (∼30 kDa) was detected, indicating that the protein likely exists as a multimer in solution. We then characterized the recombinant protein by size-exclusion chromatography coupled to multiangle laser light scattering (SEC-MALLS; Supplementary Figure S4A), which showed that CpMgm101-NoTag forms an oligomer with a molecular weight of 84.62 ± 0.17 kDa, corresponding to a trimer (theoretical MW 80.21 kDa). Similarly, the 6× His-tagged CpMgm101 had a molecular weight of 97.95 ± 0.17 kDa (theoretical MW 89.27 kDa). A precise molecular weight could not be determined for CpMgm101-Δ84 due to its instability, high tendency to aggregate and CHAPS binding to it. It may be noted here that the concentration of NaCl had no perceptible influence on the oligomeric state of the protein (Supplementary Figure S5).
To explore the possible influence of the N-terminal part of the protein on the oligomeric state of CpMgm101, we also prepared two chimeric proteins combining residues 30–96 of CpMgm101 with the C-terminal part of ScMgm101 (residues 104–269; chimera1) and residues 23–103 of ScMgm101 with residues 97–266 of CpMgm101 (chimera2). Neither chimeric protein was able to oligomerize properly. According to SEC-MALLS, chimera1 exclusively formed monomers with molecular weight 31.53 ± 0.94 kDa (Supplementary Figure S4B), while chimera2 was unstable.
CpMgm101 binds a wide variety of DNA substrates
Since CpMgm101 is involved in the maintenance of mitochondrial DNA we tested its DNA-binding properties. First, we compared its affinity for ssDNA and dsDNA substrates (Supplementary Table S3) using EMSAs under different ionic strength and pH conditions. As shown in Supplementary Figure S6, CpMgm101 efficiently binds both DNA substrates in broad pH (6.8–9.5) and NaCl concentration (0–700 mM) ranges. Like ScMgm101 (15) and Rad52 (42,43), CpMgm101 also has a slightly higher affinity for ssDNA than dsDNA (Table 1), with a Kd of 0.22 μM for ssDNA versus 0.37 μM for dsDNA. It should be noted, however, that although the affinity of CpMgm101 for ssDNA is similar to that of ScMgm101, its affinity for dsDNA is much higher. This could account for its constant association with the C. parapsilosis mitochondrial nucleoids and might also indicate that it has additional, species-specific roles within C. parapsilosis.
Table 1. Kd values in μM of CpMgm101 and CpMgm101-Δ84 for different DNA substrates.
| ssDNA | dsDNA | nick DNA | X-O | Fork | Y-form | 5′ FLAP | 3′ FLAP | 5′ over | 3′ over | |
|---|---|---|---|---|---|---|---|---|---|---|
| CpMgm101 | 0.22 | 0.37 | 0.33 | 0.18 | 0.19 | 0.07 | 0.27 | 0.06 | 0.46 | 0.13 |
| CpMgm101-Δ84 | 0.81 | 2.37 | 2.49 | 1.27 | 1.34 | 0.24 | 1.23 | 0.57 | 1.72 | 0.13 |
| ratio | 3.68 | 6.41 | 7.55 | 7.06 | 7.05 | 3.43 | 4.56 | 9.50 | 3.74 | 1 |
We also tested the affinity of CpMgm101-Δ84 for DNA. The 85–266 residue region of CpMgm101 has a high homology to the corresponding region of the ScMgm101 protein (Supplementary Figure S1) and contains a highly conserved region, predicted to possess DNA binding and annealing activities (15). We found that the binding properties of CpMgm101-Δ84 to ssDNA (Kd = 0.81 μM) and dsDNA (Kd = 2.37 μM) are much weaker than those of the full-length protein (Table 1), demonstrating that the N-terminal part of CpMgm101 may have a role in modulating its DNA-binding properties.
To characterize the preferences of CpMgm101 for different DNA structures, we tested several DNA substrates by EMSA. CpMgm101 exhibited the highest affinity for DNA molecules carrying 3′ ssDNA overhangs (Y-form, 3′ FLAP, 3′ overhang) and for substrates with complex structures (X-O and Fork) (Figure 3A,B). CpMgm101-Δ84 retained the preferences for 3′ overhangs (Y-form, 3′ FLAP, 3′ overhang) and complex structures (X-O and Fork), but generally exhibited less efficient binding. The only substrate bound similarly by both protein forms was the 3′ overhang DNA (Figure 3C; Table 1).
Figure 3.

CpMgm101 binds to a range of DNA structures with free 3′ or 5′ ends. The EMSA experiments were performed with either CpMgm101 or CpMgm101-Δ84 (concentration is expressed per monomer) and 3 nM of the corresponding DNA substrate at 30°C for 10 min (see ‘Materials and Methods’ section). Lane 1 shows a control containing the DNA probe without CpMgm101. Samples were separated in 0.6% agarose gel in 0.5× TBE. (A and B) The DNA binding properties of CpMgm101 to different DNA substrates. (1) control, (2) 0.5 μM CpMgm101, (3) 1 μM, (4) 2 μM, (5) 3 μM, (6) 5 μM, (7) 2 μM + 1 μl anti CpMgm101 IgG antibodies (B only). The addition of anti-CpMgm101 antibodies to the CpMgm101–DNA reaction mixture resulted in a supershift, this complex is too large to enter the 0.6% agarose gel and the entire DNA probe remains in the well (lane 7). (C) The DNA binding properties of CpMgm101-Δ84 to different DNA substrates. (1) control, (2) 0.5 μM, (3) 1 μM, (4) 2 μM and (5) 5 μM.
These results suggest that CpMgm101 may participate in recombination activities. To test this idea, we also determined whether CpMgm101 catalyzes ssDNA annealing and D-loop formation. To assay the annealing activity, different concentrations of CpMgm101 and yeast Rad52 (yRad52) (as a positive control) were incubated with two complementary 49-base oligonucleotides (Oligo1 and Oligo2). As shown in Figure 4A, the oligonucleotides had completely annealed to form dsDNA after 10 min of incubation in the presence of 50 nM CpMgm101, comparable to the yRad52 control reaction (Figure 4B).
Figure 4.

The single-strand annealing and strand invasion activities of CpMgm101 protein. The single-strand annealing activities of (A) CpMgm101 and (B) yRad52. The reactions were carried out using 3 nM of labeled Oligo1 and 8 nM of unlabeled Oligo2 with increasing concentrations of protein (see ‘Materials and Methods’ section). The plot shows the averaged results of three independent experiments. (C) The strand invasion activity of CpMgm101. The reactions were carried out on pBluescript replicative form I using a protein–Oligo8 complex that had been previously formed by incubating 40 nM of fluorescently labeled 90-base Oligo8 with increasing concentrations (0.38, 0.75, 1.5, 3 μM) of CpMgm101 (at 30°C) or 3 μM of hRad52 (at 37°C) proteins for 10 min (see ‘Materials and Methods’ section).
To assess the ability of CpMgm101 to carry out strand invasion, we first assembled a nucleoprotein complex containing a 90-base oligonucleotide (Oligo8 in Supplementary Table S3) and one of CpMgm101, or human Rad52 (hRad52) (as a positive controls). This complex was then incubated with a homologous, super-coiled dsDNA plasmid. As seen in Figure 4C, CpMgm101 is able to create D-loops similarly to hRad52 protein. Taken together, CpMgm101 possess strand annealing and strand invasion activities similar to Rad52 protein, supporting its functional conservation.
Electron-microscopy of CpMgm101–DNA complexes
To directly visualize a complex of CpMgm101 with mitochondrial telomeric DNA we used transmission electron microscopy (EM) and rotary shadowing. The linear mtDNA of C. parapsilosis terminates in arrays of 738-bp-long telomeric repeats with 110 nt-long single-stranded overhangs at the very 5′ end of the arrays (2). A DNA substrate was prepared which mimics the very end of the native telomere in the following manner. First a 738-bp-long EcoRI telomeric fragment was isolated followed by generation of single-stranded overhangs at each 5′ end by exonuclease III treatment, resulting in an overhang of almost 200 nt (Figure 5A). The electron micrographs show that CpMgm101 forms homogeneous ring-shaped structures at the ssDNA ends (Figure 5B–F). A closer inspection suggests that several CpMgm101 oligomers bind to the ssDNA as a filament until they reach the double-stranded region, though it is unclear how these filaments then form the loop structures. The transition from the CpMgm101-bound ssDNA to the dsDNA is probably most apparent in Figure 5C (arrow). It appears that binding is accompanied by bending of the ssDNA and the formation of ring structures 20 nm across with the DNA being wrapped around the protein. At lower protein concentrations, these ring structures continue to form, but there are fewer of them and more uncomplexed DNA. Thus the formation of these structures at the DNA ends seems to be dependent on the number of protein oligomers, the length of the ssDNA segment and possibly also on the structure of the DNA itself as reflected by the ability of CpMgm101 to bind to various replication and recombination intermediates. In particular, the EMSA results indicate that more than one protein molecule is bound to the ssDNA probe (Figure 3B). In addition, the binding of CpMgm101 to the single-stranded ends of this model telomere also induces the formation of bends and loops within the double-stranded part of the molecules (Figure 5F). It may be noted here that no large-scale structures can be seen in EM micrographs of CpMgm101 in the absence of DNA.
Figure 5.

The CpMgm101–DNA complex visualized by electron microscopy. (A) Graphical representation of the ss-ds-ss DNA template (250 ± 30 nm) with the dsDNA region at the center (150 nm) and long 5′ ssDNA overhangs at the ends (50 nm), (B–F) representative EM pictures of CpMgm101 bound to a model telomere (see ‘Materials and Methods’ section). Note the formation of circular structures at the single-stranded ends. These structures are about 20 nm across. The arrow in panel (C) indicates a junction between a Mgm101-bound ssDNA segment and an unbound dsDNA segment. The arrows in panel (F) indicate the formation of bends and loops within the double-stranded part of the DNA molecules. (G and H) control EM pictures of the DNA template alone. The scale bar is 50 nm in length.
Structural analysis of CpMgm101 by SAXS
To gain further insight into the structural details of CpMgm101 DNA binding, we used small-angle X-ray scattering (SAXS) in an attempt to determine the molecular envelope of CpMgm101 and its complex with DNA. Unfortunately, CpMgm101 shows effects consistent with either moderate aggregation or strong inter-particle attractions at every concentration studied. Because only a single CpMgm101 oligomeric species could be isolated by size-exclusion chromatography (Supplementary Figures S4 and S5), strong inter-particle attractions appear more likely. Similar behavior is often observed even in otherwise well-behaving proteins at higher concentrations (e.g. BSA (44,45) and ovalbumin (46,47)). Because these effects were present at the lowest concentration studied, the normal expedient of merging the highest and lowest concentration datasets was not helpful; moreover, extrapolation to zero concentration only diminished, but did not eliminate, these effects. This is not unexpected (48). These inter-particle effects could be eliminated, apparently completely, by measuring the X-ray scattering from a continuous series of samples as they eluted from a Superpose 6 10/300 column on an FPLC system whose elution chamber was in the path of the X-ray beam (it may be noted that this sample also lacked a His-tag). The Rg for this set was 4.59 ± 0.01 nm, Dmax was determined to be 14.90 nm and the empirical power-law relationship between MW and Vc2/Rg (49) gave a MW of 80.9 kDa, which corresponds to a trimeric particle. The scattering curve after buffer subtraction, Guinier plot, characteristic function plot, dimensionless Kratky plot and Porod-Debye plot for this dataset are shown in Supplementary Figure S7.
Bulk scattering measurements were also carried out for uncomplexed CpMgm101 and a CpMgm101–ssDNA complex at concentrations of 2, 4 and 5 mg/ml. The experimentally derived parameters for these datasets are summarized in Supplementary Table S4 together with those for the FPLC dataset; the plots for these data sets may be found in Supplementary Figures S8 and S9. It may be seen from Supplementary Table S4 the Rg and Dmax for the lowest concentration of CpMgm101 exceeds those from the FPLC dataset and that all parameters are increased in the CpMgm101–ssDNA complex.
The Kratky and Porod-Debye plots from the bulk-scattering datasets indicate that CpMgm101 is a properly folded protein. Conversely, the Porod-Debye plot for the FPLC dataset shows that the scattering intensity does not decay as s−4, but as s−3 (Supplementary Figure S7E inset). Moreover, comparing the dimensionless Kratky plot of the FPLC dataset with those of the bulk-scattering datasets (Supplementary Figures S10 and S11, panel F), shows that the peak from the FPLC dataset does not completely return to the baseline, as those from the bulk-scattering datasets do, but drops part way and then rises again. Both of these features are consistent with the presence of flexible regions within an otherwise well-folded protein (50). Since the N-terminal region of CpMgm101 is predicted to be intrinsically disordered, these features are not unexpected. Their absence in the Kratky and Porod-Debye plots suggests that the close approaches between the individual particles as a result of their attractive interactions stabilize these flexible regions.
Comparing the 2 mg/ml plots of uncomplexed and DNA-complexed CpMgm101 (Supplementary Figure S10) shows a clear increase in scattering at the lowest angles for the DNA complex, but no apparent change otherwise. At 5 mg/ml (Supplementary Figure S11), however, clear changes do occur. The most notable changes occur around s = 0.5 nm−1, and the corresponding region of the Kratky plot shows the appearance of a shoulder. A shoulder in the Kratky plot normally reflects the presence of a second domain, most commonly separated from the protein by a flexible linker. These differences between the uncomplexed and complexed forms of CpMgm101 suggest that the presence of ssDNA causes an increase in the interparticle attractive forces, possibly mediated directly by the ssDNA molecules themselves. The CpMgm101–ssDNA complex could not be cleanly separated using size-exclusion chromatography, lending some indirect support to this hypothesis.
Only the FPLC dataset could be used for modeling the shape of the scattering particle; the inter-particle interference effects prevent a reliable shape from being generated from the bulk-scattering datasets. To generate the envelope structure of the CpMgm101 scattering particle, 20 independent ab initio dummy atom models were produced using DAMMIF assuming P1 symmetry. These were filtered and averaged using DAMAVER and the resulting averaged model was refined using DAMMIN. The models produced from the FPLC dataset had an average χ2 of 1.077 ± 0.006 and superimposed with an averaged normalized spatial discrepancy of 0.92 ± 0.05; the refined averaged model had a χ2 of 1.055 (Figure 6). The final model had a rough, somewhat asymmetric C shape with an angle between the ends and the middle of the C of approximately 107°. A number of relatively small protrusions were also present, possibly representing the flexible regions of the N-terminus.
Figure 6.
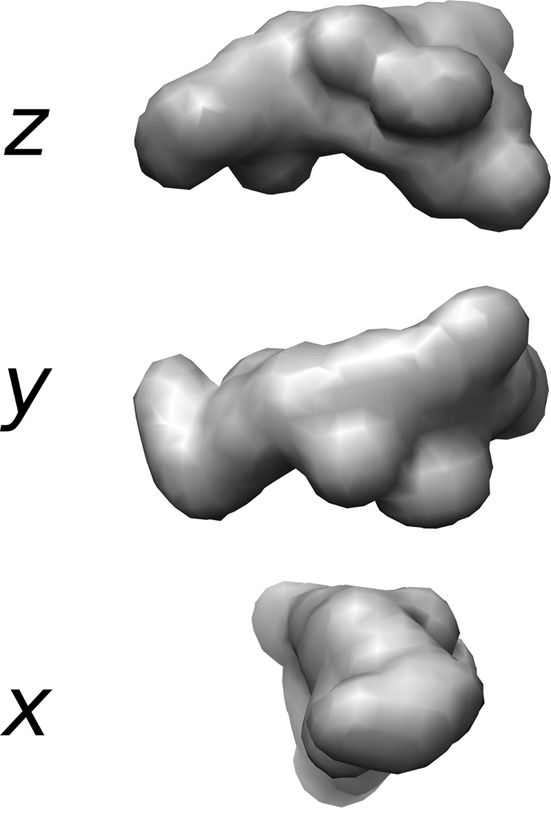
The ab initio model of CpMgm101 from SAXS data. The CpMgm101 model generated by DAMMIN from the average of 20 DAMMIF models derived from the FPLC-SAXS dataset. The view is down the z axis (chosen arbitrarily, top), the y axis (middle) and x axis (bottom). The overall structure is roughly C shaped with a central angle of approximately 107°. The surface envelopes of these bead models were drawn at a resolution of 25 Å using Chimera.
Higher symmetry models (P8, P12 and P14) were also created in an attempt to determine if CpMgm101 could be successfully modeled using the ring shape exhibited by uncomplexed ScMgm101 and observed by EM for the CpMgm101–DNA complex. None of these models fit the data well (the best χ2, for the P12 model, was >75); considering the size of the soluble particle given by SEC-MALLS, this is not unexpected.
DISCUSSION
Mgm101 is a Rad52 homolog which appears to be involved in the homologous recombination of mtDNA (15). In S. cerevisiae, it can be found in both the mitochondria, where it forms a component of the mitochondrial nucleoids and appears to be vital for mtDNA maintenance (10–12), and in the nucleus, where it takes part in the Pso2-independent pathway for repairing interstrand cross-links (16).
The protein can be divided into two functional domains: an unconserved, N-terminal segment and a conserved, Rad52-like core (18). The final 32 residues of this core domain are themselves sufficient for binding ssDNA in vitro (51). The unconserved N-terminal segment shows low homology even between proteins from closely related organisms and likely exhibits species-specific differences. For example, a chimeric Mgm101 possessing a S. cerevisiae Mgm101 N-terminus and an Acropora millepora (coral) core functions in S. cerevisiae cells much like the wild-type, but a chimera with a ScMgm101 core and an A. millepora N-terminus is functionally inactive (52). Since the N-terminus is largely intrinsically disordered (52) and since such regions frequently occur at protein–protein recognition sites, it was speculated that this difference largely arose because other mitochondrial binding partners were not able to recognize the A. millepora N-terminus. In this report, we provide evidence, based on the behavior of a CpMgm101-Δ84 mutant and two ScMgm101 and CpMgm101 chimeras formed by exchanging their N-terminal domains, that the N-terminal region is involved in the formation of Mgm101 oligomers. We also show that it affects the protein's DNA-binding properties.
Our characterization of C. parapsilosis Mgm101 reveals many similarities to as well as striking differences with ScMgm101. In the first place, CpMgm101 appears able to complement the S. cerevisiae mgm101–1ts mutation, indicating that the two proteins are functionally similar. In addition, CpMgm101 binds ssDNA with nearly the same affinity as ScMgm101. On the other hand, ScMgm101 binds to ssDNA about 5.5× more strongly than to dsDNA, but CpMgm101 ssDNA binding is only about 1.7× stronger. Furthermore, CpMgm101 is associated extensively with the mtDNA and H2O2 treatment does not cause any apparent changes in localization.
CpMgm101 is able to bind to several recombination-generated replication intermediates with affinities comparable to or better than that for ssDNA, including the Holliday junction (X–O), Fork, Y-form, 5′ flap, 3′ flap, and 3′ overhang (Table 1). The affinities for many of these drop by a large amount when the N-terminal 84 amino acids are missing. These changes can be roughly grouped into those which drop by the same relative amount as ssDNA (about 3.5–4.5×, including ssDNA, Y-form, 5′ flap and 5′ overhang), those which drop by the same relative amount as dsDNA (∼6.5–7.5×, including dsDNA, nick DNA, the Holliday junction and fork), and those which appear to be a combination of the two (∼9.5×, the 3′ flap). These changes suggest that the N-terminal segment affects the ssDNA and dsDNA bindings separately. This is consistent with the idea that Mgm101, like Rad52, has two separate DNA binding sites (53), though this has yet to be experimentally verified.
It is perhaps surprising that CpMgm101 has a higher affinity for 3′-ended ssDNA structures than 5′-ended ones given that the telomeres of the C. parapsilosis linear mtDNA terminate with 5′ extensions. This feature may reflect the evolutionary heritage of Mgm101. ScMgm101 appears to be most similar to Rad52 homologs from bacteriophages (15). Bacteriophage T4 is thought to initiate DNA replication via a recombinant process by catalyzing the invasion of a 3′ ssDNA end into a homologous DNA duplex (54). Of all the substrates tested, the 3′ overhang alone was unaffected by removal of the N-terminal segment, the evolutionarily newest part of the molecule.
ScMgm101 appears to exist in solution as a large, 14-subunit ring in the absence of DNA. Mutations which disrupt this ring lead to a non-functional protein (17). These ring structures have been suggested to be a storage form (15). Since CpMgm101 is constantly associated with the mitochondrial nucleoid, a separate storage form would either be unnecessary for it or be quite different. No large-scale structure could be detected by EM in the absence of DNA, which is consistent with the FPLC-SAXS and SEC-MALLS studies. SEC-MALLS and FPLC-SAXS both suggested that the soluble form of CpMgm101 was a trimer (Supplementary Figure S4 and Table S4). This would make it one of the smallest soluble units for a Rad52-type recombination protein yet reported. Bulk-scattering SAXS experiments seem to suggest that the actual solution form is quite a bit larger, but only the trimer could be clearly and consistently identified by SEC, indicating that these larger forms are likely to be only loosely associated, and therefore not true oligomers. Alternatively, the larger forms may be only transiently stable. Since deletion of the N-terminal segment caused CpMgm101 to aggregate, it seems likely that this segment is at least partially responsible for its quaternary form. Indeed, exchanging the N-terminal segments of CpMgm101 and ScMgm101 resulted in two chimeric proteins that were largely unable to oligomerize, showing that this segment is actually crucial.
EM structures of CpMgm101 complexed with a model mitochondrial telomere revealed the presence of ring structures at the telomere's single-stranded ends. These ring structures are 20 nm across, suggesting that each ring contains four trimers based on the Dmax for the CpMgm101–ssDNA complex detected by SAXS. Indeed, it is possible, using the model produced from the FPLC dataset, to construct a ring-shaped oligomer of the appropriate size by joining four trimers end-to-end (Supplementary Figure S12). Since two rings were observed at each end of the 200-nt ssDNA segment, it can be inferred that each CpMgm101 trimer probably binds less than 25 nt of ssDNA. The fact that this ring was not visible by SAXS suggests that the binding of relatively short ssDNA segments alone is not sufficient to trigger the formation of these rings, though it does seem sufficient for increasing the tendency of the individual trimers to associate. Either binding of very much longer segments is needed, or a ssDNA–dsDNA junction is necessary.
TEM images of a ScMgm101–ssDNA complex show that it forms condensed nucleoprotein complexes, similar to those observed for Sak from the Lactococcus lactis phage ul36 and that the 20 nm rings which appear without DNA cannot be seen (15). In contrast, our EM images of CpMgm101 show no large-scale structures in the absence of DNA, but 20 nm rings appear when CpMgm101 is complexed to DNA with single-stranded ends. At least part of this discrepancy may lie in the conditions under which the two sets of images were taken: the ScMgm101–ssDNA images reported by (15) were taken under high ssDNA, low protein conditions, while the ones reported here were taken under low ssDNA, high protein conditions. This suggests that there might be notable differences in the mechanisms of action of CpMgm101 and ScMgm101. Given that CpMgm101 can functionally complement the S. cerevisiae mgm101–1ts mutation, however, the two mechanisms must not be very dissimilar and the outline of ScMgm101's role in recombination-mediated mtDNA repair given in (55) may be equally valid for CpMgm101.
A separate question concerns whether CpMgm101 and ScMgm101 function like Rad52. To answer this question directly, we carried out both ssDNA annealing and D-loop formation assays. We found that CpMgm101 possesses both ssDNA annealing activity and strand-invasion activity comparable to Rad52 (Figure 4). Structurally, EM studies of hRad52 showed that the protein forms 10 nm rings in the absence of DNA and that in a hRad52–DNA complex, these rings are distributed along the DNA in structures that were described as ‘beads on a string’ (56). Partially as a consequence of this, the models of hRad52 function that have been proposed all assume that these rings are the DNA-binding form and are the units actively involved in homology search and annealing (57,58). Because CpMgm101 forms rings in the presence of ssDNA, it appears as though models proposed to describe the behavior of hRad52 may also be valid for CpMgm101. On the other hand, because there are no rings in the ScMgm101–ssDNA complexes, it may carry out its recombination and annealing activities using a different molecular mechanism. These hypotheses will require further investigation. In particular, it will be necessary to determine whether ScMgm101 is able to form rings under somewhat higher protein concentrations than have been previously studied.
We have shown here that CpMgm101 can bind to several recombination-generated replication intermediates with affinities comparable to or better than those for ssDNA, including the Holliday junction (X–O), Fork, Y-form, 5′ flap, 3′ flap and 3′ overhang (Table 1). We have also shown that CpMgm101, like Rad52, is able to catalyze ssDNA annealing and strand invasion, suggesting that it too can take part in homologous recombination. Given that mtDNA replication in C. parapsilosis seems to proceed through a recombination-initiated process (19) and that CpMgm101 appears capable of playing a role in homologous recombination, it seems likely that CpMgm101 may play an important role in both mitochondrial telomere maintenance and mtDNA replication in C. parapsilosis.
Figure 7 shows a scheme by which mitochondrial telomeres may be maintained through the use of t-circles in C. parapsilosis (for more detail, see (1)). Linear mtDNA molecules ending in a t-array terminate with 5′ ssDNA overhangs that are either bound by a mitochondrial telomere-binding protein (mtTBP; (7,8)), or fold into t-loop structures (59). Although the actual influence of mtTBP on t-loop formation cannot presently be determined, phosphorylation of mtTBP has been shown to inhibit its ability to bind mitochondrial telomeres in vitro (60), which may facilitate t-loop formation. These t-loops can be pinched off to form t-circles; t-circles can also be formed by homologous recombination between two repeat units. In either case, the t-circles can then undergo rolling-circle replication, thereby amplifying the t-arrays they contain (5,6). These newly replicated t-arrays can then be used to lengthen the mtDNA telomeres by recombination-dependent elongation. Each step in the above process involves an intermediate structure similar to those that CpMgm101 is able to bind to with the same affinity that it binds ssDNA. Both Mgm101 and mtTBP (7,9) are able to bind to both telomeres and to internal, single-stranded regions within the mtDNA. Although the affinity of mtTBP for telomeric overhangs has not been quantified, it is presumed to be relatively high since it belongs to the family of mitochondrial single-stranded DNA binding proteins, which exhibit Kds for ssDNA between 10−8–10−9 M (61). In any case at present it is not clear whether CpMgm101 and mtTBP can co-exist on the same oligonucleotide or if they compete for the same binding site.
Figure 7.

Potential involvement of CpMgm101 in the maintenance of mitochondrial telomeres. (A) Organization of the mitochondrial telomere of Candida parapsilosis (6). (B) A schematic illustrating the process by which mitochondrial telomeres may be replicated or extended (adapted from (1)). The irregular red objects indicate CpMgm101 in each structure to which CpMgm101 may bind. Those DNA recombination transactions potentially mediated by CpMgm101 are indicated by red arrows. Beginning at upper left, linear mtDNA telomeric ends are either protected by mtTBP (7,8) or folded into t-loops (59). The transition from an open telomere to closed t-loop structure may be facilitated by the removal of mtTBP which occurs upon phosphorylation of the protein (60). The t-loops may then be excised from the end of the mtDNA to produce t-circles. Alternatively, t-circles may be produced by homologous recombination (perhaps involving CpMgm101) within the t-arrays. These t-circles can be replicated by rolling-circle replication, and the resulting t-arrays may then be used to extend the mtDNA telomere by recombination-dependent elongation. (The dashed line shown in rolling circle replication intermediate depicts a newly synthesized lagging DNA strand).
All of these characteristics taken together suggest that CpMgm101 plays a role in the recombination-dependent replication of linear mtDNA and perhaps in mitochondrial telomere maintenance. This is supported by the ability of CpMgm101 to bind to a number of DNA substrates resembling replication or recombination intermediates, indicating that the protein may be involved in a number of recombination transactions. Specifically, CpMgm101 may promote (i) invasion of the 5′ ssDNA overhang into a dsDNA region of the mitochondrial telomere, resulting in the formation of t-loop structures; (ii) the formation and amplification of t-circles and (iii) recombination of the termini of linear mtDNA molecules with linear or circular extragenomic telomeric DNA (Figure 7). Additional experiments will, of course, be needed to verify the participation of CpMgm101 in each of these processes.
Supplementary Material
Acknowledgments
The authors would like to thank Kristina Djinović-Carugo (Max F. Perutz Laboratories, Vienna, Austria) for SEC-MALLS support, Miroslav Chovanec (Cancer Research Institute SAS, Slovakia) for plasmids and experiments assistance, BioSAXS (EMBL, Hamburg, Germany) for assistance with SAXS measurements, Karel Říha (Gregor Mendel Institute, Austrian Academy of Sciences, Vienna, Austria) for EM support, Katarína Procházková (Faculty of Natural Sciences, Comenius University, Slovakia) for fluorescence microscopy assistance, Ferdinand Molnár (University of Eastern Finland, Kuopio, Finland) for experiments assistance, Lucia Martináková for technical support, Vladena Bauerová (both of the Institute of Molecular Biology SAS, Slovakia) for SAXS assistance and Xin Jie Chen (State University of New York Upstate Medical University, NY, USA) and Geraldine Butler (Conway Institute, University College Dublin, Ireland) for yeast strains.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Howard Hughes Medical Institute [55005622 to J.N.]; Slovak Research and Development Agency [APVV-0123-10, APVV-14-0253 to J.N., APVV-0035-11 to L.T.]; Slovak Grant Agency [VEGA 2/0122/11, VEGA 2/0113/14 to E.K., VEGA 1/0333/15 to J.N., MVTS-1520 to E.K.]; Czech Science Foundation [GACR P207/12/2323, 13-26629S to L.K.]; European Regional Development Fund [Project FNUSA-ICRC, CZ.1.05/1.1.00/02.0123 to L.K.]; Federal Ministry of Economy, Family and Youth through the initiative ‘Laura Bassi Centres of Expertise’ [253275 to J.K.]; Austrian Science Fund (FWF) [P22276 to J.K.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Tomaska L., Nosek J., Kramara J., Griffith J.D. Telomeric circles: universal players in telomere maintenance? Nat. Struct. Mol. 2009;16:1010–1015. doi: 10.1038/nsmb.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nosek J., Dinouel N., Kovac L., Fukuhara H. Linear mitochondrial DNAs from yeasts: telomeres with large tandem repetitions. Mol. Gen. Genet. 1995;247:61–72. doi: 10.1007/BF00425822. [DOI] [PubMed] [Google Scholar]
- 3.Nosek J., Novotna M., Hlavatovicova Z., Ussery D.W., Fajkus J., Tomaska L. Complete DNA sequence of the linear mitochondrial genome of the pathogenic yeast Candida parapsilosis. Mol. Genet. Genomics. 2004;272:173–180. doi: 10.1007/s00438-004-1046-0. [DOI] [PubMed] [Google Scholar]
- 4.Kovac L., Lazowska J., Slonimski P.P. A yeast with linear molecules of mitochondrial DNA. Mol. Gen. Genet. 1984;197:420–424. doi: 10.1007/BF00329938. [DOI] [PubMed] [Google Scholar]
- 5.Tomaska L., Nosek J., Makhov A.M., Pastorakova A., Griffith J.D. Extragenomic double-stranded DNA circles in yeast with linear mitochondrial genomes: potential involvement in telomere maintenance. Nucleic Acids Res. 2000;28:4479–4487. doi: 10.1093/nar/28.22.4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nosek J., Rycovska A., Makhov A.M., Griffith J.D., Tomaska L. Amplification of telomeric arrays via rolling-circle mechanism. J. Biol. Chem. 2005;280:10840–10845. doi: 10.1074/jbc.M409295200. [DOI] [PubMed] [Google Scholar]
- 7.Nosek J., Tomaska L., Pagacova B., Fukuhara H. Mitochondrial telomere-binding protein from Candida parapsilosis suggests an evolutionary adaptation of a nonspecific single-stranded DNA-binding protein. J. Biol. Chem. 1999;274:8850–8857. doi: 10.1074/jbc.274.13.8850. [DOI] [PubMed] [Google Scholar]
- 8.Tomaska L., Nosek J., Fukuhara H. Identification of a putative mitochondrial telomere-binding protein of the yeast Candida parapsilosis. J. Biol. Chem. 1997;272:3049–3056. doi: 10.1074/jbc.272.5.3049. [DOI] [PubMed] [Google Scholar]
- 9.Tomaska L., Makhov A.M., Nosek J., Kucejova B., Griffith J.D. Electron microscopic analysis supports a dual role for the mitochondrial telomere-binding protein of Candida parapsilosis. J. Mol. Biol. 2001;305:61–69. doi: 10.1006/jmbi.2000.4254. [DOI] [PubMed] [Google Scholar]
- 10.Meeusen S., Tieu Q., Wong E., Weiss E., Schieltz D., Yates J.R., Nunnari J. Mgm101p is a novel component of the mitochondrial nucleoid that binds DNA and is required for the repair of oxidatively damaged mitochondrial DNA. J. Cell Biol. 1999;145:291–304. doi: 10.1083/jcb.145.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen X.J., Guan M.X., Clark-Walker G.D. MGM101, a nuclear gene involved in maintenance of the mitochondrial genome in Saccharomyces cerevisiae. Nucleic Acids Res. 1993;21:3473–3477. doi: 10.1093/nar/21.15.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuo X.M., Clark-Walker G.D., Chen X.J. The mitochondrial nucleoid protein, Mgm101p, of Saccharomyces cerevisiae is involved in the maintenance of rho(+) and ori/rep-devoid petite genomes but is not required for hypersuppressive rho(-) mtDNA. Genetics. 2002;160:1389–1400. doi: 10.1093/genetics/160.4.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meeusen S., Nunnari J. Evidence for a two membrane-spanning autonomous mitochondrial DNA replisome. J. Cell Biol. 2003;163:503–510. doi: 10.1083/jcb.200304040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaufman B.A., Newman S.M., Hallberg R.L., Slaughter C.A., Perlman P.S., Butow R.A. In organello formaldehyde crosslinking of proteins to mtDNA: identification of bifunctional proteins. Proc. Natl. Acad. Sci. U.S.A. 2000;97:7772–7777. doi: 10.1073/pnas.140063197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mbantenkhu M., Wang X., Nardozzi J.D., Wilkens S., Hoffman E., Patel A., Cosgrove M.S., Chen X.J. Mgm101 is a Rad52-related protein required for mitochondrial DNA recombination. J. Biol. Chem. 2011;286:42360–42370. doi: 10.1074/jbc.M111.307512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ward T.A., Dudasova Z., Sarkar S., Bhide M.R., Vlasakova D., Chovanec M., McHugh P.J. Components of a Fanconi-like pathway control Pso2-independent DNA interstrand crosslink repair in yeast. PLoS Genet. 2012;8:e1002884. doi: 10.1371/journal.pgen.1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nardozzi J.D., Wang X., Mbantenkhu M., Wilkens S., Chen X.J. A properly configured ring structure is critical for the function of the mitochondrial DNA recombination protein, mgm101. J. Biol. Chem. 2012;287:37259–37268. doi: 10.1074/jbc.M112.389965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zuo X., Xue D., Li N., Clark-Walker G.D. A functional core of the mitochondrial genome maintenance protein Mgm101p in Saccharomyces cerevisiae determined with a temperature-conditional allele. FEMS Yeast Res. 2007;7:131–140. doi: 10.1111/j.1567-1364.2006.00141.x. [DOI] [PubMed] [Google Scholar]
- 19.Gerhold J.M., Sedman T., Visacka K., Slezakova J., Tomaska L., Nosek J., Sedman J. Replication intermediates of the linear mitochondrial DNA of Candida parapsilosis suggest a common recombination based mechanism for yeast mitochondria. J. Biol. Chem. 2014;289:22659–22670. doi: 10.1074/jbc.M114.552828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding C., Butler G. Development of a gene knockout system in Candida parapsilosis reveals a conserved role for BCR1 in biofilm formation. Eukaryot. Cell. 2007;6:1310–1319. doi: 10.1128/EC.00136-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gietz R.D., Schiestl R.H., Willems A.R., Woods R.A. Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast. 1995;11:355–360. doi: 10.1002/yea.320110408. [DOI] [PubMed] [Google Scholar]
- 22.Kosa P., Gavenciakova B., Nosek J. Development of a set of plasmid vectors for genetic manipulations of the pathogenic yeast Candida parapsilosis. Gene. 2007;396:338–345. doi: 10.1016/j.gene.2007.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zemanova J., Nosek J., Tomaska L. High-efficiency transformation of the pathogenic yeast Candida parapsilosis. Curr. Genet. 2004;45:183–186. doi: 10.1007/s00294-003-0472-6. [DOI] [PubMed] [Google Scholar]
- 24.Berrow N.S., Alderton D., Sainsbury S., Nettleship J., Assenberg R., Rahman N., Stuart D.I., Owens R.J. A versatile ligation-independent cloning method suitable for high-throughput expression screening applications. Nucleic Acids Res. 2007;35:e45. doi: 10.1093/nar/gkm047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matulova P., Marini V., Burgess R.C., Sisakova A., Kwon Y., Rothstein R., Sung P., Krejci L. Cooperativity of Mus81.Mms4 with Rad54 in the resolution of recombination and replication intermediates. J. Biol. Chem. 2009;284:7733–7745. doi: 10.1074/jbc.M806192200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Rasband W.S. ImageJ. U. S. Bethesda, MD, USA: National Institutes of Health; 1997–2014. [Google Scholar]
- 27.Altmannova V., Eckert-Boulet N., Arneric M., Kolesar P., Chaloupkova R., Damborsky J., Sung P., Zhao X.L., Lisby M., Krejci L. Rad52 SUMOylation affects the efficiency of the DNA repair. Nucleic Acids Res. 2010;38:4708–4721. doi: 10.1093/nar/gkq195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sebesta M., Burkovics P., Haracska L., Krejci L. Reconstitution of DNA repair synthesis in vitro and the role of polymerase and helicase activities. DNA Rep. 2011;10:567–576. doi: 10.1016/j.dnarep.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blanchet C.E., Zozulya A.V., Kikhney A.G., Franke D., Konarev P.V., Shang W., Klaering R., Robrahn B., Hermes C., Cipriani F., et al. Instrumental setup for high-throughput small- and wide-angle solution scattering at the X33 beamline of EMBL Hamburg. J. Appl. Crystallogr. 2012;45:489–495. [Google Scholar]
- 30.Guinier A. La Diffraction des rayons X aux très pétits angles: application à l'etude de phénomènes ultramicroscopiques. Ann. Phys. (Paris) 1939;12:161–237. [Google Scholar]
- 31.Petoukhov M.V., Svergun D.I. Analysis of X-ray and neutron scattering from biomacromolecular solutions. Curr. Opin. Struct. Biol. 2007;17:562–571. doi: 10.1016/j.sbi.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 32.Svergun D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992;25:495–503. [Google Scholar]
- 33.R Core Team. R Foundation for Statistical Computing. Vienna, Austria: 2015. R: A language and environment for statistical computing. [Google Scholar]
- 34.Franke D., Svergun D.I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2009;42:342–346. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Volkov V.V., Svergun D.I. Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Svergun D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999;76:2879–2886. doi: 10.1016/S0006-3495(99)77443-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schrodinger L. The PyMOL molecular graphics system. Version 1.5.x. 2012 [Google Scholar]
- 38.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 39.Williamson D.H., Fennell D.J. Visualization of yeast mitochondrial DNA with the fluorescent stain ‘DAPI’. Methods Enzymol. 1979;56:728–733. doi: 10.1016/0076-6879(79)56065-0. [DOI] [PubMed] [Google Scholar]
- 40.Miyakawa I., Aoi H., Sando N., Kuroiwa T. Fluorescence microscopic studies of mitochondrial nucleoids during meiosis and sporulation in the yeast, Saccharomyces cerevisiae. J. Cell Sci. 1984;66:21–38. doi: 10.1242/jcs.66.1.21. [DOI] [PubMed] [Google Scholar]
- 41.Claros M.G., Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 42.Mortensen U.H., Bendixen C., Sunjevaric I., Rothstein R. DNA strand annealing is promoted by the yeast Rad52 protein. Proc. Natl. Acad. Sci. U.S.A. 1996;93:10729–10734. doi: 10.1073/pnas.93.20.10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ploquin M., Bransi A., Paquet E.R., Stasiak A.Z., Stasiak A., Yu X., Cieslinska A.M., Egelman E.H., Moineau S., Masson J.Y. Functional and structural basis for a bacteriophage homolog of human RAD52. Curr. Biol. 2008;18:1142–1146. doi: 10.1016/j.cub.2008.06.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang F., Skoda M.W., Jacobs R.M., Martin R.A., Martin C.M., Schreiber F. Protein interactions studied by SAXS: effect of ionic strength and protein concentration for BSA in aqueous solutions. J. Phys. Chem. B. 2007;111:251–259. doi: 10.1021/jp0649955. [DOI] [PubMed] [Google Scholar]
- 45.Zhang F., Roosen-Runge F., Skoda M.W., Jacobs R.M., Wolf M., Callow P., Frielinghaus H., Pipich V., Prevost S., Schreiber F. Hydration and interactions in protein solutions containing concentrated electrolytes studied by small-angle scattering. Phys. Chem. Chem. Phys. 2012;14:2483–2493. doi: 10.1039/c2cp23460b. [DOI] [PubMed] [Google Scholar]
- 46.Ianeselli L., Zhang F., Skoda M.W., Jacobs R.M., Martin R.A., Callow S., Prevost S., Schreiber F. Protein-protein interactions in ovalbumin solutions studied by small-angle scattering: effect of ionic strength and the chemical nature of cations. J. Phys. Chem. B. 2010;114:3776–3783. doi: 10.1021/jp9112156. [DOI] [PubMed] [Google Scholar]
- 47.Goldenberg D.P., Argyle B. Self crowding of globular proteins studied by small-angle x-ray scattering. Biophys. J. 2014;106:895–904. doi: 10.1016/j.bpj.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glatter O., Kratky O. Small Angle X-ray Scattering. London, UK: Academic Press; 1982. [Google Scholar]
- 49.Rambo R.P., Tainer J.A. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013;496:477–481. doi: 10.1038/nature12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rambo R.P., Tainer J.A. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers. 2011;95:559–571. doi: 10.1002/bip.21638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mbantenkhu M., Wierzbicki S., Wang X., Guo S., Wilkens S., Chen X.J. A short carboxyl-terminal tail is required for single-stranded DNA binding, higher-order structural organization, and stability of the mitochondrial single-stranded annealing protein Mgm101. Mol. Biol. Cell. 2013;24:1507–1518. doi: 10.1091/mbc.E13-01-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hayward D.C., Dosztanyi Z., Clark-Walker G.D. The N-terminal intrinsically disordered domain of Mgm101p is localized to the mitochondrial nucleoid. PLoS One. 2013;8:e56465. doi: 10.1371/journal.pone.0056465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kagawa W., Kagawa A., Saito K., Ikawa S., Shibata T., Kurumizaka H., Yokoyama S. Identification of a second DNA binding site in the human Rad52 protein. J. Biol. Chem. 2008;283:24264–24273. doi: 10.1074/jbc.M802204200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mosig G. Recombination and recombination-dependent DNA replication in bacteriophage T4. Annu. Rev. Genet. 1998;32:379–413. doi: 10.1146/annurev.genet.32.1.379. [DOI] [PubMed] [Google Scholar]
- 55.Chen X.J. Mechanism of homologous recombination and implications for aging-related deletions in mitochondrial DNA. Microbiol. Mol. Biol. Rev. 2013;77:476–496. doi: 10.1128/MMBR.00007-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Dyck E., Hajibagheri N.M., Stasiak A., West S.C. Visualisation of human rad52 protein and its complexes with hRad51 and DNA. J. Mol. Biol. 1998;284:1027–1038. doi: 10.1006/jmbi.1998.2203. [DOI] [PubMed] [Google Scholar]
- 57.Grimme J.M., Honda M., Wright R., Okuno Y., Rothenberg E., Mazin A.V., Ha T., Spies M. Human Rad52 binds and wraps single-stranded DNA and mediates annealing via two hRad52-ssDNA complexes. Nucleic Acids Res. 2010;38:2917–2930. doi: 10.1093/nar/gkp1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rothenberg E., Grimme J.M., Spies M., Ha T. Human Rad52-mediated homology search and annealing occurs by continuous interactions between overlapping nucleoprotein complexes. Proc. Natl. Acad. Sci. U.S.A. 2008;105:20274–20279. doi: 10.1073/pnas.0810317106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tomaska L., Makhov A.M., Griffith J.D., Nosek J. t-Loops in yeast mitochondria. Mitochondrion. 2002;1:455–459. doi: 10.1016/s1567-7249(02)00009-0. [DOI] [PubMed] [Google Scholar]
- 60.Tomaska L. Phosphorylation of mitochondrial telomere binding protein of Candida parapsilosis by camp-dependent protein kinase. Biochem. Biophys. Res. Commun. 1998;242:457–460. doi: 10.1006/bbrc.1997.7968. [DOI] [PubMed] [Google Scholar]
- 61.Tomaska L., Nosek J., Kucejova B. Mitochondrial single-stranded DNA-binding proteins: in search for new functions. Biol. Chem. 2001;382:179–186. doi: 10.1515/BC.2001.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.