


Abstract
With bacterial resistance becoming a serious threat to global public health, antimicrobial peptides (AMPs) have become a promising area of focus in antibiotic research. AMPs are derived from a diverse range of species, from prokaryotes to humans, with a mechanism of action that often involves disruption of the bacterial cell membrane. Proline-rich antimicrobial peptides (PrAMPs) are instead actively transported inside the bacterial cell where they bind and inactivate specific targets. Recently, it was reported that some PrAMPs, such as Bac71–35, oncocins and apidaecins, bind and inactivate the bacterial ribosome. Here we report the crystal structures of Bac71–35, Pyrrhocoricin, Metalnikowin and two oncocin derivatives, bound to the Thermus thermophilus 70S ribosome. Each of the PrAMPs blocks the peptide exit tunnel of the ribosome by simultaneously occupying three well characterized antibiotic-binding sites and interferes with the initiation step of translation, thereby revealing a common mechanism of action used by these PrAMPs to inactivate protein synthesis. Our study expands the repertoire of PrAMPs and provides a framework for designing new-generation therapeutics.
INTRODUCTION
The vital role of the ribosome in protein synthesis and its low mutational propensity due to the redundancy of ribosomal RNA (rRNA) genes in bacterial genomes make it an ideal target for antimicrobials. Indeed, a large number of clinically useful antibiotics, most of which are of natural origin, target the ribosome. They usually inhibit protein synthesis by binding to one of the functional sites in the ribosome (1). The knowledge of the atomic structure of the ribosome (2), and of an array of ribosome–antibiotic complexes (3–13), allows the rational design of new inhibitors that are highly active against bacterial pathogens. However, the rise of resistant bacteria represents a daunting challenge that has not been met yet with a corresponding development of effective new antibiotics. This serious public health concern has revived interest in the discovery and development of new therapeutics, especially those targeting Gram-negative microorganisms.
The sources of most of the naturally produced small-molecule antibiotics now in use are primarily bacteria and fungi. However, antimicrobial peptides (AMP) are produced as an innate immune response by many multicellular organisms (14–16). Proline-rich antimicrobial peptides (PrAMPs), which were among the first AMPs to be discovered, were identified in the late 1980s independently in honeybees (17) and cattle (18). PrAMPs, with motifs enriched in proline and arginine residues arranged in recurring patterns (19), have thus evolved in both vertebrates and invertebrates.
Unlike most other AMPs which act on bacterial membranes (20), the majority of eukaryotic PrAMPs are actively transported across the bacterial membrane into the cytoplasm by specialized transporters such as SbmA in Gram-negative bacteria (21). The first intracellular AMP target identified was the heat-shock protein DnaK (22,23). However, the findings that a DnaK null mutant retained susceptibility to PrAMPs suggested that they have other targets (24). It was recently reported that the PrAMPs oncocin and apidaecin preferentially target and inhibit the bacterial ribosome (25), and structures of oncocin bound to the 70S ribosome were also reported (26,27). These structures revealed that oncocin inhibits protein synthesis at the initiation stage by binding in the peptide exit tunnel in such a way as to interfere with the binding of the aminoacyl-tRNA in the A site (26,27).
The bovine peptide bactenecin 7 (Bac7) is the most studied mammalian PrAMP and it belongs to the cathelicidin family of innate immune effectors, which are defined by a conserved, cathelin-like proline-region followed by a highly variable C-terminal domain (28). Bac7 is a 60-residue peptide that is derived from a protein precursor (29). Previous studies showed that Bac7, and its C-terminal truncated form, Bac71–35, have a potent activity against many Gram-negative bacteria (30–32). Similar to oncocin, Bac7 was shown to bind to chaperone DnaK (33), but more recently, Bac71–35 was reported to bind bacterial ribosomal proteins and to inhibit protein synthesis (34). Other PrAMPs like Pyrrhocoricin, isolated from the insect Pyrrocoris apterus (35), and Metalnikowin, isolated from Palomena prasina (36) show similar antimicrobial properties (37,38). Pyrrhocoricin and Metalnikowin, even though isolated from different sources, are ∼70% conserved with oncocin in sequence, suggesting that they may also target the bacterial ribosome.
We report here the crystal structures of Bac71–35, Pyrrhocoricin, Metalnikowin and two oncocin derivatives bound to the Thermus thermophilus 70S ribosome at 2.7–3.0 Å resolution (Table 1 and Supplementary Table S1). The binding mode of Bac71–35 in the ribosome peptide exit tunnel is reminiscent to that of oncocin previously reported (26,27), with its N-terminal residues overlapping with the binding site for the CCA-end of A-site tRNA in the peptidyl transferase center (PTC). We observe 19 amino acid residues of Bac71–35 extending by more than 45 Å along the exit tunnel down to the constriction formed by the apical loops of ribosomal proteins uL4 and uL22. Foot-printing experiments confirm that the binding mode observed in the crystalline complex resembles the interaction between Bac71–35 and the ribosome in solution. Using a toe-printing assay, we also show that Bac71–35 and several other PrAMPs block the ribosome at the initiation codon, thereby preventing subsequent transitioning into the elongation phase of protein synthesis. We compare the Bac71–35-ribosome structure with a series of 70S-ribosome structures in complex with other PrAMPs, and delineate the molecular determinants of PrAMPs that are responsible for ribosome binding and thus for their antibiotic activity. We show that mutations of rRNA residues in the vicinity of the PrAMP binding site confer resistance to oncocin, revealing the ribosome as the key cellular target for this and likely other PrAMPs. This study reveals a common binding mode and mechanism of action of PrAMPs on the ribosome and thus provides a basis for designing improved antibacterial compounds that target the ribosome.
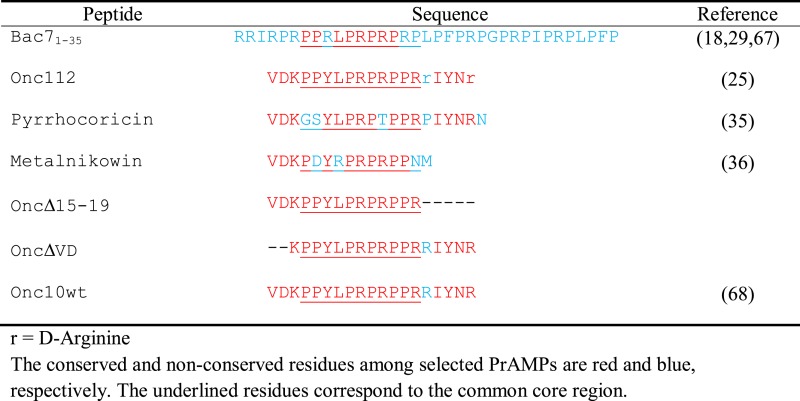
Table 1. Sequence alignment of selected PrAMPs.
|
MATERIALS AND METHODS
mRNA and tRNA
The mRNA with a Shine-Dalgarno sequence and an initiation codon in the P site was synthesized by integrated DNA technologies (Coralville, IA) with the sequence 5′ GGC AAG GAG GUA AAA AUG UUC UAA 3′. The fMet-tRNAfMet was prepared as previously described (39). All peptides used in this study were chemically synthesized by GenScript USA (Piscataway, NJ, USA).
Complex formation and crystallization
The 70S ribosomes from T. thermophilus HB8 or its mutant strain in which ribosomal protein uL9 is truncated (70S:L91–58) (40,41) were prepared and crystallized as previously described (40,42,43). Essentially, 4 μM ribosomes were incubated with 8 μM mRNA and fMet-tRNAfMet in a buffer containing 5 mM Hepes-KOH, pH 7.5, 50 mM KCl, 10 mM NH4Cl, 10 mM Mg(CH3COO)2 and 6 mM β-mercaptoethanol at 55°C for 5 min. The complex was further incubated at room temperature for 15 min in the presence of either 50 μM Bac71–35, Metalnikowin, Onc10wt, OncΔ15–19, Pyrrhocoricin or OncΔVD. We obtained the structure of Bac71–35 bound to both 70S:L91–58 and 70S wild-type ribosomes. With the 70S:L91–58 ribosomes, the complex was prepared as previously described in the presence of 5 μM elongation factor G (EF-G) fused to ribosomal protein uL9 (40) and 50 μM Bac71–35. Crystals were grown in sitting-drop trays in which 3 μl of ribosome complex was mixed with 3.5–4.5 μl reservoir solution containing 0.1 M Tris–HCl, pH 7.3 – 7.6, 2.6 – 2.9% (w/v) PEG 20000, 9–10% (v/v) 2-methyl-2,4-pentanediol, 0.15 M L-arginine and 0.5 mM β-mercaptoethanol and incubated at 19°C. Crystals were cryoprotected by gradually increasing the concentration of 2-methyl-2,4-pentanediol while all other buffer components were held constant, except that each stabilization step also contained 100 μM of peptide. The crystals were left to equilibrate overnight at 19°C and were frozen at 80K in a nitrogen stream before being plunged in liquid nitrogen.
X-ray data collection and structure refinement
X-ray diffraction data were collected at beamline 24ID-C at the Advanced Photon Source at Argonne National Laboratory (Argonne, IL, USA). The collected data were processed using the XDS software package (44), and we used PHASER from the CCP4 suite (45) to determine the initial solution for the structure by molecular replacement. The search model used was generated from the previously published high-resolution structure of the T. thermophilus 70S ribosome [PDB 1VY4 (46)] with all its ligands removed. Structures were refined with two 70S ribosomes in the asymmetric unit by rigid-body refinement, and then by five cycles of position and B-factor refinement with the PHENIX package (47). The P-site tRNA, mRNA and the peptide bound in the ribosome peptide exit tunnel were built into the Fobs–Fcalc difference Fourier electron density map using COOT (48), and the models were further refined with PHENIX (47). The final refinement statistics are provided in Supplementary Table S1.
Toe-printing analysis
Toe-printing analysis was performed using the RST2 template (49) as previously described (50). Cell free translation reactions, carried out in the PURExpress in vitro protein synthesis system (New England Biolabs, Ipswich, MA, USA) were supplemented with 50 μM thiostrepton or 100 μM of peptides which were added in water and samples (5 μl volume) were incubated for 15 min at 37°C prior to the primer extension phase of the procedure. The control reaction had no inhibitor. Primer extension was carried out for 15 min, after which the samples were processed as described (50).
Foot-printing experiments
Escherichia coli ribosomes were prepared as described (51). The wild-type T. thermophilus ribosomes used in crystallization experiments were also used in foot-printing experiments. Ribosomes (0.2 μM) were pre-incubated with 100 μM of Bac71–35 or Onc112 and then modified with dimethylsulfate (DMS) or N-cyclohexyl-N′-(2-morpholinoethyl) carbodiimide methyl-p-toluenesulfonate (CMCT) as described (52). rRNA was extracted and distribution of base modifications was analyzed by primer extension.
Determination of PrAMP's minimal inhibitory concentrations (MIC)
Minimal inhibitory concentrations (MIC) of PrAMPs were tested for E. coli strains SQ110 which contains a single rrn allele or its derivatives with a mutation in the lptD gene (SQ110 LPTD) or knock-out of the tolC gene (SQ110 DTC) (53). The A2503C and A2059G mutants were previously selected for their resistance to chloramphenicol or erythromycin, respectively (53). The A2503C/A2059G double mutant was selected by plating the A2503C mutant on the plate with the macrolide antibiotic solithromycin. For the MIC determination, cells were exponentially grown in the LB medium, diluted to A650 of 0.005 and placed in the wells of a 96-well plate. PrAMPs were added in the LB medium to the highest final concentration of 25 μM in one column of wells. After 2-fold serial dilutions, the plate was incubated overnight at 37°C with shaking and cell growth was analyzed by addition of the alamarBlue dye. When MIC was tested with the SQ110 DTC mutants, the highest concentration of Onc112 and Bac71–35 was 100 μM.
RESULTS AND DISCUSSION
Characterization of PrAMPs
The AMP Bac71–35 and a series of PrAMPs (Table 1) were chemically synthesized and used to elucidate the determinants required for their ribosome binding and antimicrobial activity. They were tested for their inhibitory activity against the SQ110 LPTD strain of E. coli, which is defective in outer membrane structure (Table 2) (53). All peptides, except OncΔVD and Metalnikowin, show detectable activity with MIC ranging between 0.75 and 25 μM, which agrees well with previous data (37,54,55). It is noteworthy that the activity of the peptides against the parental SQ110 strain was reduced in most cases (Table 2), indicating that crossing the outer membrane of Gram-negative bacteria presents a challenge for PrAMPs. As the cellular growth inhibitory activity of PrAMPs depends both on their ability to cross the cell membrane (21) and their stabilities, their inhibitory effect on protein synthesis was also assessed in vitro using a toe-printing assay (Figure 1A). While most peptides inhibited the initiation stage of protein synthesis and arrested the ribosome at the initiator codon, as previously reported for Onc112 (26), the deletion peptide OncΔVD was inactive.
Table 2. Minimal inhibitory concentration (MIC) values for selected PrAMPs against E. coli strains.
| Peptide | SQ110 (LPTD) (μM) | SQ110 (μM) |
|---|---|---|
| Bac71–35 | 0.75 | 1.5 |
| Onc112 | 0.75 | 12.5 |
| Pyrrhocoricin | 4 | >25 |
| Metalnikowin | 62.5 | >25 |
| OncΔ15–19 | 25 | >25 |
| OncΔVD | 62.5 | >25 |
| Onc10wt | 1 | 25 |
The SQ110 LPTD strain is defective in the lipopolysaccharide assembly of the outer membrane and is more sensitive to some antibiotics (53).
Figure 1.

Toe-printing (A) and foot-printing (B) experiments reveal the mode of action and verify PrAMP binding mode in solution. (A) The 20-codon synthetic RST2 ORF containing codons for all 20 amino acids (49) was translated in the PURExpress cell-free transcription-translation system by Escherichia coli ribosomes in the presence of 100 μM of PrAMPs or 50 μM of the control antibiotic thiostrepton (Ths), and the position of the stalled ribosome was determined by primer extension. U- and A- specific reactions were used as a sequencing ladder. The toeprint band (marked by an arrow in the gel and in the sequence of the gene), which occurs at position +16 counting from the first nucleotide of the codon in the ribosomal P site, places the arrested ribosome at the initiator codon (boxed on the RST2 sequence on the left from the gel). (B) Foot-printing analysis of interaction of Bac71–35 and Onc112 with the E. coli ribosome in solution. Ribosomes were pre-incubated with no PrAMP (’none’) or 50 μM of Bac71–35 or Onc112 and subjected to modification with CMCT or DMS. Control sample remained unmodified. Some of the lanes in gels shown in A and B, which contained samples irrelevant to the current study, have been computationally removed.
Structure determination of PrAMPs bound to the ribosome
To further elucidate the ribosome binding mode of all the peptides studied, each was co-crystallized with the T. thermophilus 70S ribosome bound to mRNA and initiator tRNA (Supplementary Table S1). The resulting 70S-complex structures were determined by the molecular replacement method using a high-resolution model of the vacant 70S ribosome (46). The difference Fourier maps calculated at 2.7–3.0 Å resolution using initially phased diffraction data showed clear unbiased electron density for the mRNA, tRNAfMet in the P site and the AMP located inside the peptide exit tunnel of the 50S subunit (Figure 2A Supplementary Figure S1). The absence of electron density for OncΔVD in the ribosome peptide exit tunnel (data not shown) is consistent with the toe-printing experiments (Figure 1A).
Figure 2.

The structure of PrAMPs in their ribosome-bound conformation. (A) Structure of Bac71–35 bound inside the peptide exit tunnel of the ribosome. The difference Fourier map calculated at 3.0 Å resolution using initially phased diffraction data (Fobs–Fcalc, green) contoured at ∼3σ shows clear unbiased electron density for 19 residues of Bac71–35. The 23S rRNA A-loop and ribosomal proteins uL4 and uL22 are shown. (B) The superposition of all PrAMPs bound to the ribosome reveals that the common core (middle) region overlaps almost perfectly. Bac71–35 is colored brown, Onc112 is green [PDB ID:4Z8C (27)], Metalnikowin is cyan, Onc10wt is slate, OncΔ15–19 is magenta and Pyrrhocoricin is red.
The interactions of the N-terminus of PrAMPs with the 23S rRNA are not conserved, but are crucial for ribosome binding
Nineteen residues of the Bac71–35 peptide, spanning more than 45 Å from the CCA-binding site of the aminoacyl-tRNA in the A site to the constriction of the peptide exit tunnel in the upper chamber, could be unambiguously fitted into the unbiased electron density of the difference Fourier map for the complex (Figure 2A). Compared to Onc112 (26,27), the N-terminal region of Bac71–35 extends further into the A site of the 50S subunit (Figure 3A), and would collide with the acceptor-stem of a tRNA bound in the A site if there were one (Figure 3B). The many interactions of this extended region of Bac71–35 with the 23S rRNA stabilize the peptide inside the PTC, and may also be essential for the entry of Bac71–35 into the peptide exit tunnel. Indeed, the antimicrobial activity of deletion variants of Bac71–35 is known to require the integrity of the first 16 residues of the highly cationic N-terminus, which is also known to be required for its activity against Gram-negative bacteria (32,56). In addition, it has been proposed that the loss of activity for the deletion variants of Bac71–35 is due to impaired interaction with the intra-cellular target, because the peptides remained cell-permeable (56).
Figure 3.

Conformation of the N-terminal region of PrAMPs in the ribosome. (A) The N-terminal of Bac71–35 (brown) extends further than Onc112 (green) [PDB ID: 4Z8C (27)] into the A site of the 50S subunit, establishing interactions with H89 of 23S rRNA. (B) The N-terminal of Bac71–35 is not compatible with the presence of the CCA-end of an A-site tRNA (light blue) [PDB ID: 4Y4P (69)]. (C) The Asp2 residue at the N-termini of PrAMPs Pyrrhocoricin, Metalnikowin, Onc10wt, OncΔ15–19 and Onc112 [PDB ID: 4Z8C (27)] interact with the Watson–Crick edge of nucleotide G2553 in the 23S rRNA A-loop.
The structure of Bac71–35 bound to the ribosome reveals that the N-terminal region of Bac71–35 makes several interactions with Helix 92 (H92), which forms the A-loop of the 23S rRNA, and Helix 89 (H89), which is part of the PTC (Figure 4). The epsilon nitrogen atom of residue Arg1 is within hydrogen bonding distance to the O2 atom of nucleotide U2555 and the O4′ atom of nucleotide C2556, both in H92 (Figure 4A) (E. coli nucleotide numbers are used throughout the text). Residues Arg2, Arg4 and Arg6 all make interactions with the sugar-phosphate backbone of H89 (Figure 4B-D). Finally, residue Arg2 makes a π-stacking interaction with the nucleotide base of C2573 and with Arg6, which in turn stacks on Arg4 (Figure 4E). Compared to other PrAMPs studied here and previously (26,27), the N-terminal region of Bac71–35 extends further by more than 6 Å toward H89 of 23S rRNA into the A site of the 50S subunit (Figure 3A), which may provide a clue about why this segment of the peptide is important for the antimicrobial activity of Bac71–35. The sugar-phosphate backbone of H89 has been proposed to be involved in the tRNA accommodation by mediating the first binding interactions of the aminoacyl-tRNA with the ribosome (57). By interacting closely with H89 and hindering the path to the PTC for the acceptor-stem of the aminoacyl-tRNA, the N-terminal region of Bac71–35 may be more efficient at inhibiting translation initiation than Onc112 (Table 2). Each arginine residue of the 'RRIR' motif in Bac71–35 participates in dual interactions involving the epsilon nitrogen and the amino group (Figure 4). These interactions would not be possible if arginines were substituted by lysines or other residues, thereby providing a structural basis for their high conservation in Bac71–35, bactenecin 5 (Bac5) and PR-39 (18,58). This argument is supported by previous data reporting an appreciable loss of activity for Bac71–35 N-terminal variants in which Arg1 and Arg2 residues were substituted by lysine (59). Similarly, it has been shown that arginine residues at the N-terminus of a PR-39 variant and of Bac5 are important for the antimicrobial activity of those PrAMPs (60,61).
Figure 4.

Interactions of the N-terminal (A–E) and the core (middle) (F) regions of Bac71–35 with the ribosome. Residues Arg1 (A), Arg2 (B), Arg4 (C) and Arg6 (D) establish multiple interactions with nucleotides in H89 and H92 of 23S rRNA. Each arginine residue of the N-terminal 'RRIR' motif in Bac71–35 participates in dual interactions involving the epsilon nitrogen and the amino group. (E) Four-layer π-stacking interaction formed between residues Arg2, Arg6, Arg4 and nucleotide C2573. (F) Residue Arg9 of the core region in Bac71–35 stacks with the conserved base pair formed between nucleotides C2452 and U2504 of the A-site cleft and also forms a hydrogen bond with G2061. The Leu10 residue stacks with residue Arg9.
To investigate the role of the N-terminal region in other PrAMPs, we tested an N-terminally truncated version of oncocin, OncΔVD, which lacks the first two residues (Table 1). This mutant exhibited a MIC value 80-fold higher than that of Onc112 (Table 2). Moreover, the toe-printing assay shows that OncΔVD did not block the ribosome at the initiation codon and allows translation to continue through the entire gene (Figure 1A), indicating that the mutant peptide does not inhibit the ribosome function at the concentration tested (see ‘Materials and Methods’ section). This observation correlates with our inability to observe electron density for OncΔVD in the ribosome complex (data not shown). Thus, our data provide a structural explanation for the importance of the N-terminal domain in PrAMPs for ribosome binding.
Despite the high conservation of the A-site cavity in the PTC, the structures obtained for Bac71–35 and the other PrAMPs bound to the ribosome show that their N-terminal parts interact with the ribosome in different manners. Most of them form an interaction between the Asp2 residue and the Watson–Crick edge of nucleotide G2553 of the 23S rRNA A-loop as previously observed in the Onc112–ribosome complex (Figure 3C) (26,27). In contrast, the N-terminal region of Bac71–35 is displaced toward H89 by more than 6 Å (Figure 3A), thereby positioning the carbonyl oxygen of Pro5 and the N6 atom of A2602, a universally conserved nucleotide in the PTC, within hydrogen bonding distance (Figure 4C).
The core region of PrAMPs uses a common ribosome binding mode
The superposition of the 50S subunits of ribosome–PrAMP complexes shows a similar peptide binding mode in their core regions. Residues 6 to 13 of Bac71–35 overlap perfectly with the corresponding region in other PrAMPs bound to the ribosome (Figure 2B). This segment in all peptides interacts with conserved nucleotides of the peptide exit tunnel, such as U2585 and U2506 (Figures 1B, 4F and Supplementary Figure S2), two nucleotides known to change their conformation in response to substrates binding into the PTC and the exit tunnel (62,63). This region of the ribosome is also known to bind several antibiotics like chloramphenicol (12), homoharingtonine (6,13) and hygromycin A (11). Sequence alignment of the peptides studied here shows that the core region consists of multiple proline-arginine repeats (underlined in Table 1). The tyrosine-leucine (YL) motif, which is surrounded by the proline-arginine repeats, is generally conserved but appears to accommodate variations. The sequence alignment shows that the tyrosine residue can be substituted by arginine (Table 1). In the 70S-ribosome–Bac71–35 complex, the position occupied by Arg9 is the same as the one occupied by Tyr6 in the 70S–Onc112 complex (Figure 4F and Supplementary Figure S2A) (26,27), which is also reminiscent of the position occupied by the phenylalanine residue attached to the A-site tRNA (Supplementary Figure S2B) (46). Correspondingly, both Arg9 (in Bac71–35) and Tyr6 (in Onc112) residues form similar π-stacking interactions with the conserved base pair formed between nucleotides C2452 and U2504 of the A-site cleft (Figure 4F). The high sequence conservation of this region reflects the analogous interactions that occur with the nucleotides of the peptide exit tunnel in the ribosome. In Bac71–35, the interaction of the Arg9 side chain with the A-site cleft of the PTC is stabilized by the Leu10 side chain that forms a stacking interaction with Arg9 (Figure 4F), as previously observed in the Onc112 complex (27). The crystal structures of all other PrAMPs show that this interaction is maintained and is mediated by a leucine residue, with the exception of peptide Metalnikowin where this residue is replaced by arginine (Table 1) that nevertheless forms a similar π-stacking interaction with Tyr6 (Supplementary Figure S2C).
The variable C-terminal region of PrAMPs is flexible in the upper chamber of the peptide tunnel
We did not detect any electron density in the difference Fourier map for residues 20 to 35 of Bac71–35 probably due to their flexibility (Figure 2A). In the previously reported 70S–Onc112 structure, residues 14–19 of Onc112 were also not visible (27). In the structure of peptide Pyrrhocoricin bound to the ribosome, 17 residues out of 20 are visible, which allows us to visualize the path taken by the C-terminal region of Pyrrhocoricin (Figures 2B and 5D and Supplementary Figure S1A). The superposition of ribosomes bound to Bac71–35 and Pyrrhocoricin reveals that the C-terminal region of those peptides diverge from residue 14 in Bac71–35 (11 in Pyrrhocoricin), with the main chain peptide backbone laterally displaced by more than 5 Å along the constriction formed by the loops of ribosomal proteins uL4 and uL22 (Figure 5D). The high flexibility of this region in PrAMPs indicates that the interactions of the C-terminal region with the peptide exit tunnel of the ribosome—if any—contribute less to binding than the core and N-terminal segments. Consistent with this hypothesis, a C-terminal truncation of the Bac71–35 peptide, giving Bac71–16, has been reported to retain good antimicrobial activity (32,59). However, deletion of Arg16, producing Bac71–15, abolishes activity (32,56). This effect is likely due to a loss of binding affinity to the ribosome because Bac71–15 is still transported inside the cell (56). This agrees with our structure showing that Arg16 makes multiple interactions with the ribosome (Figure 5A and C). The side chain of Arg16 forms a π-stacking interaction with His69 of protein uL4 on one side, while on the other side Arg16 is within hydrogen bonding distance to atom N1 of nucleotide A2062 (Figure 5A). The interaction between Arg16 and protein uL4 seen in this ribosome–Bac71–35 complex is substituted by Arg11 in the previous 70S–Onc112 complex structure (27), where it interacts with A2062 of the 23S rRNA (Figure 5B). Accordingly, a substitution of Arg11 in Onc112 to alanine decreases the binding affinity of oncocin to the ribosome by about 6-fold (25).
Figure 5.

Interactions of the C-terminal region of Bac71–35 with the upper chamber of the peptide exit tunnel. (A) Residue Arg16 forms a π-stacking interaction with His69 of ribosomal protein uL4 and also makes multiple interactions with the ribosome. (B) The interactions mediated by residue Arg16 in Bac71–35 (brown) are compensated by Arg11 in Onc112 (green) [PDB 4Z8C (27)]. (C) Residue Arg14 stacks with nucleotide C2586 and the main chain peptide backbone of residues 17–19 forms a stacking interaction with nucleotides C790 across the tunnel, and with residue Arg90 of ribosomal protein uL22. (D) Lateral displacement of the C-terminus of Pyrrhocoricin (red) compared with Bac71–35 (brown) by more than 5 Å along the constriction formed by the loops of proteins uL4 and uL22.
To further explore the contribution of the PrAMP's C-terminal residues to ribosome binding and activity, we designed a truncated version of Onc112 in which the last five residues are removed (OncΔ15–19). Based on the sequence alignment of other selected PrAMPs with Bac71–35 (Table 1), such truncation should not inhibit its binding to the ribosome, and therefore its antimicrobial activity. As expected, we observed electron density inside the peptide tunnel for OncΔ15–19, indicating that removal of the last five residues does indeed not affect its binding to the ribosome. Correspondingly, this oncocin mutant preserves its inhibitory activity against the ribosome as it blocks the initiation step of protein synthesis (Table 2 and Figure 1A).
In the upper chamber of the peptide exit tunnel, nucleotide A2062 adopts the same conformation in both the 70S–Bac71–35 and 70S–Onc112 complexes; however, while Arg11 of Onc112 interacts with A2062 (Figure 5B) (27), the corresponding Arg14 in Bac71–35 forms an alternative stacking interaction with nucleotide C2586, not seen previously with Onc112 (Figure 5C). Residues 16 to 19 of Bac71–35 pack along the side of the peptide exit tunnel wall formed by residues 69 and 70 of protein uL4. The main chain peptide backbone of residues 17–19 of Bac71–35 is sandwiched between nucleotide C790 across the tunnel and residue Arg90 of protein uL22 (Figure 5C). The various interactions observed between the ribosome and the C-terminal region of PrAMPs may reflect the plasticity of the upper chamber of the ribosome exit tunnel in accommodating different nascent peptide chains during translation. This correlates with recent studies of macrolide antibiotics, which bind 50S ribosomal subunit in the exact same region, revealing that some nascent polypeptide chains can escape the inhibitory effect of the antibiotic in a sequence dependent manner (64).
Taken together, our results and the available data on truncated variants of Bac71–35 suggest that the binding determinants for Bac71–35 and other PrAMPs reside within the first 14–15 residues of the peptide, with the main contribution to ribosome binding being provided by the residues of the N-terminal and middle segments of the peptides.
While crystallographic studies can provide critical insights into the interactions of inhibitors with the ribosome in the crystalline state, the intrinsic dynamics of the ribosome in solution could hypothetically lead to alternative or additional binding modes. Therefore, in order to verify that the binding of Bac71–35 and Onc112 inferred from the X-ray analysis adequately reflects interaction of PrAMPs with the ribosome in solution, we carried out foot-printing experiments, in which we identified rRNA nucleotides protected by PrAMPs from chemical modification in the E. coli or T. thermophilus ribosomes (Figure 1B, and Supplementary Figures S4 and S5). Consistent with our structural data (Figure 4F and Supplementary Figure S2A), we observed strong protection of U2506, U2585 and A2451 by both peptides in the ribosome from mesophylic and thermophilic bacteria. Interestingly, Bac71–35 completely protected U2609 from modification with carbodiimide (CMCT), but Onc112 afforded only partial protection in the E. coli ribosome, indicating differences in PrAMPs binding to the upper chamber of the peptide exit tunnel. Correspondingly, Bac71–35 provides increased accessibility of A2062 to DMS modification in E. coli over Onc112 (Figure 1B), which is in agreement with differences in interaction of Onc112 and Bac71–35 with A2062 (Figure 5B).
Ribosomal mutations affect the activity of PrAMPs
The primary intra-cellular target for PrAMPs has long been thought to be the chaperone DnaK because PrAMPs can bind to DnaK and inhibit its activity (33,38,55,65). However, the sensitivity of an E. coli strain lacking DnaK to PrAMPs suggests another key target (24). The new data presented here highlight the importance of a different target for oncocins and Bac71–35—the ribosome (25–27,34). However, neither structural, nor biochemical data can distinguish between the primary target of antibiotic action, whose inhibition results in growth arrest, and a fortuitous binding. Therefore, in order to test whether the ribosome is indeed a target for PrAMPs in the bacterial cell, we tested whether rRNA mutations in the PrAMP binding site can render E. coli resistant to Bac71–35 or Onc112. Guided by antibiotic binding studies (66), we used the previously selected resistant mutants with alterations at 23S rRNA residues A2503 and A2059 (Table 3) (53). For Bac71–35, the selected mutations did not provide any clear advantage over the wild-type strain, however the A2503C or A2059C mutations in the peptide exit tunnel increased resistance against Onc112 by about 4-fold. When present together in the A2503C/A2059G double mutant, the mutations increased E. coli resistance to Onc112 by more than 15-fold. Both of these residues interact with A2062 (Supplementary Figure S2) which in turn forms interactions with the peptide (Figure 5B). Because of the enhanced stacking between Onc112 and A2062 compared to Bac71–35 (Figure 5B), any mutation which affects the position of A2062 is expected to have a greater effect on the binding of Onc112 than Bac71–35. The results of mutational analysis suggest that the proper positioning of A2062 influences the activity of Onc112 and establish the ribosome as the immediate cellular target of Onc112 and likely other PrAMPs.
Table 3. Ribosomal RNA mutations confer resistance to Onc112.
| SQ110 DTCa | MIC [μmol L-1] | |
|---|---|---|
| Bac71–35 | Onc112 | |
| “wild-type"b | 0.75 | 3 |
| 2503C | 0.75 | 12.5 |
| 2059C | 0.75 | 12.5 |
| 2059C | 0.75 | 12.5 |
| 2503C/2059G | 0.75 | 50 |
aThe E. coli strain SQ110 DTC and its derivatives used in these experiments carry a single chromosomal rrn allele and lack the tolC transporter gene (53).
bThe parental SQ110 DTC strain lacking rRNA mutations is designated “wild type".
CONCLUSION
We have established the molecular determinants required for ribosome binding of several PrAMPs. Our data show that the variable N-terminal domain makes different types of interactions with the ribosome, providing a rationale for the observed sequence diversity of the N-terminal domain in PrAMPs. For all selected PrAMPs, the location occupied by their N-terminal region is sterically incompatible with the simultaneous binding of the aminoacyl-tRNA in the ribosomal A site, thereby interfering with the initiation step of protein synthesis. The conserved middle region of all PrAMPs presented here superposes surprisingly well and correspondingly forms homologous interactions with the peptide tunnel. This region of the peptide tunnel is known to be the binding site of several antibiotics, including chloramphenicol, homoharingtonine, hygromycin A and other antibiotics, and also to accommodate the amino acid attached to the aminoacyl-tRNA bound in the A site. The variable C-terminal region interacts with different elements of the ribosome and overlaps with the binding site of macrolide and streptogramin B antibiotics. All PrAMPs presented in this study, including Onc112 reported previously (26,27), bind in the ‘reverse’ orientation inside the peptide exit tunnel of the ribosome. This mode of interaction appears to be required for the ribosome binding and protein synthesis inhibitory activity by these PrAMPs. It is noteworthy that all of these PrAMPs are derived through proteolysis of precursor polypeptides, which are synthesized on ribosomes (15,29). Presumably, during the transit of precursor PrAMPs through the ribosome peptide exit tunnel, which occurs in the canonical direction, the polypeptides do not form specific contacts with the ribosome that would otherwise stall protein synthesis. This provides the cell with an elegant strategy of using its own protein synthesis machinery to synthesize ribosome-targeted AMPs.
The binding in the peptide exit tunnel by different PrAMPs and their common mechanism of action of protein synthesis inhibition afforded by interaction with multiple rRNA residues encompassing three antibiotic-binding sites indicates that it has been conserved throughout evolution, suggesting an efficient way to inactivate the bacterial ribosome. The common ribosome binding and mechanism of translation inhibition by the multiple PrAMPs reported here establishes a structural basis for the design of new and more effective antibiotics.
ACCESSION NUMBERS
The atomic coordinates and structure factors have been deposited in the Protein Data Bank (5HAU) for the T. thermophilus 70S ribosome in complex with Bac71–35; (5HD1) for the T. thermophilus 70S ribosome in complex with Pyrrhocoricin; (5HCP) for the T. thermophilus 70S ribosome in complex with Metalnikowin; (5HCQ) for the T. thermophilus 70S ribosome in complex with OncΔ15–19; and (5HCR) for the T. thermophilus 70S ribosome in complex with Onc10wt.
Supplementary Material
Acknowledgments
We are thankful to P. B. Moore and N. Vázquez-Laslop for advice and critical reading of the manuscript. We thank the staff of the North East Collaborative Access Teams (NE-CAT) at the Advanced Photon Source (beamline 24-ID) for technical assistance with data collection. We thank the staff at the Richards Center at Yale University for computational support, R. L. Grodzicki for preparation of the fMet-tRNAfMet and Teresa Szal (UIC) for the double mutant E. coli strain.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Howard Hughes Medical Institute and National Institutes of Health [GM022778 to T.A.S.]; National Institutes of Health [GM106386 to A.S.M.]; National Institute of General Medical Sciences [P41GM103403 to the North East Collaborative Access Teams (NE-CAT) at the Advanced Photon Source (beamline 24-ID)]; U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory [DE-AC02-06CH11357]. Funding for open access charge: Howard Hughes Medical Institute.
Conflict of interest statement. None declared.
REFERENCES
- 1.McCoy L.S., Xie Y., Tor Y. Antibiotics that target protein synthesis. Wiley Interdiscip. Rev. RNA. 2011;2:209–232. doi: 10.1002/wrna.60. [DOI] [PubMed] [Google Scholar]
- 2.Selmer M., Dunham C.M., Murphy F.V.t., Weixlbaumer A., Petry S., Kelley A.C., Weir J.R., Ramakrishnan V. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 2006;313:1935–1942. doi: 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- 3.Amunts A., Fiedorczuk K., Truong T.T., Chandler J., Peter Greenberg E., Ramakrishnan V. Bactobolin A binds to a site on the 70S ribosome distinct from previously seen antibiotics. J. Mol. Biol. 2015;427:753–755. doi: 10.1016/j.jmb.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polikanov Y.S., Szal T., Jiang F., Gupta P., Matsuda R., Shiozuka M., Steitz T.A., Vazquez-Laslop N., Mankin A.S. Negamycin interferes with decoding and translocation by simultaneous interaction with rRNA and tRNA. Mol. Cell. 2014;56:541–550. doi: 10.1016/j.molcel.2014.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Polikanov Y.S., Osterman I.A., Szal T., Tashlitsky V.N., Serebryakova M.V., Kusochek P., Bulkley D., Malanicheva I.A., Efimenko T.A., Efremenkova O.V., et al. Amicoumacin a inhibits translation by stabilizing mRNA interaction with the ribosome. Mol. Cell. 2014;56:531–540. doi: 10.1016/j.molcel.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garreau de Loubresse N., Prokhorova I., Holtkamp W., Rodnina M.V., Yusupova G., Yusupov M. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 2014;513:517–522. doi: 10.1038/nature13737. [DOI] [PubMed] [Google Scholar]
- 7.Bulkley D., Brandi L., Polikanov Y.S., Fabbretti A., O'Connor M., Gualerzi C.O., Steitz T.A. The antibiotics dityromycin and GE82832 bind protein S12 and block EF-G-catalyzed translocation. Cell Rep. 2014;6:357–365. doi: 10.1016/j.celrep.2013.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bulkley D., Johnson F., Steitz T.A. The antibiotic thermorubin inhibits protein synthesis by binding to inter-subunit bridge B2a of the ribosome. J. Mol. Biol. 2012;416:571–578. doi: 10.1016/j.jmb.2011.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blaha G.M., Polikanov Y.S., Steitz T.A. Elements of ribosomal drug resistance and specificity. Curr. Opin. Struct. Biol. 2012;22:750–758. doi: 10.1016/j.sbi.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stanley R.E., Blaha G., Grodzicki R.L., Strickler M.D., Steitz T.A. The structures of the anti-tuberculosis antibiotics viomycin and capreomycin bound to the 70S ribosome. Nat. Struct. Mol. Biol. 2010;17:289–293. doi: 10.1038/nsmb.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polikanov Y.S., Starosta A.L., Juette M.F., Altman R.B., Terry D.S., Lu W., Burnett B.J., Dinos G., Reynolds K.A., Blanchard S.C., et al. Distinct tRNA accommodation intermediates observed on the ribosome with the antibiotics hygromycin A and A201A. Mol. Cell. 2015;58:832–844. doi: 10.1016/j.molcel.2015.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bulkley D., Innis C.A., Blaha G., Steitz T.A. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc. Natl. Acad. Sci. U.S.A. 2010;107:17158–17163. doi: 10.1073/pnas.1008685107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gurel G., Blaha G., Moore P.B., Steitz T.A. U2504 determines the species specificity of the A-site cleft antibiotics: the structures of tiamulin, homoharringtonine, and bruceantin bound to the ribosome. J. Mol. Biol. 2009;389:146–156. doi: 10.1016/j.jmb.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li W., Tailhades J., O'Brien-Simpson N.M., Separovic F., Otvos L., Jr, Hossain M.A., Wade J.D. Proline-rich antimicrobial peptides: potential therapeutics against antibiotic-resistant bacteria. Amino Acids. 2014;46:2287–2294. doi: 10.1007/s00726-014-1820-1. [DOI] [PubMed] [Google Scholar]
- 15.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 16.Yi H.Y., Chowdhury M., Huang Y.D., Yu X.Q. Insect antimicrobial peptides and their applications. Appl. Microbiol. Biotechnol. 2014;98:5807–5822. doi: 10.1007/s00253-014-5792-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Casteels P., Ampe C., Jacobs F., Vaeck M., Tempst P. Apidaecins: antibacterial peptides from honeybees. EMBO J. 1989;8:2387–2391. doi: 10.1002/j.1460-2075.1989.tb08368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gennaro R., Skerlavaj B., Romeo D. Purification, composition, and activity of two bactenecins, antibacterial peptides of bovine neutrophils. Infect. Immun. 1989;57:3142–3146. doi: 10.1128/iai.57.10.3142-3146.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scocchi M., Tossi A., Gennaro R. Proline-rich antimicrobial peptides: converging to a non-lytic mechanism of action. Cell Mol. Life Sci. 2011;68:2317–2330. doi: 10.1007/s00018-011-0721-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nguyen L.T., Haney E.F., Vogel H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011;29:464–472. doi: 10.1016/j.tibtech.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 21.Runti G., Lopez Ruiz Mdel C., Stoilova T., Hussain R., Jennions M., Choudhury H.G., Benincasa M., Gennaro R., Beis K., Scocchi M. Functional characterization of SbmA, a bacterial inner membrane transporter required for importing the antimicrobial peptide Bac7(1–35) J. Bacteriol. 2013;195:5343–5351. doi: 10.1128/JB.00818-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otvos L., Jr Antibacterial peptides isolated from insects. J. Pept. Sci. 2000;6:497–511. doi: 10.1002/1099-1387(200010)6:10<497::AID-PSC277>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 23.Mattiuzzo M., Bandiera A., Gennaro R., Benincasa M., Pacor S., Antcheva N., Scocchi M. Role of the Escherichia coli SbmA in the antimicrobial activity of proline-rich peptides. Mol. Microbiol. 2007;66:151–163. doi: 10.1111/j.1365-2958.2007.05903.x. [DOI] [PubMed] [Google Scholar]
- 24.Berthold N., Hoffmann R. Cellular uptake of apidaecin 1b and related analogs in Gram-negative bacteria reveals novel antibacterial mechanism for proline-rich antimicrobial peptides. Protein Pept. Lett. 2014;21:391–398. doi: 10.2174/09298665113206660104. [DOI] [PubMed] [Google Scholar]
- 25.Krizsan A., Volke D., Weinert S., Strater N., Knappe D., Hoffmann R. Insect-derived proline-rich antimicrobial peptides kill bacteria by inhibiting bacterial protein translation at the 70 s ribosome. Angew. Chem. Int. Ed. Engl. 2014;53:12236–12239. doi: 10.1002/anie.201407145. [DOI] [PubMed] [Google Scholar]
- 26.Seefeldt A.C., Nguyen F., Antunes S., Perebaskine N., Graf M., Arenz S., Inampudi K.K., Douat C., Guichard G., Wilson D.N., et al. The proline-rich antimicrobial peptide Onc112 inhibits translation by blocking and destabilizing the initiation complex. Nat. Struct. Mol. Biol. 2015;22:470–475. doi: 10.1038/nsmb.3034. [DOI] [PubMed] [Google Scholar]
- 27.Roy R.N., Lomakin I.B., Gagnon M.G., Steitz T.A. The mechanism of inhibition of protein synthesis by the proline-rich peptide oncocin. Nat. Struct. Mol. Biol. 2015;22:466–469. doi: 10.1038/nsmb.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomasinsig L., Zanetti M. The cathelicidins–structure, function and evolution. Curr. Protein Pept. Sci. 2005;6:23–34. doi: 10.2174/1389203053027520. [DOI] [PubMed] [Google Scholar]
- 29.Scocchi M., Romeo D., Zanetti M. Molecular cloning of Bac7, a proline- and arginine-rich antimicrobial peptide from bovine neutrophils. FEBS Lett. 1994;352:197–200. doi: 10.1016/0014-5793(94)00954-6. [DOI] [PubMed] [Google Scholar]
- 30.Marlow V.L., Haag A.F., Kobayashi H., Fletcher V., Scocchi M., Walker G.C., Ferguson G.P. Essential role for the BacA protein in the uptake of a truncated eukaryotic peptide in Sinorhizobium meliloti. J. Bacteriol. 2009;191:1519–1527. doi: 10.1128/JB.01661-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Podda E., Benincasa M., Pacor S., Micali F., Mattiuzzo M., Gennaro R., Scocchi M. Dual mode of action of Bac7, a proline-rich antibacterial peptide. Biochim. Biophys. Acta. 2006;1760:1732–1740. doi: 10.1016/j.bbagen.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 32.Benincasa M., Scocchi M., Podda E., Skerlavaj B., Dolzani L., Gennaro R. Antimicrobial activity of Bac7 fragments against drug-resistant clinical isolates. Peptides. 2004;25:2055–2061. doi: 10.1016/j.peptides.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 33.Zahn M., Kieslich B., Berthold N., Knappe D., Hoffmann R., Strater N. Structural identification of DnaK binding sites within bovine and sheep bactenecin Bac7. Protein Pept. Lett. 2014;21:407–412. doi: 10.2174/09298665113206660111. [DOI] [PubMed] [Google Scholar]
- 34.Mardirossian M., Grzela R., Giglione C., Meinnel T., Gennaro R., Mergaert P., Scocchi M. The host antimicrobial peptide Bac71–35 binds to bacterial ribosomal proteins and inhibits protein synthesis. Chem. Biol. 2014;21:1639–1647. doi: 10.1016/j.chembiol.2014.10.009. [DOI] [PubMed] [Google Scholar]
- 35.Cociancich S., Dupont A., Hegy G., Lanot R., Holder F., Hetru C., Hoffmann J.A., Bulet P. Novel inducible antibacterial peptides from a hemipteran insect, the sap-sucking bug Pyrrhocoris apterus. Biochem. J. 1994;300:567–575. doi: 10.1042/bj3000567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chernysh S., Cociancich S., Briand J.P., Hetru C., Bulet P. The inducible antibacterial peptides of the hemipteran insect Palomena prasina: Identification of a unique family of proline-rich peptides and of a novel insect defensin. J. Insect Physiol. 1996;42:81–89. [Google Scholar]
- 37.Narayanan S., Modak J.K., Ryan C.S., Garcia-Bustos J., Davies J.K., Roujeinikova A. Mechanism of Escherichia coli resistance to Pyrrhocoricin. Antimicrob. Agents Chemother. 2014;58:2754–2762. doi: 10.1128/AAC.02565-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kragol G., Lovas S., Varadi G., Condie B.A., Hoffmann R., Otvos L., Jr The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding. Biochemistry. 2001;40:3016–3026. doi: 10.1021/bi002656a. [DOI] [PubMed] [Google Scholar]
- 39.Junemann R., Wadzack J., Triana-Alonso F.J., Bittner J.U., Caillet J., Meinnel T., Vanatalu K., Nierhaus K.H. In vivo deuteration of transfer RNAs: overexpression and large-scale purification of deuterated specific tRNAs. Nucleic Acids Res. 1996;24:907–913. doi: 10.1093/nar/24.5.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin J., Gagnon M.G., Bulkley D., Steitz T.A. Conformational changes of elongation factor G on the ribosome during tRNA translocation. Cell. 2015;160:219–227. doi: 10.1016/j.cell.2014.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gagnon M.G., Lin J., Bulkley D., Steitz T.A. Crystal structure of elongation factor 4 bound to a clockwise ratcheted ribosome. Science. 2014;345:684–687. doi: 10.1126/science.1253525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Polikanov Y.S., Blaha G.M., Steitz T.A. How hibernation factors RMF, HPF, and YfiA turn off protein synthesis. Science. 2012;336:915–918. doi: 10.1126/science.1218538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gagnon M.G., Seetharaman S.V., Bulkley D., Steitz T.A. Structural basis for the rescue of stalled ribosomes: structure of YaeJ bound to the ribosome. Science. 2012;335:1370–1372. doi: 10.1126/science.1217443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 1993;26:795–800. [Google Scholar]
- 45.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A., et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Polikanov Y.S., Steitz T.A., Innis C.A. A proton wire to couple aminoacyl-tRNA accommodation and peptide-bond formation on the ribosome. Nat. Struct. Mol. Biol. 2014;21:787–793. doi: 10.1038/nsmb.2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 49.Orelle C., Szal T., Klepacki D., Shaw K.J., Vazquez-Laslop N., Mankin A.S. Identifying the targets of aminoacyl-tRNA synthetase inhibitors by primer extension inhibition. Nucleic Acids Res. 2013;41:e144. doi: 10.1093/nar/gkt526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vazquez-Laslop N., Thum C., Mankin A.S. Molecular mechanism of drug-dependent ribosome stalling. Mol. Cell. 2008;30:190–202. doi: 10.1016/j.molcel.2008.02.026. [DOI] [PubMed] [Google Scholar]
- 51.Shimizu Y., Kanamori T., Ueda T. Protein synthesis by pure translation systems. Methods. 2005;36:299–304. doi: 10.1016/j.ymeth.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 52.Merryman C., Noller H.F. Footprinting and modification-interference analysis of binding sites on RNA. In: CWJ S, editor. RNA:Protein Interactions, A Practical Approach. NY: Oxford University Press; 1998. pp. 237–253. [Google Scholar]
- 53.Orelle C., Carlson S., Kaushal B., Almutairi M.M., Liu H., Ochabowicz A., Quan S., Pham V.C., Squires C.L., Murphy B.T., et al. Tools for characterizing bacterial protein synthesis inhibitors. Antimicrob. Agents Chemother. 2013;57:5994–6004. doi: 10.1128/AAC.01673-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benincasa M., Zahariev S., Pelillo C., Milan A., Gennaro R., Scocchi M. PEGylation of the peptide Bac7(1–35) reduces renal clearance while retaining antibacterial activity and bacterial cell penetration capacity. Eur. J. Med. Chem. 2015;95:210–219. doi: 10.1016/j.ejmech.2015.03.028. [DOI] [PubMed] [Google Scholar]
- 55.Knappe D., Zahn M., Sauer U., Schiffer G., Strater N., Hoffmann R. Rational design of oncocin derivatives with superior protease stabilities and antibacterial activities based on the high-resolution structure of the oncocin-DnaK complex. Chembiochem. 2011;12:874–876. doi: 10.1002/cbic.201000792. [DOI] [PubMed] [Google Scholar]
- 56.Sadler K., Eom K.D., Yang J.L., Dimitrova Y., Tam J.P. Translocating proline-rich peptides from the antimicrobial peptide bactenecin 7. Biochemistry. 2002;41:14150–14157. doi: 10.1021/bi026661l. [DOI] [PubMed] [Google Scholar]
- 57.Sanbonmatsu K.Y., Joseph S., Tung C.S. Simulating movement of tRNA into the ribosome during decoding. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15854–15859. doi: 10.1073/pnas.0503456102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Agerberth B., Lee J.Y., Bergman T., Carlquist M., Boman H.G., Mutt V., Jornvall H. Amino acid sequence of PR-39. Isolation from pig intestine of a new member of the family of proline-arginine-rich antibacterial peptides. Eur. J. Biochem. 1991;202:849–854. doi: 10.1111/j.1432-1033.1991.tb16442.x. [DOI] [PubMed] [Google Scholar]
- 59.Guida F., Benincasa M., Zahariev S., Scocchi M., Berti F., Gennaro R., Tossi A. Effect of size and N-terminal residue characteristics on bacterial cell penetration and antibacterial activity of the proline-rich peptide Bac7. J. Med. Chem. 2015;58:1195–1204. doi: 10.1021/jm501367p. [DOI] [PubMed] [Google Scholar]
- 60.Chan Y.R., Zanetti M., Gennaro R., Gallo R.L. Anti-microbial activity and cell binding are controlled by sequence determinants in the anti-microbial peptide PR-39. J. Invest. Dermatol. 2001;116:230–235. doi: 10.1046/j.1523-1747.2001.01231.x. [DOI] [PubMed] [Google Scholar]
- 61.Raj P.A., Edgerton M. Functional domain and poly-L-proline II conformation for candidacidal activity of bactenecin 5. FEBS Lett. 1995;368:526–530. doi: 10.1016/0014-5793(95)00712-i. [DOI] [PubMed] [Google Scholar]
- 62.Voorhees R.M., Weixlbaumer A., Loakes D., Kelley A.C., Ramakrishnan V. Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nat. Struct. Mol. Biol. 2009;16:528–533. doi: 10.1038/nsmb.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sothiselvam S., Liu B., Han W., Ramu H., Klepacki D., Atkinson G.C., Brauer A., Remm M., Tenson T., Schulten K., et al. Macrolide antibiotics allosterically predispose the ribosome for translation arrest. Proc. Natl. Acad. Sci. U.S.A. 2014;111:9804–9809. doi: 10.1073/pnas.1403586111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kannan K., Vazquez-Laslop N., Mankin A.S. Selective protein synthesis by ribosomes with a drug-obstructed exit tunnel. Cell. 2012;151:508–520. doi: 10.1016/j.cell.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 65.Zahn M., Berthold N., Kieslich B., Knappe D., Hoffmann R., Strater N. Structural studies on the forward and reverse binding modes of peptides to the chaperone DnaK. J. Mol. Biol. 2013;425:2463–2479. doi: 10.1016/j.jmb.2013.03.041. [DOI] [PubMed] [Google Scholar]
- 66.Dunkle J.A., Xiong L., Mankin A.S., Cate J.H. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc. Natl. Acad. Sci. U.S.A. 2010;107:17152–17157. doi: 10.1073/pnas.1007988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frank R.W., Gennaro R., Schneider K., Przybylski M., Romeo D. Amino acid sequences of two proline-rich bactenecins. Antimicrobial peptides of bovine neutrophils. J. Biol. Chem. 1990;265:18871–18874. [PubMed] [Google Scholar]
- 68.Knappe D., Piantavigna S., Hansen A., Mechler A., Binas A., Nolte O., Martin L.L., Hoffmann R. Oncocin (VDKPPYLPRPRPPRRIYNR-NH2): a novel antibacterial peptide optimized against gram-negative human pathogens. J. Med. Chem. 2010;53:5240–5247. doi: 10.1021/jm100378b. [DOI] [PubMed] [Google Scholar]
- 69.Polikanov Y.S., Melnikov S.V., Soll D., Steitz T.A. Structural insights into the role of rRNA modifications in protein synthesis and ribosome assembly. Nat. Struct. Mol. Biol. 2015;22:342–344. doi: 10.1038/nsmb.2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.