

Abstract
RIZ1 is an estrogen receptor (ER) coactivator but is also a histone lysine methyltransferase that methylates lysine 9 of histone H3, an activity known to repress transcription. We show here that target organs of mice deficient in RIZ1 exhibit decreased response to female sex hormones. RIZ1 interacted with SRC1 and p300, suggesting that the coactivator function of RIZ1 may be mediated by its interaction with other transcriptional coactivators. In the presence of estrogen, RIZ1 binding to estrogen target genes became less direct and followed the binding of ER to DNA and RIZ1 methyltransferase activity on H3-Lys 9 was inhibited, indicating derepression may play a role in estrogen induction of gene transcription. Reducing RIZ1 level correlated with decreased induction of pS2 gene by estrogen in MCF7 cells. The data suggest that a histone methyltransferase is required for optimal estrogen response in female reproductive tissues and that estrogen-bound ER may turn a transcriptional repressor into a coactivator.
Female sex steroid hormones, such as estrogen (E2) and progesterone, play an essential role in various tissues and in numerous physiological processes, including the control of puberty, sexual behavior, bone homeostasis, mammopoiesis, and reproductive functions. Decreased steroid levels and/or responses are associated with aging and its associated syndromes, such as osteoporosis, cardiovascular disease, and Alzheimer's disease. Altered hormone responses are involved in the development and progression of breast cancer, the most common malignancy inflicted upon women, with more than 180,000 new cases each year in the United States alone.
The biological actions of E2 and progesterone are mainly mediated by their receptors that are ligand-dependent transcription factors. Upon binding of hormones, the receptors bind to their cognate DNA response elements on target genes and recruit coactivators and general transcription factors to form an active transcriptional complex, resulting in enhancement of target gene expression (13, 34). Three major classes of coactivators or coactivator complexes have been described. One class appears to function as histone/protein acetyltransferases (HATs) or to interact with HATs, which include CBP/p300 (7), SRC-1 (NCoA-1/p160) (21, 37), SRC-2 (TIF2/GRIP1) (16, 46), and SRC-3 (AIB1/pCIP/RAC3/ACTR/TRAM-1) (4, 9, 28, 44). Some HAT complexes also contain an RNA coactivator SRA (26). A physiological role for a HAT coactivator in hormone action is demonstrated by a mouse model deficient in SRC-1 showing partial hormone resistance (50, 52). The second class is the DRIP/TRAP protein complex (11, 38). Finally, recent studies indicate that histone/protein methyltransferases (HMTs) are potential coactivators. CARM1 and PRMT1 are arginine HMTs that methylate arginine residues on histones and other proteins such as p300 (8, 24, 43, 48, 53). RIZ1 and NSD1 are members of a superfamily of lysine HMTs (1, 17).
The RIZ (PRDM2) gene was originally isolated in a functional screening for proteins that bind to the Rb tumor suppressor (6). It has also been independently isolated as a DNA-binding protein MTB-Zf (35), a GATA3-binding protein G3B (41), and an estrogen receptor (ER)-binding protein (1). Two products of the gene exist owing to alternative promoter usage: RIZ1, which contains a PR (for PRDI-BF1 and RIZ1) domain, and RIZ2, which lacks the domain but is otherwise identical to RIZ1 (30). The PR domain was initially found as a stretch of ∼130-residue that shows homology between RIZ1 and a transcriptional repressor PRDI-BF1 (6, 18). This domain was later found to be related to the SET domain (20). The SET domain is the catalytic motif of lysine HMTs (39). RIZ1 has methyltransferase activity toward the lysine 9 residue of histone H3 (22), an activity known to be linked with transcriptional repression. RIZ1 has been largely studied as a tumor suppressor gene (19, 23, 42). RIZ1 is commonly silenced or inactivated in human cancers. Mouse gene knockout models show that RIZ1 inactivation can cause tumor susceptibility (42).
RIZ has LXXLL motifs that mediate estrogen-dependent binding to the ligand-binding domain of ER (1). RIZ1 also has transcriptional coactivator functions, as assayed in vitro by cotransfection studies (42). Here, we characterized the RIZ1 knockout mice to address the role of RIZ1 in sex hormone action in vivo.
MATERIALS AND METHODS
Plasmids and transient transfections.
The human RIZ1 and RIZ2 expression vectors and RIZ1 mutant vectors have been described (42). Expression vectors of RIZ proteins and expression vectors containing full-length mammalian ERα, progesterone receptor (PR), retinoic acid receptor α (RARα), retinoid X receptor α (RXRα), vitamin D receptor (VDR), androgen receptor (AR), thyroid receptor (TR), glucocorticoid receptor (GR), GRIP-1, and SRC-1 were cotransfected into CV-1 cells with an appropriate reporter construct containing a synthetic hormone response element linked to the tk-CAT reporter. The reporter TREpal-tk-CAT containing a synthetic response element for RAR, RXR, and TR (55) was used to evaluate the effects of RARα, RXRα, and TR, the reporter GRE-tk-CAT (54) was used for GR and AR, the reporter ERE-tk-CAT (27) was used for ERα, the reporter PRE-tk-cat was used for PR (33), and the reporter VDRE-tk-CAT (2) was used for VDR.
A calcium phosphate precipitation procedure was used for transient transfection as described previously (27). Briefly, 0.5 to 1.0 × 105 cells/well were seeded in 24-well plates, and 50 to 400 ng of RIZ plasmids, 100 ng of expression vectors for nuclear hormone receptors (NHRs), 100 ng of reporter plasmid, and 100 ng of a β-Gal expression vector were mixed with carrier DNA to 1 μg of total DNA/well. Transfections of MCF-7 cells (2 × 105 cells/well in six-well plates) used the Effectene transfection reagent (Qiagen), according to the manufacturer's instructions. Cells were treated with or without the indicated hormone (50 nM for estradiol [E2] and 100 nM for R5020 [synthetic P agonist], all-trans-retinoic acid, 9-cis-retinoic acid, 1,25 dihydroxyvitamin D3, dihydrotestosterone, triiodothyronine, and dexamethasone; all were from Sigma except R5020, which was from NEN) for 24 h, and chloramphenicol acetyltransferase (CAT) activity was measured as described previously (27). CAT values were normalized for transfection efficiency by the corresponding β-Gal activity.
Immunoprecipitation and immunoblotting.
MCF-7 and T47-D breast cancer cells were grown in 15-cm dishes in Dulbecco modified Eagle medium (DMEM) with 5% fetal calf serum and 2 mM l-glutamine. Subconfluent cells (4 × 106) were then cultured in DMEM without phenol red, serum, or hormones for an additional 3 days, during which time the medium was changed twice daily. Cells were incubated for 24 h with or without hormones (50 nM for E2 and 100 nM for R5020) and infected with an adenovirus vector containing RIZ1 (AdRIZ1; at a concentration of 1010 viral particles/15-cm dish) or the empty vector (AdNull) (14). Cells were then grown for an additional 48 h in DMEM without phenol red supplemented with 5% charcoal-treated fetal calf serum (Omega Scientific). Cells were harvested and proceeded to immunoprecipitation and immunoblotting essentially as described previously (6, 30). Antibodies used included RIZ monoclonal antibody 2D7 (6), monoclonal antibodies versus ERα (sc-8005; Santa Cruz Biotechnology), progesterone receptor (PR; 1A6; Dako), SRC-1 (M-342; Santa Cruz Biotechnology), or p300 (N-15; Santa Cruz Biotechnology).
Mice.
RIZ1−/− mice were generated in our laboratory as described previously (42). To examine reproductive functions, female RIZ1−/− mice were bred with male RIZ1+/− mice, female RIZ1+/− mice were bred with male RIZ1−/− mice, and breedings between heterozygous mutants were also performed. The number of litters was recorded, and all mice were routinely measured for body weight throughout the studies.
Analysis of gene expression.
For PR staining, the polyclonal rabbit anti-PR antibody (A0098; Dako) was used at a dilution of 1/100, essentially as described previously (45). Tissues were fixed in Bouin's solution and processed by routine methods for embedding in paraffin and sectioning (5 μm). The sections were subjected to treatment by using the target retrieval solution (Dako), all performed according to the manufacturer's instructions. Sections were then incubated with the primary antibody followed with a biotinylated secondary antibody. The localization of the primary antibody was visualized with the imidazole-diaminobenzidine reaction producing a brown stain, followed by hematoxylin counterstaining and routine processing for bright-field microscopy analysis.
For reverse transcription-PCR (RT-PCR) analysis of RIZ gene expression, tissues from 7-week-old RIZ1+/+ mice were pulverized in liquid nitrogen, and total RNA was isolated by using the Trizol reagent (Gibco-BRL). cDNA was synthesized by using the first-strand cDNA synthesis kit (Gibco-BRL). The oligonucleotide pairs RP260 (5′-CTC ATT CAT CTA AGA AAG GTG G-3′)-RP259 (5′-TGA TTC CAG GTC ACT TCA GG-3′) and RP170 (5′-GAA GCC AAA GGC CTC TCA TC-3′)-K05 (5′-AGA CTC TGG CTG AGG TAC C-3′) were used in standard PCR conditions encompassing 30 cycles at an annealing temperature of 59°C to amplify the RIZ1+2 and RIZ1 specific fragments, respectively.
Analysis of target organs in response to hormone treatments.
To investigate E2-mediated increments in proliferation and hyperemia of uteri and changes in the cellular organization of the vaginal epithelium (52), 8-week-old female RIZ−/− and RIZ+/+ mice were ovariectomized (OVXed). At day 15 to 17 after ovariectomy (OVX), mice were treated with subcutaneous (s.c.) injections of E2 (0.8 ng/g/day; Sigma) or with the vehicle (0.1 ml of corn oil) alone for 3 days. The mice were then sacrificed at day 18, the uterine wet weight was measured, and vaginal tissue was collected. The tissues were processed as described above, and vaginal specimens were stained with routine hematoxylin and eosin, and uterine specimens were prepared for PR expression. The thickness of the vaginal epithelium and the cornified layer was photographed and measured as described in Fig. 4 by using the Spot 3.2.4 software (Diagnostic Instruments).
FIG. 4.

Mammopoiesis in RIZ1 deficient mice. Whole mounts of the fourth mammary gland of mice with the indicated genotypes were prepared and stained as described in Materials and Methods. (A and B) Seven-week-old virgin mice; (C and D) mice pregnant for the first time; (E and F) higher magnification of the ducts and alveolar structures of the mammary glands of the pregnant mice; (G and H) mammary glands from mice treated with hormone pellets containing P and E2 as described in Materials and Methods; (I and J) higher magnification of the mammary ducts and alveolar structures from G and H, respectively. Scale bars that apply to both genotypes are inserted.
To evaluate the P- and E2-mediated decidual response, we used a previously described protocol (32, 52). Briefly, 8-week-old female mice were OVXed and at days 10 to 12 after OVX, mice were treated with s.c. injections of E2 in corn oil (100 ng/day) and then treated with P (1 mg/day; Sigma) plus E2 (6.7 ng/day) from days 16 to 23. At 6 h after the third P+E2 injection on day 18, the left uterine horn (the right horn served as a control) was traumatically stimulated by insertion of a burred needle proximal to the cervix and longitudinally scratching the entire length of the uterine horn antimesometrially. The mice were sacrificed on day 23, 6 h after the last P+E2 injection, and the wet weights of the left and right uterine horns were measured.
To measure the effects on mammopoiesis by normal development, pregnancy, and female sex hormone treatment, the following protocols were used (51, 52). To determine the effects of normal pubertal development on mammary gland growth, 7-week-old virgin female mice were sacrificed. In addition, pregnant female mice at day 19 of first pregnancy were sacrificed. To examine the effects of E2+P treatments on mammary gland development, 8-week-old female mice were OVXed and, at 14 days after OVX, they were treated with s.c. 21-day releasing hormone pellets containing 0.1 mg of E2 and 10 mg of P4 or s.c. placebo pellets (Innovative Research of America). On day 35, the mice were sacrificed, and the uterus was wet weighed and analyzed microscopically to ensure actual hormone release. In all cases, whole mounts were prepared from the fourth mammary gland and carmine-stained according to standard procedures (10), and the relationship between mammary ducts and mammary fat pad and the extent of branching, as well as number of branches, were investigated microscopically.
In order to evaluate the response to testosterone, 12-week-old male mice were orchiectomized, and the testicles were wet weighed (52). At 9 days after orchiectomy, mice were treated during days 9 to 15 with s.c. injections of testosterone (3 mg/kg/day; Sigma) or vehicle alone (0.1 ml of corn oil). On day 16, mice were sacrificed, and the prostate and the pars prostate of the urethra (for technical reasons) were removed and wet weighed.
ChIP analysis.
The procedure of Shang et al. was used for chromatin immunoprecipitation (ChIP) assays (40). Briefly, MCF7 cells were grown to 95% confluence in phenol red-free Dulbecco comodified Eagle medium (i.e., DMEM) supplemented with 10% charcoal-dextran-stripped fetal bovine serum for 3 days. After the addition of hormone for 45 min, cells were washed twice with phosphate-buffered saline and cross-linked with 1% formaldehyde at room temperature for 10 min. The cells were then processed for ChIP assays as described by Shang et al. (40). Antibodies used included anti-ER (Santa Cruz), anti-dimethyl-H3-K9 (Upstate), anti-dimethyl-H3-K4 (Upstate), anti-acetyl-H3-K9 (Upstate), and anti-RIZ1 antibody KG-7.1S or monoclonal antibody MAB1045 (Abgent, San Diego, Calif.). The primers used were as follows: for PCR amplification of human pS2 gene promoter, pS2Pf (GGC CAT CTC TCA CTA TGA ATC ACT TCT GC) and pS2Pr (GGC AGG CTC TGT TTG CTT AAA GAG CG); and for PCR amplification of GAPDH gene promoter, GAPDHPF2 (5′-AAA AGC GGG GAG AAA GTA GG) and GAPDHPR2 (5′-GTC GAA CAG GAG GAG CAG AG). RIZ1 knockout embryo fibroblasts were similarly processed for ChIP analysis. PCR primer sequences were as follows: mG6PDpf, 5-CAT CGG GGA AGG CGT AAG GGC GG; mG6PDpr, 5-GGG GGC TAA GCT TCG CTG GCC CAT; mcdc25Cpf, 5-CGC CCT GAG CAA CTG CAA TGT AAC; and mcdc25Cpr, 5-CTT CAG AGT CTT CAC CGA GGG AG. These primers cover the promoter regions of these genes.
Knockdown assays.
RIZ1 knockdown small interfering RNA (siRNA) vector pSiRIZ1 was constructed by inserting into pSilencer2.0-U6 (Ambion), a DNA oligonucleotide containing RIZ1 target sequence (upper strand, 5′-GATC GGTCC TAAAG AAGAC GAAG TTCAAGAG CTTC GTCTT CTTTA GGACC TTTTTT GGAA-3′; bottom strand, 5′-AGCT TTCC AAAAAA GGTCC TAAAG AAGAC GAAG CTCTTGAA CTTCG TCTTC TTTAG GACC-3′). MCF7 cells that were in phenol red-free DMEM supplemented with 10% charcoal-dextran-stripped fetal bovine serum for 1 day were transfected with siRNA vectors by Lipofectamine (Invitrogen). Cells were cultured for 2 days after transfection in phenol red-free DMEM supplemented with 10% charcoal-dextran-stripped fetal bovine serum. Cells were then treated with E2 for 3 h before they were harvested for RNA analysis of pS2 gene expression or for 45 min before harvesting for ChIP analysis. The primers used for RT-PCR analysis of pS2 gene were as follows: pS2RTPCR-f, 5′-ATGGCCACCATGGAGAACAA; and pS2RTPCR-r, 5′-TAAAACAGTGGCTCCTGGCG. The primers for amplification of human beta-actin were as follows: bActin1, 5′-GTGGGGCGCCCCAGGCACCA-3′; and bActin2, 5′-CTCCTTAATGTCACGCACGATTTC-3′. Western blot analyses with anti-RIZ1 rabbit serum (Abcam) and anti-PARP (poly-ADP-ribose polymerase) antibody (Santa Cruz Biotech) were performed with nuclear extracts derived from cells at 2 days posttransfection.
Statistical analysis.
Student’s unpaired t test was used for statistical evaluation of means, with a P of <0.05 considered to be significant. All results are expressed as means ± the standard error of the mean (SEM) or means ± the standard deviation, when stated.
Methylation assays.
Methylation reactions (30 to 40 μl) contained 20 mM Tris-HCl (pH 8.0), 0.2 M NaCl, 0.4 mM EDTA, immunoprecipitation products, free histones (10 to 20 μg), and 3 μl (1.65 μCi and 21 pmol) of [methyl-3H]-adenosylmethionine (Amersham-Pharmacia). Nuclear extracts were prepared according to standard procedures and used for immunoprecipitation. Antisera used for immunoprecipitation were anti-KG7.1S and preimmune serum as described previously (6).
RESULTS
RIZ1 is a coactivator of ER and PR.
RIZ1 has been shown to be a coactivator of ER and can directly interact with ER (1, 42). To better understand the coactivator function of RIZ1, we compared the effects of RIZ1 on ER with its effects on several other NHRs. As shown in Fig. 1A, RIZ1 enhanced ER and PR function in a ligand- and dose-dependent manner. In comparison, RIZ1 had no major effects on the activities of androgen receptor, thyroid hormone receptor, glucocoticoid receptor, vitamin D receptor (not shown), retinoic acid receptor α (not shown), and retinoic X receptor α (not shown). Consistent with RIZ1 effects on ER and PR, RIZ1 formed protein complexes with these receptors as detected by immunoprecipitation with anti-receptor antibodies (Fig. 1B, lanes 2 to 5) or control preimmune antibody (Fig. 1B, lane 1), followed by Western blot analysis with anti-RIZ1 antibodies. Thus, RIZ1 seems to be a specific coactivator of ERs and PRs. The fact that RIZ1 has activator function on nonphysiological-chromatin templates such as plasmid DNA suggests that RIZ1's activator function may not involve its histone/chromatin methylation activity. RIZ1 has been shown to be widely expressed (6). By RT-PCR analysis, we confirmed that RIZ1 was expressed in various target tissues of sex steroid hormones (Fig. 1C).
FIG. 1.

RIZ1 is a selective coactivator of ER and PR. (A) Promoter reporter analysis. The indicated amounts of RIZ1 and various NHR expression vectors were cotransfected with a reporter construct containing appropriate hormone receptor response element into CV-1 cells. Cells were treated with or without the indicated hormone for 24 h and assayed for CAT activity as described in Materials and Methods. The data shown represent the means ± the SEM of three independent experiments. (B) RIZ1 forms hormone-dependent protein complexes in vivo with ERα and PR proteins. T47-D (for PR) and MCF-7 (for ER) cells were infected with adenovirus expressing RIZ1. Cell extracts treated with or without hormones were prepared and immunoprecipitated (IP) with preimmune serum (PI, lane 1) or monoclonal antibodies to PR (PR, lanes 2 and 3) or ERα (lanes 4 and 5) as indicated. The immunoprecipitated products were analyzed by immunoblot analysis with anti-RIZ1 antibody. (C) RT-PCR analysis of RIZ1 expression in target organs of sex steroid hormones. Total RNAs isolated from the indicated tissues of mice were used for RT-PCR analysis as described in Materials and Methods.
Partial hormone resistance in target organs of RIZ1-deficient female mice.
The RIZ1 mutant mice lack RIZ1, but not RIZ2, and therefore lack specifically any PR-domain related functions of the gene (42). These animals are viable and fertile and show no gross developmental defects (42). To determine the physiological role of RIZ1 in steroid hormone action, uterine growth in response to E2 was first examined in OVXed mice (31). Wild-type or heterozygous RIZ1 mutant mice responded to E2 treatment with a 5.3 ± 0.5- or a 5.4 ± 0.4-fold increase, respectively, in uterine wet weight. The uteri of homozygous RIZ1 mutants showed a smaller increase of ca. 3.7 ± 0.4-fold (P < 0.001 for +/+ versus −/− animals; Fig. 2A and B). Uterine response to mechanical traumatization (decidual stimulation) is mainly a PR-dependent process (32). We treated OVXed mice with a high dose of progesterone and a low dose of E2, followed by mechanical stimulation of the left uterine horn of each animal. The unstimulated left uterine horn served as a control. The decidual response resulted in an increase in the uterine horn size and was consistently observed in the stimulated left uterine horn in wild-type mice. Only a partial response was observed in the uterine horn of the RIZ1-null mutant (Fig. 2C). These results suggest that RIZ1 is required for maximal uterine response to E2 and progesterone in vivo.
FIG. 2.

Uterine and vaginal responses to female sex steroid hormones in RIZ1 mutant mice. Eight-week-old females with the indicated genotypes were OVXed and then treated for 3 days with E2 or the vehicle (V) alone from days 15 to 17 post-OVX. Uterine and vaginal tissues were collected on day 18 and analyzed as follows. (A and B) Impaired uterine growth stimulated by E2 in RIZ1 mutant mice. The uterine wet weight was measured, and the ratio of uterine weight to body weight was calculated for the indicated number of mice and is presented as the mean ± the SEM. RIZ1−/− mice showed a significantly reduced response to E2 treatment compared to RIZ1+/+ mice (P < 0.001). (C) RIZ1 mutant mice show a reduced uterine response to a decidual stimulus. The decidual response was measured as described in Materials and Methods. After treatment with E2 and progesterone, and mechanical stimulation of the left uterine horn, the ratio of the weights of the stimulated (L) horn to the unstimulated (control; R) horn was calculated for the indicated number of mice and is presented as the mean ± the SEM. (D and E) Vaginal responses. Vagina tissues were collected and fixed for hematoxylin and eosin staining as described in Materials and Methods. The thickness of the entire vaginal epithelium and the cornified layer were measured at seven randomly selected areas at the maximum depth of the vaginal epithelium in each mouse, and data are presented as the means ± the SEM for seven mice in each group.
We also examined vaginal cornification in response to a 3-day treatment of E2 in OVXed mice. The hormone produces an increased vaginal thickening and cornification of the epithelium (31), which was consistently seen in the wild-type mice (Fig. 2D and E). RIZ1-null female mice, however, failed to show a maximal such response, suggesting that RIZ1 is required for vaginal response to E2 in vivo.
Next, we analyzed the epithelial and stromal compartments of the uterus for E2-induced changes in PR expression. Such a regulation via ER is highly compartment specific and mimics the changes in the uterus during the estrous cycle (45). PR staining in OVXed RIZ1+/+ and RIZ1−/− mice treated with vehicle was apparently similar and showed strong immunoreactivity in almost all cells of both the luminal epithelium (LE) and the glandular epithelium (GE), whereas only a fraction of stromal and myometrial cells were positive (Fig. 3B, data not shown). As expected, E2 treatment of wild-type animals had a dual effect on PR expression, reducing the levels in the LE and increasing the levels in the stromal and myometrial compartments. Immunoreactivity in the GE was unchanged, a finding consistent with previous findings showing that E2 alone does not regulate PR expression in this compartment (Fig. 3) (45). Upon E2 treatment of RIZ1-deficient animals, the decrease of PR levels in the LE was ∼8-fold less than in wild-type tissues, and the increase of the receptor level in the stroma and myometrium was ∼2-fold less than normal. These findings suggest that RIZ1 plays a role in the E2 regulation of PR expression in the uterus.
FIG. 3.

Impaired regulation of uterine PR expression by E2 in RIZ1 mutant mice. Uteri were collected and immunohistochemically analyzed for PR expression. (A) Estrogen-treated uteri (top panel, low magnification; bottom panel, high magnification). Randomly selected areas were photographed, and more than 1,000 cells from each compartment of the uteri of four mice in each group were counted. (B) Data are presented as the means ± the SEM. Inserted scale bars apply to both genotypes.
Steroid hormones play an important role in mammopoiesis (15). Both E2 and progesterone are essential for alveolar development during pregnancy. In 7-week-old wild-type females, mammary ducts had started to grow into the mammary fat pad but with no apparent differences in the age-matched RIZ1-null mutants (Fig. 4A and B). However, when stimulated by pregnancy, alveolar structures in wild-type mammary glands were highly developed and appeared on all ductal sections, filling the interductal spaces. In the RIZ1 mutant mammary glands, alveoli were much less developed in terms of number and size of alveoli, and fewer alveoli were observed at the ends of ducts at the same stage of pregnancy (Fig. 4C through F). Although mammary glands of RIZ1-null mutants can still produce milk (data not shown), our results suggest that RIZ1 is required for normal mammary ductal elongation and alveolar development in vivo during pregnancy (Fig. 4C through F). We also examined mammary gland development in response to E2 and progesterone treatment in OVXed adult mice. E2 and progesterone stimulated a complex ductal arborization and extensive alveolar formation in mammary glands of wild-type mice, which mimics a stage of mammopoiesis in early pregnancy. In the mammary glands of RIZ1 mutant mice, only partial ductal growth was observed after hormone treatment (Fig. 4G through J). Thus, RIZ1 seems to be required for efficient proliferation and differentiation of the mammary gland in response to E2 and progesterone.
Normal steroid hormone response in RIZ1-deficient male mice.
Although RIZ1 is expressed in target tissues of male steroid hormones such as testis (6), it appears not to serve as coactivator for androgen receptor (Fig. 1A). To study this further, we measured prostate growth in orchiectomized male mice after they were treated with androgen. Similar to female mice, no difference in total body weight was noted in RIZ1-null mutant male animals (Table 1). Eight days after orchiectomization, the prostates in both wild-type and mutant animals regressed. Injection of testosterone for 7 days stimulated prostate growth in wild-type animals, as well as in RIZ1-null animals. Wild-type and RIZ1-null mutant animals had similar ratios of the weight of the prostate and urethra to body weight (1.19 ± 0. 03 versus 1.14 ± 0.03; n = 10 and 9 for +/+ and −/− mice, respectively; P = 0.23). They also had similar ratio of testis weight to body weight (7.7 ± 0. 4 versus 7.4 ± 0.4; n = 14 for both genotypes; P = 0.58). Thus, tissue responses to testosterone are not significantly affected in mice lacking RIZ1, a finding consistent with the lack of effect of the gene on androgen receptor transactivation functions (Fig. 1A).
TABLE 1.
Body weight analysis of live animals
| Mouse phenotype | Mean body wt (g) ± SD (no. of mice weighed) at:
|
|||
|---|---|---|---|---|
| 5 wk
|
8 wk (female) | 12 wk (female) | ||
| Female | Male | |||
| RIZ1−/− | 16.83 ± 1.64 (11) | 19.24 ± 2.25 (7) | 21.28 ± 2.70 (32) | 24.27 ± 2.21 (13) |
| RIZ1+/− | 16.83 ± 2.01 (15) | 20.00 ± 2.56 (15) | 20.49 ± 2.70 (12) | 24.51 ± 3.22 (23) |
| RIZ1+/+ | 18.52 ± 1.55 (5) | 20.61 ± 2.72 (7) | 20.37 ± 2.13 (31) | 21.71 ± 3.49 (13) |
Reduced litter size by RIZ1-deficient female mice.
We next sought to determine whether RIZ1 mutant mice might have less-efficient reproductive abilities. Female RIZ1−/− mice were bred with male RIZ1+/− mice and, as controls, female RIZ1+/− mice were bred with male RIZ1−/− mice. Also, breedings between heterozygous mutants were conducted. The average litter size from breeding RIZ−/− females and RIZ1+/− males was 6.6 ± 3.2 (standard deviation; n = 35 litters), but the average litter sizes were 8.4 ± 2.0 (n = 20 litters) for litters of breeding RIZ1+/− females with RIZ1−/− males (P < 0.01) and 8.0 ± 2.2 (n = 50 litters) for litters of breeding between heterozygous mice (P < 0.02). The results indicated a slightly compromised reproductive ability associated with female but not male mice deficient in RIZ1, a finding consistent with the above results showing defective tissue responses to steroids in female mice but not in male animals.
RIZ1 interacts with proteins in the steroid hormone receptor coactivator complex.
The partial resistance of RIZ1-deficient mice to female sex steroid hormones was similar to that observed in the SRC-1-null animals (52). We next analyzed whether RIZ1 may act cooperatively with proteins of the SRC-1/p160 family members in enhancing ER transactivation. In the presence of either SRC-1 or GRIP-1, RIZ1 enhanced the ER transactivation activity in a dose-dependent manner (Fig. 5A). Consistent with these findings, RIZ1-SRC-1 protein complexes could be detected by immunoprecipitation with RIZ1 or SRC-1 antibody (Fig. 5B, lanes 2 to5) but not with control preimmune antibody (Fig. 5B, lane 1), followed by Western blot analysis with anti-RIZ1 or anti-SRC-1 antibody. In addition, RIZ1 physically interacted with p300 acetyltransferase, as detected by immunoprecipitation with p300 antibody but not with control preimmune antibody, followed by anti-RIZ1 Western blotting (Fig. 5B, compare lane 1 with lanes 6 and 7). These findings suggest that RIZ1 may cooperate with HAT class coactivators.
FIG. 5.

RIZ1 interacts with HAT-class coactivators. (A) RIZ1 enhances the coactivator functions of members of the SRC-1 (p160) family. The indicated amounts of RIZ1, ERE-tk-CAT, ERα, and SRC-1 or GRIP-1 expression vectors were cotransfected into CV-1 cells. Cells were treated with or without E2 and assayed for CAT activity. Expression of β-Gal was used to control transfection efficiency. The data shown represent the means ± the SEM of three independent experiments. (B) RIZ1 forms protein complexes with SRC-1 and p300. T47-D cells were infected with AdRIZ1. Cell extracts treated with or without E2 were prepared and immunoprecipitated (IP) followed by immunoblot analysis. As a control, a preimmune serum (PI) was also used in the immunoprecipitation experiment (lane 1).
RIZ1 binding to estrogen target gene.
RIZ1 has been shown to methylate H3-K9 (22), which implicates a role in transcriptional repression and thus appears to be in conflict with RIZ1's role as an ER coactivator. To help us understand this paradox, we examined binding of RIZ1 to estrogen target gene pS2 promoter in MCF7 cells by ChIP analysis. Expression of pS2 gene and binding of ER to pS2 gene promoter are known to be induced by estrogen in MCF7 cells (40). As a positive control, we confirmed E2-induced ER-binding to pS2 promoter. Unlike ER, RIZ1 was found present on the pS2 promoter in the absence of E2 treatment (Fig. 6A). The binding of RIZ1 to pS2 promoter was reduced by E2 treatment. To determine histone modification status of the pS2 promoter, we examined the effects of E2 on H3-K9 and H3-K4 methylation and on H3-K9 acetylation. E2 treatment decreased H3-K9 methylation but did not significantly affect H3-K4 methylation. Furthermore, E2 treatment enhanced H3-K9 acetylation, a finding consistent with the known recruitment of HAT class coactivators (40). We found no detectable amount of precipitated chromatin corresponding to a control gene promoter, the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene promoter. These results show a correlation between decreased binding of RIZ1 and reduced H3-K9 methylation, suggesting a role for derepression in the induction of pS2 gene transcription by estrogen.
FIG. 6.

ChIP analysis of estrogen target genes. (A) Soluble chromatin was prepared from MCF7 cells not treated or treated with E2 for 45 min. Immunoprecipitation was performed with antibodies against RIZ1, dimethylated H3-K9, dimethylated H3-K4, acetylated H3-K9, and ER. DNA extractions were amplified by using primer sets that cover the pS2 gene promoter region or the GAPDH gene promoter. (B) Time course analysis of RIZ1 binding to the pS2 gene promoter. MCF7 cells treated with E2 for different periods of time, as indicated at the top of each lane, were processed for ChIP analysis. Immunoprecipitation was performed with antibodies to RIZ1, ER, and SRC1 and control immunoglobulin G as indicated. (C) ChIP analysis was performed on RIZ1 knockout mouse embryonic fibroblasts by using anti-dimethyl-H3 K9 antibody and anti-RIZ1 antibody. Immunoprecipitated chromatin DNA was analyzed by PCR with primers in the G6pd promoter region and in the cdc25c promoter region.
RIZ1 is a zinc finger protein and has DNA-binding activity (1, 35). The results presented in Fig. 6A suggest that RIZ1 may bind to pS2 promoter DNA directly in the absence of E2 and function to repress pS2 gene expression. After E2 treatment, the direct binding of RIZ1 to DNA may be disrupted by ER, and the interaction of ER-bound RIZ1 with DNA may become indirect or ER mediated. The reduced binding of RIZ1 to DNA in the presence of E2 may reflect a lower efficiency in DNA-protein cross-linking (in the ChIP assay) of an indirect interaction between DNA and RIZ1 versus a direct interaction. To further test whether RIZ1 binding to DNA may become ER-mediated after E2 treatment, we examined the time course profile of RIZ1 binding to pS2 promoter (Fig. 6B). We confirmed that ER binding to pS2 promoter was cyclical, being on the promoter at 45 and 135 min but off the promoter at 0 and 90 min of E2 treatment (Fig. 6B), as previously reported (40). Furthermore, a known ER coactivator that requires ER for loading onto promoter, SRC1, showed similar cyclical binding to DNA as ER (Fig. 6B), a finding consistent with an earlier report (40). We found that RIZ1 binding to the promoter was also cyclical in the presence of E2, being on the promoter at 45 and 135 min but off the promoter at 90 min (Fig. 6B). The amounts of DNA bound by RIZ1 at 45 and 135 min were similar but were less than that at 0 min, suggesting that RIZ1 binding to DNA maybe switched by E2 from direct binding to indirect binding. The indirect binding of RIZ1 to DNA may be mediated by RIZ1-bound ER, since RIZ1 binding to DNA coincided with ER binding to DNA after E2 treatment.
To further confirm RIZ1 binding to estrogen target genes, we examined the binding of RIZ1 to estrogen target genes in the RIZ1 knockout mouse embryo fibroblasts. Since pS2 gene is not a well-characterized estrogen target gene in the mouse, we selected the known mouse estrogen target gene glucose-6-phosphate dehydrogenase (G6PD) for analysis. As shown in Fig. 6C, binding of RIZ1 to G6PD promoter was detected in wild-type but not in RIZ1 knockout cells. Also, H3-K9 methylation at G6PD was reduced in RIZ1 knockout cells. As a control, cdc25c gene was not bound by RIZ1 and did not show H3-K9 methylation changes in the knockout cells.
To determine the significance of RIZ1 binding to pS2 promoter, we sought to determine by using the siRNA approach whether the expression and methylation state of pS2 changes when RIZ1 is knocked down. As shown by immunoblot analysis in Fig. 7A, transfection of a RIZ1 siRNA-expressing vector decreased RIZ1 protein levels in MCF7 cells relative to a control vector transfection. As demonstrated by RT-PCR analysis (Fig. 7B), induction of pS2 gene by E2 was reduced in RIZ1 siRNA transfected cells versus control transfected cells. Also, siRNA knockdown decreased H3-K9 methylation of pS2 promoter, which correlates with reduced RIZ1 binding to pS2 promoter, as shown by ChIP analysis (Fig. 7C). The results suggest that RIZ1 plays a coactivator role in the estrogen-induced transcription of pS2 gene.
FIG. 7.

Knockdown of RIZ1 by siRNA affects expression and methylation of pS2 gene. (A) Western blot analysis of equal amounts of nuclear extracts derived from siRNA and control vector-transfected MCF7 cells. Antibodies used are indicated on the left side of the panels. Equal loading is indicated by Western blot of PARP (lower panel). (B) RT-PCR analysis of pS2 and beta-actin expression with RNA derived from siRNA and control vector-transfected MCF7 cells treated with E2 and without E2. Amplification was done on three different concentrations of each RNA (twofold serial dilution). (C) pS2 gene ChIP analysis of siRNA and control vector-transfected MCF7 cells treated with or without E2. Antibodies used for immunoprecipitation are indicated at the top of the panel.
Effect of estrogen on RIZ1 enzyme activity.
We next examined estrogen effects on the H3 methylation activity of RIZ1. Nuclear extracts from control and E2-treated MCF-7 cells were immunoprecipitated by RIZ1 serum or preimmune serum. Methylation of H3 was detected in the immunoprecipitates by RIZ1 serum but not by preimmune serum (Fig. 8A, lanes 1 and 3). E2 treatment decreased H3 methylation by RIZ1 (Fig. 8A, lane 4). RNase protection analysis did not reveal altered RIZ1 gene expression by E2 treatment of MCF-7 cells (not shown). Furthermore, E2 also similarly inhibited the H3 methylation activity of overexpressed RIZ1 protein in AdRIZ1 virus-infected MCF-7 cells (Fig. 8A, lanes 5 to 6); cells infected with control virus showed results similar to those observed with noninfected cells (not shown). That E2 did not affect RIZ1 protein expression was confirmed by RIZ1 immunoblot analysis of the immunoprecipitates used for HMT assays of Fig. 7A (Fig. 8B).
FIG. 8.
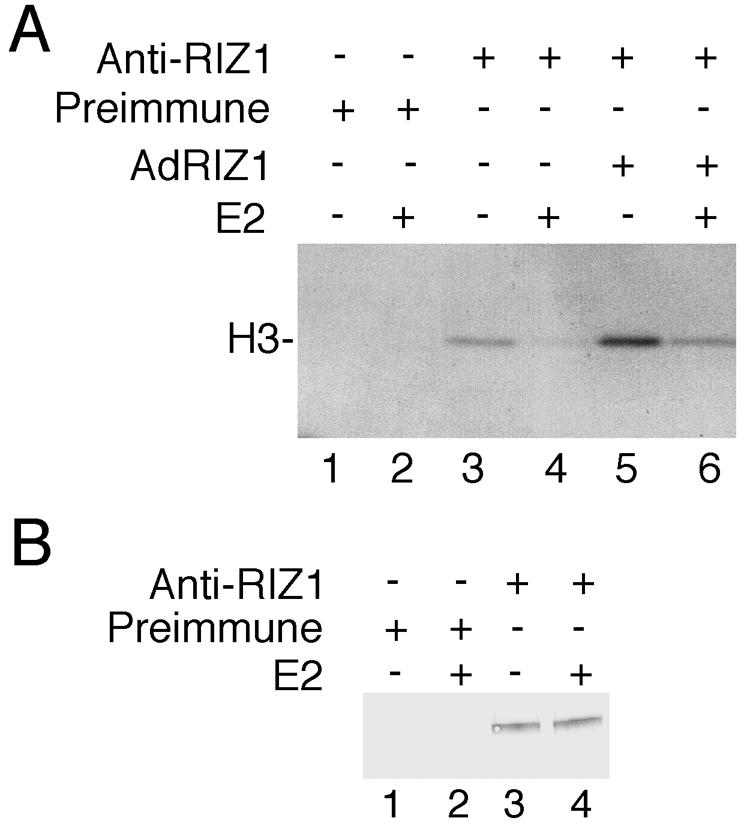
Regulation of RIZ1 methyltransferase activity by estrogen. The same amounts of nuclear extracts (500 μg) from E2-treated or control MCF-7 cells or from AdRIZ1-infected cells were immunoprecipitated with preimmune serum or RIZ1 serum anti-KG7.1S. (A) The immunoprecipitates were then assayed for HMT activity by using free histones as substrates. (B) Also, the immunoprecipitates from AdRIZ1-infected cells were analyzed by immunoblot analysis with RIZ1 monoclonal antibody 2D7. Panel A shows fluorography, indicating methylation of histone H3. Panel B shows that E2 treatment did not alter RIZ1 protein amounts.
DISCUSSION
In this study, we show that female mice lacking RIZ1 display an impaired response to E2 and progesterone and reduced reproductive abilities. RIZ1 has coactivator activity for ER and PR but not for other NHRs examined. The binding of RIZ1 to estrogen target genes and the HMT activity of RIZ1 were regulated by E2, which correlated with reduced H3-K9 methylation of target gene promoters. RIZ1 is also required for maximal induction of pS2 gene in MCF7 cells by estrogen. Furthermore, RIZ1 interacted with the other coactivators SRC-1 and p300. The results suggest a coactivator role of RIZ1 in estrogen action. It is tempting to speculate a model of RIZ1 action, as illustrated in Fig. 9. In the absence of E2, RIZ1 binds directly to estrogen target genes and represses transcription through its H3-K9 methylation activity. Methylation on H3-K9 precludes acetylation and hence gene activation, in spite of RIZ1 binding to other coactivators. In the presence of estrogen, the direct binding of RIZ1 to DNA is disrupted by ER, and the ER-bound RIZ1 no longer binds DNA directly and is recruited to promoter by ER to serve as a coactivator through its ability to interact with other coactivator proteins. The reduction in H3-K9 methylation may be a result of replacement of H3 by the histone variant H3.3 (3). Disruption of direct DNA binding of RIZ1 and inhibition of RIZ1's HMT activity on H3 by E2 may prevent methylation of H3.3. We show that RIZ1 interacts specifically with female sex hormone receptors. This is unlike the SRC-1/p160 family members, which interact with a wider spectrum of nuclear hormone receptors. Therefore, we may expect more subtle phenotypes for RIZ1-deficient mice versus SRC-deficient animals. Indeed, this is the case even when we compare them to mice deficient in SRC-1, which show the mildest phenotypes among animals deficient in an SRC member (12, 49, 51, 52). Although SRC-1- and RIZ1-deficient female animals show similar reduced sensitivities to female sex hormones, the SRC-1 mutation, unlike RIZ1, also affects mammary duct growth during puberty in virgin mice and hormone response in males. The partial hormone resistance phenotypes observed in RIZ1- or SRC-1-deficient mice likely reflect overlapping functions of RIZ1- or SRC-1-related proteins or it may be related to an additive effect of RIZ1 and SRC-1 on ER function.
FIG. 9.

Model of RIZ1 in estrogen action. In the absence of estrogen, RIZ1 sits on estrogen target genes through direct protein-DNA interaction and represses transcription through methylation of H3-K9. In the presence of estrogen, ER binds to RIZ1 and switches it from direct binding to DNA into indirect binding as mediated by ER. The ER-bound RIZ1 has lower H3-K9 methylation activity and cooperates with other coactivators to stimulate transcription.
The partial reduction in tissue response to steroids in RIZ1-deficient mice was accompanied by a corresponding reduction in target gene expression. E2 induction of PR gene expression in the stroma and myometrium compartments of the uterus was reduced in RIZ1-deficient mice (Fig. 3). The result suggests a physiological role of RIZ1 in facilitating ER-transcriptional activation function and that such a coactivator function is likely responsible for the observed tissue response to E2.
Although the overall tissue response to steroids is only partially reduced by RIZ1 deficiency, this does not necessarily indicate that RIZ1 is unable to serve a unique and nonredundant function in facilitating NHR function. It is possible that, at the molecular level, RIZ1 may be uniquely involved in controlling expression of certain target genes. Indeed, estrogen repressed PR gene expression nearly 20-fold in the luminal epithelial cells of the uterus in the presence of RIZ1 but only 2-fold in its absence (Fig. 3). Although ER appears to use RIZ1 as a coactivator, this result suggests that it may also use it as a corepressor in certain circumstances.
Recent progress in understanding chromatin methylation in gene transcription suggests that activation of silenced genes may involve a two-step process. Silenced genes are associated with methylation of H3-K9, which acts to repress transcription (5, 25, 29, 36). The first step in gene activation may involve derepression and removal of H3-K9 methylation or of such methylated H3, leading to a transcription-competent chromatin state. The second step may be the assembly of transcription initiation complex. The first step may be necessary but insufficient for gene activation. The second step may not take place without the first step, since methylation of H3-K9 may preclude activation-associated histone modifications such as acetylation (47). Extensive studies in the past have produced a large body of knowledge on the second step. Little is known about the first step, and our study here suggests that disruption of the DNA binding of a H3-K9 methyltransferase may be necessary for the derepression of silenced genes.
In summary, our study here suggests that an HMT (RIZ1) is a target of estrogen and is required for efficient female sex hormone action in vivo. Altered HMT function may be involved in clinical syndromes characterized by an impaired female sex hormone homeostasis such as cancer, osteoporosis, cardiovascular disease, and Alzheimer's disease.
Acknowledgments
T.C. was supported in part by the Swedish Society of Medical Research, the Swedish Medical Research Council, and the Wennergren Foundation. This study was supported by NIH grants CA76146 (to S.H.) and CA60988 to X.-K.Z. and by grants from the Tobacco Related Disease Research Program (TRDRP-7RT0026 [to S.H.]) and the Cancer Research Program of California (CCRP-1I0023 [to S.H.]).
REFERENCES
- 1.Abbondanza, C., N. Medici, V. Nigro, V. Rossi, L. Gallo, G. Piluso, A. Belsito, A. Roscigno, P. Bontempo, A. A. Puca, A. M. Molinari, B. Moncharmont, G. A. B. Puca, and G. A. Puca. 2000. The retinoblastoma-interacting zinc-finger protein RIZ is a downstream effector of estrogen action. Proc. Natl. Acad. Sci. USA 97:3130-3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agadir, A., G. Lazzaro, Y. Zheng, X. K. Zhang, and R. Mehta. 1999. Resistance of HBL100 human breast epithelial cells to vitamin D action. Carcinogenesis 20:577-582. [DOI] [PubMed] [Google Scholar]
- 3.Ahmad, K., and S. Henikoff. 2002. The histone variant H3.3 marks active chromatin by replication-independent nucleosome assembly. Mol. Cell 9:1191-1200. [DOI] [PubMed] [Google Scholar]
- 4.Anzick, S. L., J. Kononen, R. L. Walker, D. O. Azorsa, M. M. Tanner, X. Y. Guan, G. Sauter, O. P. Kallioniemi, J. M. Trent, and P. S. Meltzer. 1997. AIB1, a steroid receptor coactivator amplified in breast and ovarian cancer. Science 277:965-968. [DOI] [PubMed] [Google Scholar]
- 5.Bannister, A. J., P. Zegerman, J. F. Partridge, E. A. Miska, J. O. Thomas, R. C. Allshire, and T. Kouzarides. 2001. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410:120-124. [DOI] [PubMed] [Google Scholar]
- 6.Buyse, I. M., G. Shao, and S. Huang. 1995. The retinoblastoma protein binds to RIZ, a zinc finger protein that shares an epitope with the adenovirus E1A protein. Proc. Natl. Acad. Sci. USA 92:4467-4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chakravarti, D., V. J. LaMorte, M. C. Nelson, T. Nakajima, I. G. Schulman, H. Juguilon, M. Montminy, and R. M. Evans. 1996. Role of CBP/P300 in nuclear receptor signalling. Nature 383:99-103. [DOI] [PubMed] [Google Scholar]
- 8.Chen, D., H. Ma, H. Hong, S. S. Koh, S. M. Huang, B. T. Schurter, D. W. Aswad, and M. R. Stallcup. 1999. Regulation of transcription by a protein methyltransferase. Science 284:2174-2177. [DOI] [PubMed] [Google Scholar]
- 9.Chen, H., R. J. Lin, R. L. Schiltz, D. Chakravarti, A. Nash, L. Nagy, M. L. Privalsky, Y. Nakatani, and R. M. Evans. 1997. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell 90:569-580. [DOI] [PubMed] [Google Scholar]
- 10.Evans, J. A., U. Wagner, C. M. Santos, and L. Hennighausen. 2000. The interactive web-based histology atlas system. Oncogene 19:989-991. [DOI] [PubMed] [Google Scholar]
- 11.Fondell, J. D., H. Ge, and R. G. Roeder. 1996. Ligand induction of a transcriptionally active thyroid hormone receptor coactivator complex. Proc. Natl. Acad. Sci. USA 93:8329-8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gehin, M., M. Mark, C. Dennefeld, A. Dierich, H. Gronemeyer, and P. Chambon. 2002. The function of TIF2/GRIP1 in mouse reproduction is distinct from those of SRC-1 and p/CIP. Mol. Cell. Biol. 22:5923-5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glass, C. K., and M. G. Rosenfeld. 2000. The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev. 14:121-141. [PubMed] [Google Scholar]
- 14.He, L., J. X. Yu, L. Liu, I. M. Buyse, M.-S. Wang, Q.-C. Yang, A. Nakagawara, G. M. Brodeur, Y. E. Shi, and S. Huang. 1998. RIZ1, but not the alternative RIZ2 product of the same gene, is underexpressed in breast cancer, and forced RIZ1 expression causes G2-M cell cycle arrest and/or apoptosis. Cancer Res. 58:4238-4244. [PubMed] [Google Scholar]
- 15.Hennighausen, L., and G. W. Robinson. 1998. Think globally, act locally: the making of a mouse mammary gland. Genes Dev. 12:449-455. [DOI] [PubMed] [Google Scholar]
- 16.Hong, H., K. Kohli, M. J. Garabedian, and M. R. Stallcup. 1997. GRIP1, a transcriptional coactivator for the AF-2 transactivation domain of steroid, thyroid, retinoid, and vitamin D receptors. Mol. Cell. Biol. 17:2735-2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang, N., E. vom Baur, J. M. Garnier, T. Lerouge, J. L. Vonesch, Y. Lutz, P. Chambon, and R. Losson. 1998. Two distinct nuclear receptor interaction domains in NSD1, a novel SET protein that exhibits characteristics of both corepressors and coactivators. EMBO J. 17:3398-3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang, S. 1994. Blimp-1 is the murine homolog of the human transcriptional repressor PRDI-BF1. Cell 78:9. [DOI] [PubMed] [Google Scholar]
- 19.Huang, S. 2002. Histone methyltransferases, diet nutrients, and tumor suppressors. Nat. Rev. Cancer. 2:469-476. [DOI] [PubMed] [Google Scholar]
- 20.Huang, S., G. Shao, and L. Liu. 1998. The PR domain of the Rb-binding zinc finger protein RIZ1 is a protein binding interface and is related to the SET domain functioning in chromatin-mediated gene expression. J. Biol. Chem. 273:15933-15940. [DOI] [PubMed] [Google Scholar]
- 21.Kamei, Y., L. Xu, T. Heinzel, J. Torchia, R. Kurokawa, B. Gloss, S. C. Lin, R. A. Heyman, D. W. Rose, C. K. Glass, and M. G. Rosenfeld. 1996. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell 85:403-414. [DOI] [PubMed] [Google Scholar]
- 22.Kim, K.-C., L. Geng, and S. Huang. 2003. Inactivation of a histone methyltransferase by mutations in human cancers. Cancer Res. 63:7619-7623. [PubMed] [Google Scholar]
- 23.Kim, K.-C., and S. Huang. 2003. Histone methyltransferases in tumor suppression. Cancer Biol. Ther. 2:491-499. [DOI] [PubMed] [Google Scholar]
- 24.Koh, S. S., D. Chen, Y. H. Lee, and M. R. Stallcup. 2001. Synergistic enhancement of nuclear receptor function by p160 coactivators and two coactivators with protein methyltransferase activities. J. Biol. Chem. 276:1089-1098. [DOI] [PubMed] [Google Scholar]
- 25.Lachner, M., D. O'Carroll, S. Rea, K. Mechtler, and T. Jenuwein. 2001. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410:116-120. [DOI] [PubMed] [Google Scholar]
- 26.Lanz, R. B., N. J. McKenna, S. A. Onate, U. Albrecht, J. Wong, S. Y. Tsai, M. J. Tsai, and B. W. O'Malley. 1999. A steroid receptor coactivator, SRA, functions as an RNA and is present in an SRC-1 complex. Cell 97:17-27. [DOI] [PubMed] [Google Scholar]
- 27.Lee, M. O., Y. Liu, and X. K. Zhang. 1995. A retinoic acid response element that overlaps an estrogen response element mediates multihormonal sensitivity in transcriptional activation of the lactoferrin gene. Mol. Cell. Biol. 15:4194-4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li, H., P. J. Gomes, and J. D. Chen. 1997. RAC3, a steroid/nuclear receptor-associated coactivator that is related to SRC-1 and TIF2. Proc. Natl. Acad. Sci. USA 94:8479-8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Litt, M., M. Simpson, M. Gaszner, C. D. Allis, and G. Felsenfeld. 2001. Correlation between histone lysine methylation and developmental changes at the chicken beta-globan locus. Science 293:2453-2455. [DOI] [PubMed] [Google Scholar]
- 30.Liu, L., G. Shao, G. Steele-Perkins, and S. Huang. 1997. The retinoblastoma interacting zinc finger gene RIZ produces a PR domain lacking product through an internal promoter. J. Biol. Chem. 272:2984-2991. [DOI] [PubMed] [Google Scholar]
- 31.Lubahn, D. B., J. S. Moyer, T. S. Golding, J. F. Couse, K. S. Korach, and O. Smithies. 1993. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc. Natl. Acad. Sci. USA 90:11162-11166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lydon, J. P., F. J. DeMayo, C. R. Funk, S. K. Mani, A. R. Hughes, C. A. Montgomery, Jr., G. Shyamala, O. M. Conneely, and B. W. O'Malley. 1995. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 9:2266-2278. [DOI] [PubMed] [Google Scholar]
- 33.McKay, L. I., and J. A. Cidlowski. 1998. Cross-talk between nuclear factor-κB and the steroid hormone receptors: mechanisms of mutual antagonism. Mol. Endocrinol. 12:45-56. [DOI] [PubMed] [Google Scholar]
- 34.McKenna, N. J., R. B. Lanz, and B. W. O'Malley. 1999. Nuclear receptor coregulators: cellular and molecular biology. Endocrine Rev. 20:321-344. [DOI] [PubMed] [Google Scholar]
- 35.Muraosa, Y., K. Takahashi, M. Yoshizawa, and S. Shibahara. 1996. cDNA cloning of a novel protein containing two zinc-finger domains that may function as a transcription factor for the human heme-oxygenase-1 gene. Eur. J. Biochem. 235:471-479. [DOI] [PubMed] [Google Scholar]
- 36.Noma, K. I., C. D. Allis, and S. I. S. Grewal. 2001. Transistions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 293:1150-1155. [DOI] [PubMed] [Google Scholar]
- 37.Onate, S. A., S. Y. Tsai, M. J. Tsai, and B. W. O'Malley. 1995. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 270:1354-1357. [DOI] [PubMed] [Google Scholar]
- 38.Rachez, C., Z. Suldan, J. Ward, C. P. Chang, D. Burakov, H. Erdjument-Bromage, P. Tempst, and L. P. Freedman. 1998. A novel protein complex that interacts with the vitamin D3 receptor in a ligand-dependent manner and enhances VDR transactivation in a cell-free system. Genes Dev. 12:1787-1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rea, S., F. Elsenhaber, D. O'Carroll, B. Strahl, S. Zu-Wen, S. Manfred, S. Opravil, K. Mechtler, C. Ponting, C. Allis, and T. Jenuwein. 2000. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406:593-599. [DOI] [PubMed] [Google Scholar]
- 40.Shang, Y., X. Hu, J. DiRenzo, M. A. Lazar, and M. Brown. 2000. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 103:843-852. [DOI] [PubMed] [Google Scholar]
- 41.Shapiro, V. S., P. Lee, and A. Winoto. 1995. Identification and cloning of the G3B cDNA encoding a 3′ segment of a protein binding to GATA-3. Gene 163:329-330. [DOI] [PubMed] [Google Scholar]
- 42.Steele-Perkins, G., W. Fang, X. H. Yang, M. Van Gele, T. Carling, J. Gu, I. M. Buyse, J. Fletcher, J. Liu, R. Bronson, R. Chadwick, A. de la Chapelle, X. K. Zhang, F. Speleman, and S. Huang. 2001. Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein-methyltransferase superfamily. Genes Dev. 15:2250-2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strahl, B. D., S. D. Briggs, C. J. Brame, J. A. Caldwell, S. S. Koh, H. Ma, R. G. Cook, J. Shabanowitz, D. F. Hunt, M. R. Stallcup, and C. D. Allis. 2001. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr. Biol. 11:1-5. [DOI] [PubMed] [Google Scholar]
- 44.Takeshita, A., G. R. Cardona, N. Koibuchi, C. S. Suen, and W. W. Chin. 1997. TRAM-1, A novel 160-kDa thyroid hormone receptor activator molecule, exhibits distinct properties from steroid receptor coactivator-1. J. Biol. Chem. 272:27629-27634. [DOI] [PubMed] [Google Scholar]
- 45.Tibbetts, T. A., M. Mendoza-Meneses, B. W. O'Malley, and O. M. Conneely. 1998. Mutual and intercompartmental regulation of estrogen receptor and progesterone receptor expression in the mouse uterus. Biol. Reprod. 59:1143-1152. [DOI] [PubMed] [Google Scholar]
- 46.Voegel, J. J., M. J. Heine, C. Zechel, P. Chambon, and H. Gronemeyer. 1996. TIF2, a 160-kDa transcriptional mediator for the ligand-dependent activation function AF-2 of nuclear receptors. EMBO J. 15:3667-3675. [PMC free article] [PubMed] [Google Scholar]
- 47.Wang, H., R. Cao, L. Xia, H. Erdjument-Bromage, C. Borchers, P. Tempst, and Y. Zhang. 2001. Purification and functional characterization of a histone H3-lysine 4-specific methyltransferase. Mol. Cell 8:1207-1217. [DOI] [PubMed] [Google Scholar]
- 48.Wang, H., Z. Q. Huang, L. Xia, Q. Feng, H. Erdjument-Bromage, B. Strahl, S. Briggs, C. D. Allis, J. Wong, P. Tempst, and Y. Zhang. 2001. Methylation of histone H4 at arginine 3 facilitates transcriptional activation by nuclear hormone receptor. Science 293:853-857. [DOI] [PubMed] [Google Scholar]
- 49.Wang, Z., D. W. Rose, O. Hermanson, F. Liu, T. Herman, W. Wu, D. Szeto, A. Gleiberman, A. Krones, K. Pratt, R. Rosenfeld, C. K. Glass, and M. G. Rosenfeld. 2000. Regulation of somatic growth by the p160 coactivator p/CIP. Proc. Natl. Acad. Sci. USA 97:13549-13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weiss, R. E., J. Xu, G. Ning, J. Pohlenz, B. W. O'Malley, and S. Refetoff. 1999. Mice deficient in the steroid receptor co-activator 1 (SRC-1) are resistant to thyroid hormone. EMBO J. 18:1900-1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu, J., L. Liao, G. Ning, H. Yoshida-Komiya, C. Deng, and B. W. O'Malley. 2000. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proc. Natl. Acad. Sci. USA 97:6379-6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu, J., Y. Qiu, F. J. DeMayo, S. Y. Tsai, M. J. Tsai, and B. W. O'Malley. 1998. Partial hormone resistance in mice with disruption of the steroid receptor coactivator-1 (SRC-1) gene. Science 279:1922-1925. [DOI] [PubMed] [Google Scholar]
- 53.Xu, W., H. Chen, K. Du, H. Asahara, M. Tini, B. M. Emerson, M. Montminy, and R. M. Evans. 2001. A transcriptional switch mediated by cofactor methylation. Science 294:2507-2511. [DOI] [PubMed] [Google Scholar]
- 54.Zhang, X. K., J. M. Dong, and J. F. Chiu. 1991. Regulation of alpha-fetoprotein gene expression by antagonism between AP-1 and the glucocorticoid receptor at their overlapping binding site. J. Biol. Chem. 266:8248-8254. [PubMed] [Google Scholar]
- 55.Zhang, X. K., B. Hoffmann, P. B. Tran, G. Graupner, and M. Pfahl. 1992. Retinoid X receptor is an auxiliary protein for thyroid hormone and retinoic acid receptors. Nature 355:441-446. [DOI] [PubMed] [Google Scholar]