

CONSPECTUS
The [FeFe] hydrogenases (H2ases) catalyze the redox reaction that interconverts protons and H2. This area of biocatalysis has attracted attention because the metal-based chemistry is unusual, and the reactions have practical implications. The active site consists of a [4Fe–4S] cluster bridged to a [Fe2(μ-dithiolate)(CN)2(CO)3]z center (z = 1− and 2−). The dithiolate cofactor is [HN(CH2S)2]2−, called the azadithiolate ([adtH]2−). Although many derivatives of Fe2(SR)2(CO)6–xLx are electrocatalysts for the hydrogen evolution reaction (HER), most operate by slow nonbiomimetic pathways. Biomimetic hydrogenogenesis is thought to involve intermediates, wherein the hydride substrate is adjacent to the amine of the adtH, being bonded to only one Fe center.
Formation of terminal hydride complexes is favored when the diiron carbonyl models contain azadithiolate. Although unstable in the free state, the adt cofactor is stable once it is affixed to the Fe2 center. It can be prepared by alkylation of Fe2(SH)2(CO)6 with formaldehyde in the presence of ammonia (to give adtH derivatives) or amines (to give adtR derivatives). Weak acids protonate Fe2(adtR)(CO)2(PR3)4 to give terminal hydrido (term-H) complexes. In contrast, protonation of the related 1,3-propanedithiolate (pdt2−) complexes Fe2(pdt)(CO)2(PR3)4 requires strong acids. The amine in the azadithiolate is a kinetically fast base, relaying protons to and from the iron, which is a kinetically slow base. The crystal structure of the doubly protonated model [(term-H)Fe2(HadtH)(CO)2(dppv)2]2+ confirms the presence of both ammonium and terminal hydrido centers, which interact through a dihydrogen bond (dppv = cis-C2H2(PPh2)2). DFT calculations indicate that this H---H interaction is sensitive to the counterions and is strengthened upon reduction of the diiron center. For the monoprotonated models, the hydride [(term-H)Fe2(adtH)(CO)2(dppv)2]+ exists in equilibrium with the ammonium tautomer [Fe2(HadtH)(CO)2(dppv)2]+. Both [(term-H)Fe2(HadtH)(CO)2(dppv)2]2+ and [(term-H)Fe2(adtH)(CO)2(dppv)2]+ are highly active electrocatalysts for HER. Catalysis is initiated by reduction of the diferrous center, which induces coupling of the protic ammonium center and the hydride ligand. In contrast, the propanedithiolate [(term-H)Fe2(pdt)(CO)2(dppv)2]+ is a poor electrocatalyst for HER.
Oxidation of H2 has been demonstrated, starting with models for the oxidized state (“Hox”), for example, [Fe2(adtH)-(CO)3(dppv)(PMe3)]+. Featuring a distorted Fe(II)Fe(I) center, this Hox model reacts slowly with high pressures of H2 to give [(μ-H)Fe2(adtH)(CO)3(dppv)(PMe3)]+. Highlighting the role of the proton relay, the propanedithiolate [Fe2(pdt)-(CO)3(dppv)(PMe3)]+ is unreactive toward H2. The Hox-model + H2 reaction is accelerated in the presence of ferrocenium salts, which simulate the role of the attached [4Fe–4S] cluster. The redox-complemented complex [Fe2(adtBn)(CO)3(dppv)-(FcP*)]n+ catalyzes both proton reduction and hydrogen oxidation (FcP* = (C5Me5)Fe(C5Me4CH2PEt2)).
Graphical abstract
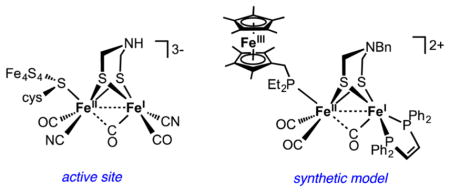
1. INTRODUCTION TO THE [FeFe]-H2ases
The crystallographic characterization1 of the [FeFe]-hydrogenases (H2ases) from Desulfovibrio desulf uricans and Clostridium pasteurianum coincided roughly with a surge of interest in the “hydrogen economy.” These two developments led to intense scrutiny of these unusual enzymes by many scientific communities, including geneticists, biophysicists, organometallic chemists, and computational scientists.2–4 This Account presents an organometallic chemist’s perspective on this area, emphasizing contributions from the author’s laboratory. We have focused on replicating the structure and function of the active site, especially the role of the cofactors and hydride-containing intermediates.
To the eyes of an organometallic chemist, the active site of the [FeFe]-H2ases combines the familiar and unfamiliar. The presence of Fe–Fe bonds as well as CO and CN− ligands are conventional motifs. Unusual are the geometry of the Fe2 site, a 1e-redox active catalytic center, and a cofactor (azadithiolate ligand) that modulates catalysis at Fe. The active site consists of a Fe6 entity called the H-cluster, wherein a [4Fe–4S] cubane is linked via a cysteinyl thiolate to an Fe2(SR)2(CN)2(CO)3 core with a Fe---Fe distance of 2.60 Å. Typical [FeFe]-H2ases, although notably not that from the alga Chlamydomonas reinhardtii, feature several auxiliary Fe–S clusters, whose presence not only underscores the mechanistic importance of electron-transfer but also complicates their study.3 The nature of the dithiolate cofactor that bridges the Fe2 subunit has posed the greatest challenge to the definition of the active site. The dithiolate cofactor was eventually identified by reconstitution experiments using synthetic models developed in our laboratory, as described below.
The [FeFe]-, [FeNi]-, and [Fe]-H2ases all feature Fe-CO-SR centers at their active sites.2 Because no evolutionary relationship exists between these three enzymes, one must conclude that the Fe-SR-CO ensemble is particularly well suited for the activation of H2.3 The reducing and oxidizing equivalents that drive H2ases are supplied by [4Fe–4S] clusters, so the question naturally arises, why do [4Fe–4S]-containing proteins not function as H2ases? The answer is that, compared to high-spin alternatives (cf. [4Fe–4S] clusters), low-spin Fe centers (cf. iron carbonyls) better stabilize metal-hydrides, as is usually required to interconvert H2 and protons.
The [FeFe]-H2ases can be isolated in two active states (Scheme 1), Hox and Hred. With one unpaired electron, Hox is especially well characterized spectroscopically. It features a Lewis acidic Fe(I) center poised to bind H2. Hred is the reduced state with an S = 0 ground state (i.e., all electrons are paired). Available data support two descriptions for Hred: Fe(II)Fe(0) and Fe(I)Fe(I). Protonation of Hred or H2 activation at Hox (concomitant with electron transfer) would give a diferrous hydride Fe(II)Fe(II)-H−, a presumed catalytic intermediate.5 A catalytically relevant super-reduced state has also been identified in the enzyme from C. reinhardtii, featuring a reduced [4Fe–4S]+ cluster and possibly an Fe–H bond.2,6
Scheme 1. Active Site of [FeFe]-H2ase in Two Catalytically Significant Statesa.
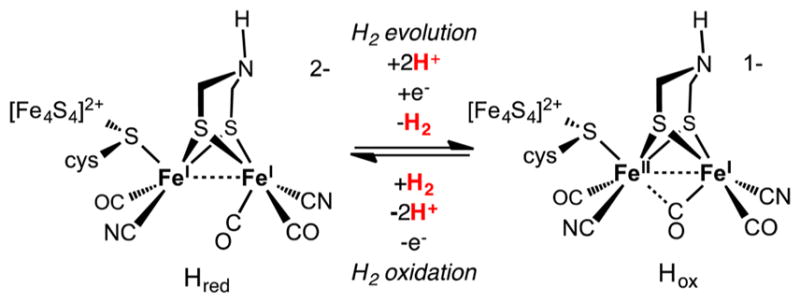
aThe charges of the complexes ignore the [4Fe–4S] site.
2. ACID–BASE REACTIONS OF Fe2(SR)2(CO)6–XLX
The similarity of the two-iron subunit of the [FeFe]-H2ases and classical organoiron complexes7 sparked the development of synthetic models.8 Species like Fe2(SEt)2(CO)6, which have been known for decades, resist protonation9 and undergo redox reluctantly. In contrast, the active site of the [FeFe]-H2ases exhibits both acid–base and redox reactivity. The basicity of classical diiron(I) dithiolate carbonyls can be elevated upon replacement of some CO ligands with better Lewis bases, for example, cyanide, which can be introduced according to eq 1.10
| (1) |
The resulting complex remains electron-precise (i.e., satisfying the 18e rule as does its precursor), but the diiron dicyanide center is far more electron-rich, as indicated by νCO, the intense C–O vibrational bands observed by FT-IR spectroscopy. Thus, (νCO)avg for [Fe2(pdt)(CN)2(CO)4]2− is 1924 vs 2015 cm−1 for Fe2(pdt)(CO)6 (ligand abbreviations in Scheme 2).
Scheme 2.
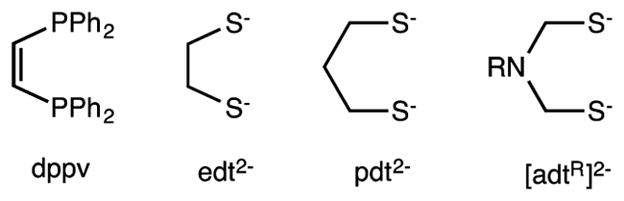
Abbreviations for Some Ligands Used in Synthetic Models
Being more basic, substituted (i.e., electron-rich) diiron(I) dithiolates readily protonate to give hydrido derivatives. The process entails formal oxidation of Fe(I)Fe(I) to Fe(II)Fe(II)-H−, as indicated by an increase in νCO by ~90 cm−1. Spanning the Fe–Fe bond is a bridging hydride, which is labeled μ-H. The complex [(μ-H)Fe2(pdt)(CN)2(CO)4]− has a pKaMeCN of 16, indicating that it is less acidic than a typical pyridinium (pKaMeCN = 12.3) and more acidic than a tertiary ammonium salt (pKaMeCN = 18.8). The Fe2 center of the reduced active site might be expected to have a pKa near that of neutral water, its source of protons. Typical for low-spin ferrous (d6) complexes, the diferrous hydrido complexes are stereochemically more rigid than their [Fe(I)]2 precursors. Thus, three isomers of [(μ-H)Fe2(pdt)(CN)2(CO)4]− are distinguishable by NMR spectroscopy. 11
For mechanistic analysis, most fruitful synthetic models contain organophosphorus ligands, not cyanide. The complexes Fe2(pdt)(CO)6–x(PR3)x form well-behaved hydrides for x ≥ 2. Studies on these phosphine derivatives uncovered the first evidence for terminal hydrides. In these species, the hydride ligand (i.e., substrate) occupies a coordination site cis to both sulfur ligands. Terminal hydrides are of particular interest because they are implicated in the catalytic cycle by biophysical5,12 and computational studies.13 The more prevalent μ-hydrides are also competent electrocatalysts for hydrogen evolution,14 but they probably do not utilize biomimetic mechanisms,7,15 although the matter is debated.16
Models with terminal hydrides can be stabilized by steric congestion, which slows their inevitable isomerization to the μ-hydride.17 The terminal hydrides [(term-H)Fe2(pdt)-(CO)2(dppv)2]+ and [(term-H)Fe2(pdt)(CO)2(PMe3)4]+ have half-lives of minutes at room temperature.18,19 In contrast, [(term-H)Fe2(pdt)(CO)4(dppv)]+ isomerizes in seconds at −80 °C, and [(term-H)Fe2(pdt)(CO)4(PMe3)2]+ has never been detected despite strenuous efforts.20 In Nature, terminal-to-bridge isomerization is prevented by strong hydrogen bonding between the protein and at least one of the FeCN sites.21 Although Fe–H species are likely intermediates in the biological mechanism,5,16 they have not been confirmed in native enzymes.
The protonation of Fe2(pdt)(CO)2(PR3)4 has been studied in depth. Sulfur is implicated as the kinetic site of protonation by strong acids. The resulting SH-containing intermediate rearranges to Fe–H species.22 The recurring pattern is that protonation of metal complexes and clusters occurs first at nonmetal sites, even weakly basic ones, followed by more basic metal centers.23
Studies on Fe2(dithiolate)(CO)2(PMe3)4 ([dithiolate]2− = [edt]2− and [pdt]2−) also provide insights into the formation of μ-hydrides. Although a nuisance for modeling the [FeFe]-H2ase, μ-hydrides are important because they are pervasive in multimetallic clusters. Bridging hydride ligands occur in [NiFe]-H2ases, and perhaps other bioorganometallic centers,24 including the FeMoco cluster in nitrogenase.25 In the diiron dithiolates, a major pathway to μ-hydrides entails intramolecular rearrangement of terminal hydrides (Scheme 3). The isomerization of the terminal to μ-hydride is far faster for the edt derivatives, indicating the influence of steric bulk of the dithiolate on the behavior of the underlying diiron center.26,27 Isomerization of term-H to μ-H follows first order kinetics. H/D exchange is not observed when the isomerization is conducted in the presence of D2O. Computational analysis suggests that the isomerization proceeds via a intermediates with trigonal prismatic geometry at the FeH center.28 This isomerization is probably impossible for the enzyme owing to the strong interaction of the distal FeCN center with the protein.21
Scheme 3.
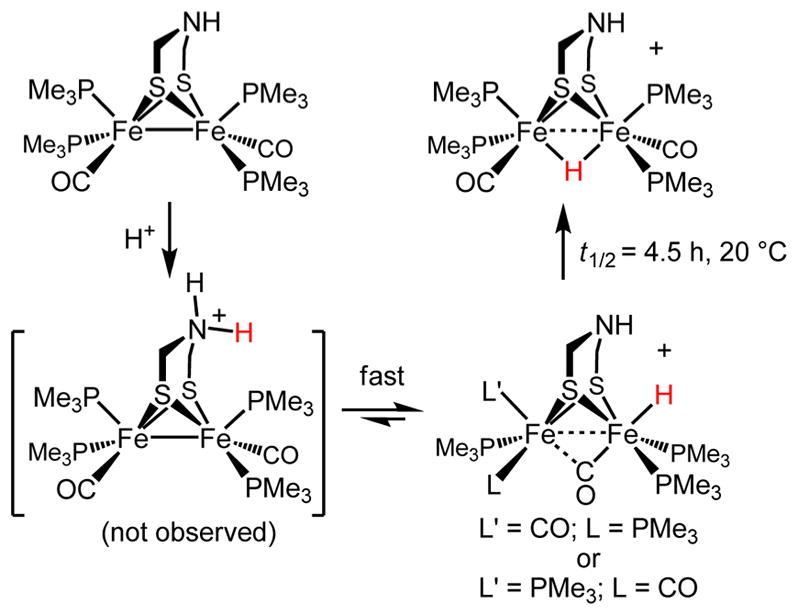
Protonation of Fe2(adtH)(CO)2(PMe3)4 and Isomerization of the Resulting Hydrido Cations
A second pathway to μ-hydrides was detected for Fe2(dithiolate)(CO)2(PMe3)4 ([dithiolate]2− = [edt]2−, [pdt]2−). The main finding is that in concentrated solutions μ-hydrides appear at temperatures too low for intramolecular isomerization. Analysis of the reaction kinetics suggests that the S-protonated species [Fe2(Hdithiolate)(CO)2(PMe3)4]+ intermolecularly protonates the Fe–Fe bond of [Fe2(dithiolate)-(CO)2(PMe3)4].19
3. REDOX REACTIONS OF Fe2(SR)2(CO)6–XLX AND DERIVED HYDRIDES
In addition to exhibiting acid–base reactivity, the diiron subunit of the H-cluster is redox active. The classical diiron dithiolato carbonyls Fe2(SR)2(CO)6 resist oxidation, although they can be reduced to Fe(0)–Fe(I) derivatives. Such reduced diiron complexes exhibit fascinating chemistry,7,15 but they are probably not relevant to the enzyme. Diiron dithiolates with mild and biological plausible redox couples result from replacement of some of the CO ligands in Fe2(SR)2(CO)6 by stronger bases (Figure 1). These substituted species undergo at least quasi-reversible one-electron oxidation, the glaring exception being the cyanide derivatives, which oxidize at mild potentials but always irreversibly.29 It is this irreversibility that forced modeling efforts to refocus on phosphine-based complexes.
Figure 1.

Redox potentials of diiron hydrides (red), Fe(I)Fe(I) complexes (black), and selected reference compounds (blue). Redox couples are at least quasi-reversible, recorded in CH2Cl2 or MeCN solution with quaternary ammonium salts of BF4− or BArF4− as electrolytes.
Protonation at these electron-rich diiron complexes strongly impacts the redox properties. The hydrido complexes exhibit highly negative reduction potentials (Figure 1). For the couple [(term-H)Fe2(pdt)(CO)2(dppv)2]+/0, the site of reduction can avoid the FeH center,30 and thus, this couple is 200 mV milder than the isomeric [(μ-H)Fe2(pdt)(CO)2(dppv)2]+/0.17
4. DIIRON AZADITHIOLATES Fe2(adtR)(CO)6–XLX
Traditionally, Fe2(SR)2(CO)6 compounds arise from thermal reactions of iron(0) carbonyls with thiols, although sometimes disulfides or even thioethers are employed.7 Synthesis of azadithiolato complexes Fe2(adtR)(CO)6, however, require specialized methods (Scheme 4). Free azadithiols (RN-(CH2SH)2) and azadithiolates ([RN(CH2S)2]2−) are unstable, 31 so these azadithiolates must be constructed on the diiron framework.
Scheme 4.
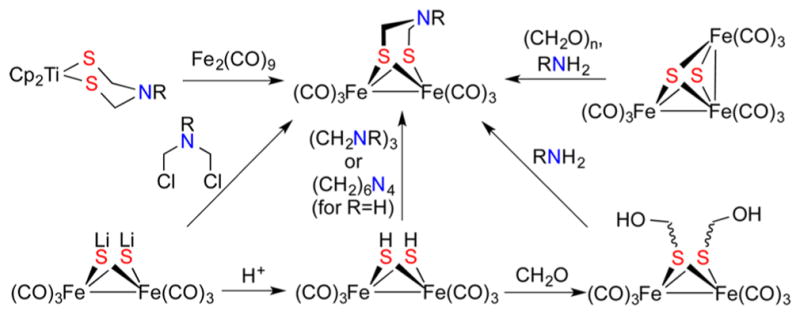
Routes to Diiron Azadithiolates (adtR’s) (Reproduced with permission from ref 31. Copyright 2015 American Chemical Society)
A widely used route entails the four-component reaction of Fe2(SH)2(CO)6 with formaldehyde and an amine. This kind of S-centered reactivity has long been a topic of our research.32 In terms of its mechanism, this reaction proceeds by the condensation of FeSH and CH2O groups to give Fe2(SCH2OH)2(CO)6, which has been crystallized.33 Treatment of Fe2(SCH2OH)2(CO)6 with ammonia and primary amines gives Fe2(adtR)(CO)6. The N-substituted derivatives Fe2(adtR)(CO)6 are also readily prepared by alkylation of Li2Fe2S2(CO)6 with bis(chloromethyl)amines, for example, MeN(CH2Cl)2.34 These N-substituted derivatives also arise from the condensation of Fe2(SH)2(CO)6 and imine trimers (RNCH2)3.
In terms of structure, the two Fe(CO)3 centers in Fe2(adtH)(CO)6 are nonequivalent because of the folded conformation of the adt. An analogous situation applies to the pdt derivatives. The flipping occurs with a barrier of about 10 kcal/mol, although it appears that the adtR derivatives are slightly more rigid than analogous pdt complexes. Two isomers are possible for Fe2(adtH)(CO)6, depending on the orientation of the NH group (Scheme 5). In the axial isomer, the N-substituent projects toward the Fe-center, whereas in the equatorial isomer, the lone pair projects toward the diiron subsite. DFT calculations predict that the equatorial isomer is stabilized by 5.7 kcal/mol by an anomeric interaction between the lone pair orbital on nitrogen and the C–S σ* orbitals. With N-alkyl derivatives, the substituents are typically axial for hexacarbonyls but often equatorial when the diiron center bears bulky ligands in an apical site. The amine is planar when the substituent R can participate in π-bonding (R = aryl groups,35 acyls, alkynyl).
Scheme 5.
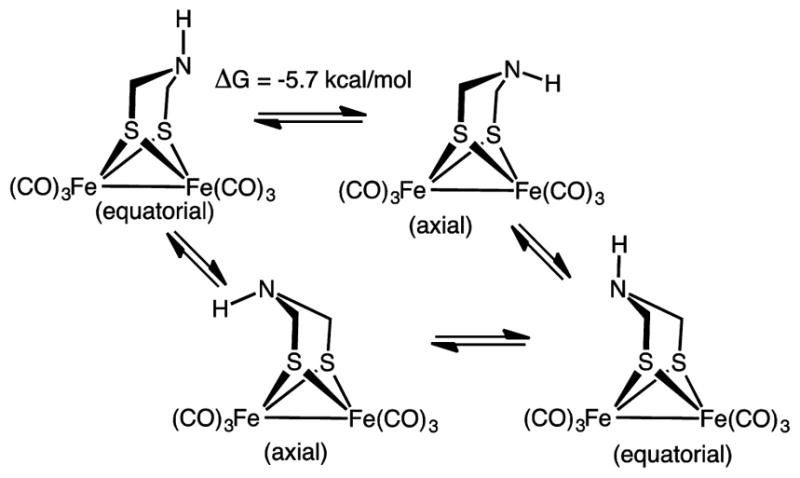
Stereodynamics of Fe2(adtH)(CO)6
Just like other diiron dithiolates, Fe2(adtH)(CO)6 undergoes CO substitution by donor ligands. For example, treatment with cyanide salts gives [Fe2(adtH)(CN)2(CO)4]2−.36 This species can be incorporated in the apo-H2ase from C. reinhardtii to give fully active enzyme. This “artificial maturation” experiment can be extended to many analogous but these derivatives are far less catalytically active.37 These elegant experiments verified that azadithiolate is the dithiolate cofactor in the active site.
The acid–base behavior of the diiron dithiolato complexes is obviously enriched by the presence of the [adtR]2− ligand. The parent Fe2(adtH)(CO)6 undergoes reversible protonation at nitrogen, signaled by a νCO shift of ~15 cm−1 (vs ~65 cm−1 for protonation at Fe).38 The basicity of the amine, pKaMeCN = 8,39 is much greater than that of the Fe centers, pKaMeCN for which is <2.6 (pKaMeCN of CF3SO3H). The relative basicity of the Fe vs amine is strongly affected by substitution of the CO ligands. For Fe2(adtH)(CO)4(PMe3)2, the pKaMeCN of the amine has increased to 10.2, but the Fe–Fe bond is still more basic, having increased by about 12 units. Protonation of complexes of the type Fe2(adtR)(CO)4(PR3)2 first gives the ammonium species, which subsequently converts to the μ-hydride.40 These experiments establish that the amine, not the Fe centers, is the superior kinetic base, reflecting the small reorganizational penalties for their protonation.23 The pKaMeCN value of [(term-H)Fe2(pdt)(CO)2(PR3)4]+ and [(μ-H)Fe2(pdt)(CO)2(PR3)4]+ are estimated respectively at 16 and >19. At equilibrium, the terminal hydride complex is never observed in any model complex.
5. HYDROGEN EVOLUTION CATALYZED BY Fe2(adtR)(CO)2(dppv)2
With respect to catalytic hydrogen evolution, an informative catalyst is Fe2(adtH)(CO)2(dppv)2. This greenish-brown, air-sensitive solid is conveniently soluble in organic solvents. In terms of spectroscopic (IR and variable temperature 31P NMR) and redox properties, this adtH complex and the pdt analogue are very similar as are the corresponding hydrides. Since the electronic structures of the pdt and adtH systems are so similar,41 the thermodynamics for their protonations are the same and differences can be attributed to kinetic factors. Overall, the pdt complex allows for perfect control experiments in studying the role of the second coordination sphere because the first coordination spheres are identical.
The Fe sites in Fe2(adtH)(CO)2(dppv)2 are more basic than the amine. In CH2Cl2 solution, the equilibrium is delicately balanced, as indicated by the finding that the addition of BF4−, a weak hydrogen-bond acceptor, shifts the equilibrium toward the ammonium tautomer (eq 2). With the use of BArF4− counterions (BArF4− = B(3,5-C6H3(CF3)2)4−), this complication is absent, and the hydride is the exclusive species.
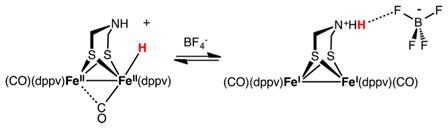 |
(2) |
Treatment of Fe2(adtH)(CO)2(dppv)2 with 2 equiv of HBF4· Et2O gives the doubly protonated species [(term-H)-Fe2(HadtH)(CO)2(dppv)2](BF4)2 wherein one Fe and amine are protonated (Figure 2). Crystals were of sufficient quality that the NH and FeH centers were located and refined. The NH---HFe distance of 1.88(7) Å indicates dihydrogen bonding.42 Careful analysis of this dication by DFT revealed that the experimental H---H distance is affected by the interaction of the N–H centers with two BF4− anions. Upon removal these anions in silico, the H---H separation contracts to 1.40 Å, consistent with a strong dihydrogen bond.43
Figure 2.
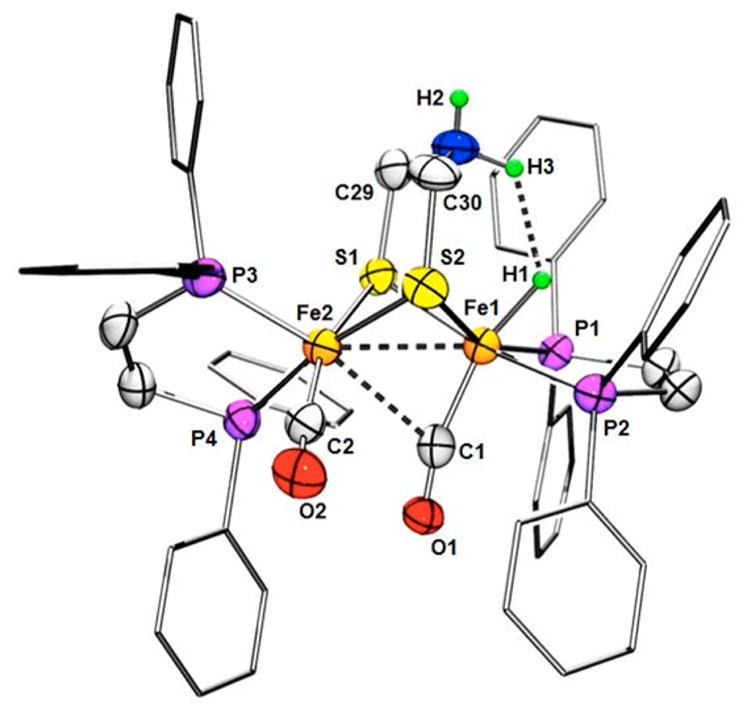
Structure of the dication [(term-H)Fe2(HadtH)-(CO)2(dppv)2]2+.
The ammonium-hydrides [(term-H)Fe2(HadtH)(CO)2L4]2+ (L4 = (dppv)2 and (PMe3)4) do not eliminate H2.19,38 The nonreactivity is understandable: loss of H2 would generate a highly unsaturated dication, which electrochemical measurements confirm are high energy species.44,45 In contrast to the stability of [(term-H)Fe2(HadtH)(CO)2L4]2+, the ammonium hydrides of nickel(II) [HNi(P2N2)(HP2N2)]2+ eliminate H2 spontaneously (P2N2 = diaminodiphosphine).46 Protonation of [(term-H)Fe2(edt)(CO)2(PMe3)4]+ does give H2, but this reaction requires the presence of MeCN, which enables formation of [Fe2(edt)(CO)2(NCMe)(PMe3)4]2+.18
Although the diferrous adt-hydrides are thermally stable, they release H2 upon reduction. Cyclic voltammetry studies established the irreversibility of the couple [(term-H)Fe2(adtH)-(CO)2(dppv)2]+/0. This irreversibility is attributed to hydrogen release, following rapid proton transfer. Computational studies indicate reduction (i) is localized on the proximal (nonhydride-bearing) iron center and (ii) induces coupling of the N-Hδ+ and Fe-Hδ− centers to give an intermediate corresponding to the weakly bound H2 adduct of the Hox state.47 In contrast, the [(μ-H)Fe2(adtH)(CO)2(dppv)2]+/0 and the [(term-H)Fe2(pdt)-(CO)2(dppv)2]+/0 couples are at least quasi-reversible, as are the analogous (PMe3)4 complexes.
The complex [(term-H)Fe2(HadtH)(CO)2s(dppv)2]2+ is a very fast electrocatalyst. In the presence of strong acids such as HBF4·Et2O or trifluoroacetic acid, a catalytic wave appears at −1.22 V associated with a turnover frequency of 58 000 s−1 (Figure 3). In stark contrast, the isomeric bridging hydrido complexes [(μ-H)Fe2(adtH)(CO)2(dppv)2]+, are slow catalysts (~20 s−1) and operate at even higher overpotentials (Table 2).
Figure 3.

Left: Mechanisms for H2 production with strong (solid lines, characterized by DFT calculations) and weak acids (dashed lines). (Reproduced with permission from ref 47. Copyright 2014 American Chemical Society.) Right: Cyclic voltammograms of a 0.5 mM solution of [(term-H)Fe2(adtH)(CO)2(dppv)2]+ (0 °C, 0.125 M [Bu4N]BArF4, CH2Cl2, scan rate =1.0 V/s, glassy carbon working electrode, Pt counter electrode, Ag wire pseudo reference electrode, Fc internal standard) recorded with increasing equivalents of CF3CO2H.
Table 2.
Selected Electrocatalytic Properties of Fe2(adtH)- and Fe2(pdt)-Based Models
| catalyst | acid | k (s−1) | overpotential (V)a |
|---|---|---|---|
| [(term-H)Fe2(adtH)(CO)2(dppv)2]+ | ClCH2CO2H | 5000 | 0.71 |
| [(term-H)Fe2(HadtH)(CO)2(dppv)2]2+ | CF3CO2H | 58 000 | 0.51 |
| [(μ-H)Fe2(adtH)(CO)2(dppv)2]+ | ClCH2CO2H | 20 | 0.90 |
| [(term-H)Fe2(pdt)(CO)2(dppv)2]+ | HBF4·Et2O | 5 | 1.32 |
| [(μ-H)Fe2(pdt)(CO)2(dppv)2]+ | ClCH2CO2H | 3 | 0.95 |
Overpotential = E°HA/H2 – Ecat, where E°HA/H2 is the standard reduction potential of the acid.48
6. MODELS FOR THE HOX STATE: H2 OXIDATION
The Hox state is a well-characterized, almost iconic state of the [FeFe]-H2ases (Scheme 1). With a mixed valence Fe(II)–Fe(I) core, Hox is 1e-oxidized relative to the usual diiron(I) dithiolates. In models and the proteins alike, the Fe(I) center features a vacant coordination site adjacent to the adt cofactor (Figure 4). As the oxidized state, Hox is poised for activation of H2. Models for this state are of the type [Fe2(dithiolate)-(CO)4–xLx]+, where L = PR3 or carbene ligand, and x = 2, 3, 4.49
Figure 4.
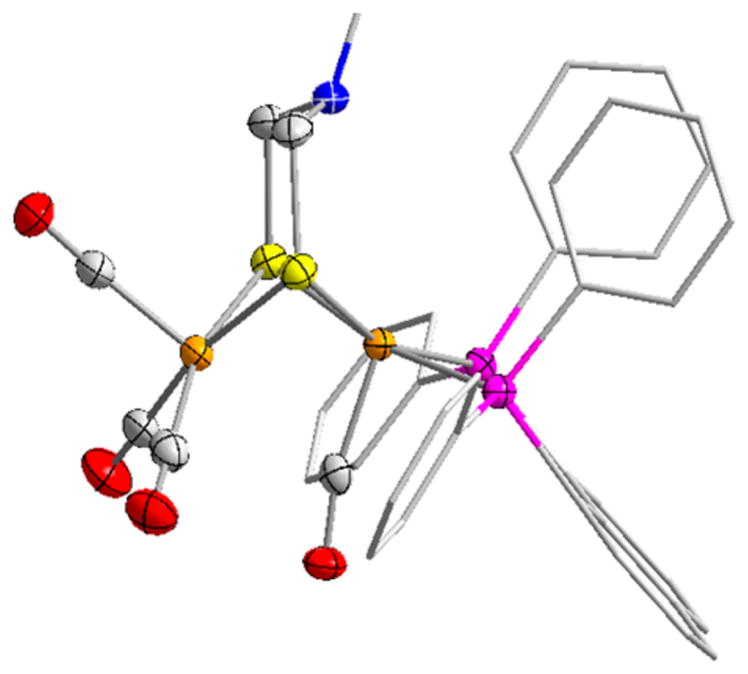
Structure of [Fe2(adtBn)(CO)4(naphthalene-1,8-(PPh2)2)]+, a synthetic model for the diiron subunit in the Hox state (Ph on adt omitted).
Initial experiments on the oxidation of H2 entailed studies on the hydrogenation of [Fe2(dithiolate)(CO)3(dppv)(PMe3)]+, where [dithiolate]2− = [pdt]2− and [adtH]2−. Consistent with their electrophilicity, these cations are labile in solution as their BF4− salts, but the corresponding BArF4− salts give stable CH2Cl2 solutions. The salt [Fe2(adtH)(CO)3(dppv)(PMe3)]-BArF4 reacts with H2, but slowly (18h, 1800 psi, 25 °C). The product is [(μ-H)Fe2(adtH)(CO)3(dppv)(PMe3)]+, as expected since [(term-H)Fe2(adt)(CO)3(dppv)(PMe3)]+, the anticipated kinetic product, isomerizes extremely rapidly near room temperature.26 In contrast to the efficient hydrogenation of the adtH complex, the propanedithiolate [Fe2(pdt)(CO)3(dppv)-(PMe3)]BArF4 was recovered unchanged after prolonged treatment with high pressures of hydrogen. These experiments established that adtR assists in the activation of H2.50
The slowness of the reaction between Hox models and H2 can be rationalized in terms of thermodynamics and electron counting. To activate H2, the catalytic center must compensate for the 76 kcal/mol (in MeCN solution46) required to cleave H2 into H+ and H−. This energetic price is paid in two ways:46 the protonation of the amine of the adt and the coordination of the hydride at Fe. The contribution from deprotonation by the amine is 1.35(pKa), which translates to ~20 kcal/mol. So the major contributor to heterolysis of H2 derives from the affinity of the diiron center for H−, i.e. its hydricity, which must be around 55 kcal/mol. Such hydricity is achievable for ferrous complexes,51 but unlikely for weakly electrophilic Fe(I) centers as exist in simple models for Hox. From the perspective of traditional electron counting, the coordination of H− to the 17e− Fe(I) center is unfavorable. On the other hand, oxidation of such Hox models to the diferrous state induces coordination of the amine to Fe, saturating its coordination sphere and thus ruining any chance of heterolysis.44
The above considerations led to proposition that H2 heterolysis is synchronized with oxidation of the diiron center.52 According to this hypothesis, the distal Fe center “turns on” its electrophilicity only in the presence of substrate H2. The hypothesized coupling of H2 activation and redox is supported by the observation that [Fe2(adtR)(CO)3(dppv)(PMe3)]+ activates 1 atm H2 at room temperature in the presence of decamethylferrocenium (Fc*+), a mild oxidant, and P(o-tolyl)3, a weak non-nucleophilic base. Note that Fc*+ does not oxidize [Fe2(adtR)(CO)3(dppv)(PMe3)]+; instead, it intercepts a diiron hydrido intermediate. The rate of hydrogen oxidation is first order both in H2 and in the Hox model. No reaction occurs when the adtR is replaced by pdt (eqs 3–5).52
slow:
| (3) |
fast:
| (4) |
control:
| (5) |
To achieve catalytic oxidation of H2, the model requires complementation by a redox cofactor. To fulfill this function, the diiron center was fitted with phosphine ligand linked to decamethylferrocene. Called FcP*, the ligand (C5Me5)Fe-(C5Me4CH2PEt2) is a rough functional approximation of an [4Fe–4S] cluster, although about 700 mV less reducing (Figure 1).53 Oxidation of Fe2(adtBn)(CO)3(dppv)(FcP*) occurs first at the FeFe site (ΔνCO ~ 60 cm−1) and second at FcP* (ΔνCO ~ 5 cm−1).54 Incidentally, the dication [Fe2(adtBn)-(CO)3(dppv)(FcP*)]2+ (as well as Hox) is a rare example of species with iron in three different oxidation states (Fe(III), Fe(II), and Fe(I)). In the presence of excess P(o-tolyl)3 and Fc+, the dication catalyzes the oxidation H2 (1 atm), before it deactivates to [(μ-H)Fe2(adtBn)(CO)3(dppv)(FcP*)]+.
Design of catalysts for H2 oxidation was guided by the fact that the H2/H+ couple requires two redox equivalents, only one of which can be provided by the diiron center (the Hox/Hred couple, Scheme 1). In our models, the second redox equivalent is supplied by ferrocenium reagents, whereas in the protein the [4Fe–4S] cluster serves this role. The same principles apply to hydrogen evolution (next section). These results underscore the functional role of the [4Fe–4S] component of the H-cluster. 6
Although H2 oxidation proceeds, the rates are modest, being a few turnovers per hour vs thousands per second for the enzyme. The problem is attributed to the low affinity of the Hox models for H2, which correlates with their low affinity for CO,49 a potent inhibitor of these enzymes. Some mono-Fe complexes are faster catalysts for H2 oxidation, with rates of 2 s−1,43 suggesting that this performance gap can be closed.
7. BIDIRECTIONAL CATALYSIS
While H2ases are expressed for either the production or oxidation of hydrogen, these enzymes characteristically mediate both reactions.3 Thus, far this Account describes complexes that are electrocatalysts for H2 production and others that oxidize H2. It turns out that the H2-oxidizing catalysts are also active for hydrogen evolution, thereby establishing bidirectionality in these models.
Analogous to other Fe2(dithiolate)(CO)3(PR3)3 complexes, Fe2(adtBn)(CO)3(dppv)(FcP*) reacts with one equiv of [H(OEt2)2]BArF4 to give [(μ-H)Fe2(adtBn)(CO)3(dppv)-(FcP*)]+. Curiously, when the acid is added rapidly, H2 is produced (Scheme 6). Furthermore, when excess [H(OEt2)2]-BArF4 is added to a mixture of the model complex and the electron donor Fc*, H2 is produced catalytically according to the stoichiometry 2Fc* + 2H+ → 2Fc*+ + H2. Subsequent to H2 evolution, the main organometallic product is [Fe2(HadtBn)-(CO)3(dppv)(FcP*)]2+. EPR and FT-IR spectroscopic evidence indicates that this dication contains a ferrocenium group and a Fe(I)Fe(I) core. This species is conveniently slow to rearrange to the μ-hydride because its ferrocenium substituent lowers the basicity of the diiron subunit. In the ammonium species [Fe2(HadtBn)(CO)3(dppv)(FcP*)]+, the FcP* group is more reducing than the Fe2 center. In other words, N-protonation induces the electron hole to shift from the FeFe core to FcP* (Figure 5). Electron-transfer is fast and so does not require covalent attachment of the electron donor to the catalytic center. Thus, Fe2(adtBn)(CO)3(dppv)(PMe3) also catalyzes the reduction of acids by Fc* to H2, provided that the acid is added to a preformed solution of the catalyst and the electron donor Fc*. The order of addition is important, lest μ-hydrides form irreversibly.58
Scheme 6.

Competition between μ-Hydride Formation and H2 Evolution in the Protonation of Fe2(adtBn)(CO)3(dppv)(FcP*)58
Figure 5.

Catalytic cycle for H2 oxidation and production by models containing the redox complement (FcP*) and proton relay (adtBn). The central species, a resting state, arises by comproportionation.58
The adtR cofactor in the redox-complemented H2 evolution catalyst does not relay a proton to the Fe(I)Fe(I) site. The individual Fe sites (not the Fe–Fe bond!) in Fe2(adtR)-(CO)3(PR3)3 are less basic than the amine (see Table 1). For this reason, the rate-determining step in H2 evolution is probably protonation of the weakly basic Fe center of [Fe2(HadtR)(CO)3(PR3)3]+, hence the requirement for strong acids. Upon formation of the ammonium diferrous hydride, electron transfer from the FcP* center would induce heterolytic coupling of the N-Hδ+ and Fe-Hδ− centers to give H2. This electron transfer out-competes the isomerization of the term-hydride to μ-hydride, which occurs in seconds even at −90 °C.26
Table 1.
Effect of Coligands on the Relative Basicities of the Three Basic Sites in adt-Containing Diiron Carbonyls. Terminal hydrides are only detected for the (PR3)4 case.
| reaction: H+ + | relative basicities of three sites |
|---|---|
| Fe2(adtH)(CO)6 | N > Fe–Fe > Feterm |
| Fe2(adtH)(CO)4(PR3)2 | Fe–Fe > N > Feterm |
| Fe2(adtH)(CO)3(PR3)3 | Fe–Fe > N > Feterm |
| Fe2(adtH)(CO)2(PR3)4 | Fe–Fe > Feterm ~ N |
| [Fe2(adtH)(CN)2(CO)4]2− | Fe–Fe > Feterm ~ N |
8. SUMMARY AND CLOSING COMMENTS
The catalysts described above utilize bioinspired motifs to mediate the interconversion of H+ and H2. The observed reactivity can be attributed to the combined effect of these factors:
The adt cofactor, which compensates for otherwise slow rates of proton transfer at metals.23
A low-spin, redox-active Fe2(SR)2 core sufficiently basic to form Fe–H species. To enhance the basicity of the diiron center, Nature employs cyanide, whereas synthetic models often rely on phosphine ligands.
A source of redox equivalents to complement the [Fe2(SR)2(CO)6–xLx]+/0 couple. Redox equivalents can be supplied by electrodes or artificial electron donors/acceptors.
Prevention of μ-hydride formation. Model complexes rely on steric congestion to slow isomerization, but Nature exploits the embrace of the protein.21
Further work on FeFe-H2ase modeling would naturally focus on catalysts that operate faster and at lower overpotentials. These goals might be met in part with models featuring preorganized catalytic centers. Indeed, chemists are learning how to control the geometry at the substrate-bearing Fe center55 and to fine-tune proton relays.5,56 Finally, these exquisitely multifunctional complexes57 could in principle be repurposed to effect other reactions, such as hydrogenation and atom-transfer processes.
Acknowledgments
This research was supported by the National Institutes of Health (Grant GM61153). We are indebted to the contributions from computational colleagues in Milan (Luca De Gioia and Giuseppe Zampella), Magdeburg (Matthias Stein), and Urbana (Mioy Huynh and Sharon Hammes-Schiffer).
Biography
Thomas B. Rauchfuss studied at the University of Puget Sound, Washington State University, and the Australian National University, before starting his independent career at the University of Illinois at Urbana–Champaign where he is Larry Faulkner Professor of Chemistry. His research interests are synthetic inorganic and organometallic chemistry.
Footnotes
Published as part of the Accounts of Chemical Research special issue “Synthesis in Biological Inorganic Chemistry”.
Notes
The author declares no competing financial interest.
References
- 1.Fontecilla-Camps JC, Volbeda A, Cavazza C, Nicolet Y. Structure/Function Relationships of [NiFe]- and [FeFe]-Hydrogenases. Chem Rev. 2007;107:4273–4303. doi: 10.1021/cr050195z. [DOI] [PubMed] [Google Scholar]
- 2.Lubitz W, Ogata H, Rüdiger O, Reijerse E. Hydrogenases. Chem Rev. 2014;114:4081–4148. doi: 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- 3.Peters JW, Schut GJ, Boyd ES, Mulder DW, Shepard EM, Broderick JB, King PW, Adams MWW. [FeFe]-[NiFe]-hydrogenase Diversity Mechanism and Maturation. Biochim Biophys Acta Mol Cell Res. 2014;1853:1350–1369. doi: 10.1016/j.bbamcr.2014.11.021. [DOI] [PubMed] [Google Scholar]
- 4.Schollhammer P, Weigand W, editors. Bioinspired Catalysis. Wiley-VCH; Weinheim: 2014. [Google Scholar]
- 5.Mulder DW, Ratzloff MW, Bruschi M, Greco C, Koonce E, Peters JW, King PW. Investigations on the Role of Proton-Coupled Electron Transfer in Hydrogen Activation by [FeFe]-Hydrogenase. J Am Chem Soc. 2014;136:15394–15402. doi: 10.1021/ja508629m. [DOI] [PubMed] [Google Scholar]
- 6.Mulder DW, Ratzloff MW, Shepard EM, Byer AS, Noone SM, Peters JW, Broderick JB, King PW. EPR and FTIR Analysis of the Mechanism of H2 Activation by [FeFe]-Hydrogenase HydA1 from Chlamydomonas reinhardtii. J Am Chem Soc. 2013;135:6921–6929. doi: 10.1021/ja4000257. [DOI] [PubMed] [Google Scholar]
- 7.Tard C, Pickett CJ. Structural and Functional Analogues of the Active Sites of the [Fe]-, [NiFe]-, and [FeFe]-Hydrogenases. Chem Rev. 2009;109:2245–2274. doi: 10.1021/cr800542q. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt M, Contakes SM, Rauchfuss TB. First Generation Analogues of the Binuclear Site in the Fe-Only Hydrogenases: Fe2(μ-SR)2(CO)4(CN)22−. J Am Chem Soc. 1999;121:9736–9737. [Google Scholar]; Le Cloirec A, Davies SC, Evans DJ, Hughes DL, Pickett CJ, Best SP, Borg S. A Di-Iron Dithiolate Possessing Structural Elements of the Carbonyl/Cyanide Sub-Site of the H-Centre of Fe-Only Hydrogenase. Chem Commun. 1999:2285–2286. [Google Scholar]; Lyon EJ, Georgakaki IP, Reibenspies JH, Darensbourg MY. Carbon Monoxide and Cyanide Ligands in a Classical Organometallic Complex Model for Fe-Only Hydrogenase. Angew Chem, Int Ed. 1999;38:3178–3180. [PubMed] [Google Scholar]
- 9.Matthews SL, Heinekey DM. A Carbonyl-Rich Bridging Hydride Complex Relevant to the Fe-Fe Hydrogenase Active Site. Inorg Chem. 2010;49:9746–9748. doi: 10.1021/ic1017328. [DOI] [PubMed] [Google Scholar]
- 10.Mack AE, Rauchfuss TB. (1, 3-Propanedithiolato)-hexacarbonyldiiron and Cyanide Derivatives. Inorg Synth. 2011;35:142–147. [Google Scholar]
- 11.Boyke CA. PhD Thesis. University Illinois; Urbana-Champaign: 2006. [Google Scholar]; Manor BC, Ringenberg MR, Rauchfuss TB. Borane-Protected Cyanides as Surrogates of H-Bonded Cyanides in [FeFe]-Hydrogenase Active Site Models. Inorg Chem. 2014;53:7241–7247. doi: 10.1021/ic500470z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fontecilla-Camps JC, Amara P, Cavazza C, Nicolet Y, Volbeda A. Structure-function relationships of anaerobic gas-processing metalloenzymes. Nature. 2009;460:814–822. doi: 10.1038/nature08299. [DOI] [PubMed] [Google Scholar]
- 13.Bruschi M, Greco C, Kaukonen M, Fantucci P, Ryde U, De Gioia L. Influence of the [2Fe]H Subcluster Environment on the Properties of Key Intermediates in the Catalytic Cycle of [FeFe] Hydrogenases: Hints for the Rational Design of Synthetic Catalysts. Angew Chem, Int Ed. 2009;48:3503–3506. doi: 10.1002/anie.200900494. [DOI] [PubMed] [Google Scholar]
- 14.Tschierlei S, Ott S, Lomoth R. Spectroscopically Characterized Intermediates of Catalytic H2 Formation by [FeFe] Hydrogenase Models. Energy Environ Sci. 2011;4:2340–2352. [Google Scholar]; Felton GAN, Mebi CA, Petro BJ, Vannucci AK, Evans DH, Glass RS, Lichtenberger DL. Review of Electrochemical Studies of Complexes Containing the Fe2S2 Core Characteristic of [FeFe]-Hydrogenases Including Catalysis by These Complexes of the Reduction of Acids to Form Dihydrogen. J Organomet Chem. 2009;694:2681–2699. [Google Scholar]
- 15.Gloaguen F, Rauchfuss TB. Small Molecule Mimics of Hydrogenase: Hydrides and Redox. Chem Soc Rev. 2009;38:100–108. doi: 10.1039/b801796b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chernev P, Lambertz C, Bruenje A, Leidel N, Sigfridsson KGV, Kositzki R, Hsieh CH, Yao S, Schiwon R, Driess M, Limberg C, Happe T, Haumann M. Hydride Binding to the Active Site of [FeFe]-Hydrogenase. Inorg Chem. 2014;53:12164–12177. doi: 10.1021/ic502047q. [DOI] [PubMed] [Google Scholar]
- 17.Barton BE, Rauchfuss TB. Terminal Hydride in [FeFe]-Hydrogenase Model Has Lower Potential for H2 Production Than the Isomeric Bridging Hydride. Inorg Chem. 2008;47:2261–2263. doi: 10.1021/ic800030y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Vlugt JI, Rauchfuss TB, Whaley CM, Wilson SR. Characterization of a Diferrous Terminal Hydride Mechanistically Relevant to the Fe-Only Hydrogenases. J Am Chem Soc. 2005;127:16012–16013. doi: 10.1021/ja055475a. [DOI] [PubMed] [Google Scholar]
- 19.Zaffaroni R, Rauchfuss TB, Gray DL, De Gioia L, Zampella G. Terminal vs Bridging Hydrides of Diiron Dithiolates: Protonation of Fe2(dithiolate)(CO)2(PMe3)4. J Am Chem Soc. 2012;134:19260–19269. doi: 10.1021/ja3094394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu C, Peck JNT, Wright JA, Pickett CJ, Hall MB. Density Functional Calculations on Protonation of the [FeFe]-Hydrogenase Model Complex Fe2(μ-pdt)(CO)4(PMe3)2 and Subsequent Isomerization Pathways. Eur J Inorg Chem. 2011:1080–1093. [Google Scholar]
- 21.Knoerzer P, Silakov A, Foster CE, Armstrong FA, Lubitz W, Happe T. Importance of the Protein Framework for Catalytic Activity of [FeFe]-Hydrogenases. J Biol Chem. 2012;287:1489–1499. doi: 10.1074/jbc.M111.305797. [DOI] [PMC free article] [PubMed] [Google Scholar]; Finkelmann AR, Stiebritz MT, Reiher M. Inaccessibility of the μ-Hydride Species in [FeFe] Hydrogenases. Chem Sci. 2014;5:215–221. [Google Scholar]
- 22.Zaffaroni R, Rauchfuss TB, Fuller A, De Gioia L, Zampella G. Contrasting Protonation Behavior of Diphosphido vs Dithiolato Diiron(I) Carbonyl Complexes. Organometallics. 2013;32:232–238. [Google Scholar]
- 23.Kramarz KW, Norton JR. Slow Proton Transfer Reactions in Organometallic and Bioinorganic Chemistry. Prog Inorg Chem. 1994;42:1–65. [Google Scholar]
- 24.Ogata H, Nishikawa K, Lubitz W. Hydrogens detected by subatomic resolution protein crystallography in a NiFe hydrogenase. Nature. 2015;520:571–574. doi: 10.1038/nature14110. [DOI] [PubMed] [Google Scholar]
- 25.Hoffman BM, Lukoyanov D, Dean DR, Seefeldt LC. Nitrogenase: A Draft Mechanism. Acc Chem Res. 2013;46:587–595. doi: 10.1021/ar300267m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barton BE, Zampella G, Justice AK, De Gioia L, Rauchfuss TB, Wilson SR. Isomerization of the Hydride Complexes [HFe2(SR)2(PR3)x(CO)6–x]+ (x = 2, 3, 4) Relevant to the Active Site Models for the [FeFe]-Hydrogenases. Dalton Trans. 2010;39:3011–3019. doi: 10.1039/b910147k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singleton ML, Bhuvanesh N, Reibenspies JH, Darensbourg MY. Synthetic Support of De Novo Design: Sterically Bulky [FeFe]-Hydrogenase Models. Angew Chem, Int Ed. 2008;47:9492–9495. doi: 10.1002/anie.200803939. [DOI] [PubMed] [Google Scholar]
- 28.Zampella G, Fantucci P, De Gioia L. DFT Characterization of the Reaction Pathways for Terminal- to μ-Hydride Isomerisation in Synthetic Models of the [FeFe]-Hydrogenase Active Site. Chem Commun. 2010:8824–8826. doi: 10.1039/c0cc02821e. [DOI] [PubMed] [Google Scholar]
- 29.Gloaguen F, Lawrence JD, Schmidt M, Wilson SR, Rauchfuss TB. Synthetic and Structural Studies on [Fe2(SR)2(CN)x(CO)6–x]x− as Active Site Models for Fe-Only Hydrogenases. J Am Chem Soc. 2001;123:12518–12527. doi: 10.1021/ja016071v. [DOI] [PubMed] [Google Scholar]
- 30.Wang W, Nilges MJ, Rauchfuss TB, Stein M. Isolation of a Mixed Valence Diiron Hydride: Evidence for a Spectator Hydride in Hydrogen Evolution Catalysis. J Am Chem Soc. 2013;135:3633–3639. doi: 10.1021/ja312458f. [DOI] [PubMed] [Google Scholar]
- 31.Angamuthu R, Chen CS, Cochrane TR, Gray DL, Schilter D, Ulloa OA, Rauchfuss TB. N-Substituted Derivatives of the Azadithiolate Cofactor from the [FeFe]-Hydrogenases: Stability and Complexation. Inorg Chem. 2015;54:000. doi: 10.1021/acs.inorgchem.5b00290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giolando DM, Rauchfuss TB. Alkylidenebis(perthiolates): A New Class of Organosulfur Ligands Prepared from (RC5H4)2TiS5. Organometallics. 2002;3:487–489. [Google Scholar]
- 33.Stanley JL, Rauchfuss TB, Wilson SR. Studies on the Condensation Pathway to and Properties of Diiron Azadithiolate Carbonyls. Organometallics. 2007;26:1907–1911. doi: 10.1021/om0611150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawrence JD, Li H, Rauchfuss TB, Bénard M, Rohmer MM. Diiron Azadithiolates as Models for the Iron-only Hydrogenase Active Site: Synthesis, Structure, and Stereoelectronics. Angew Chem, Int Ed. 2001;40:1768–1771. doi: 10.1002/1521-3773(20010504)40:9<1768::aid-anie17680>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; Lawrence JD, Li H, Rauchfuss TB. Beyond Fe-Only Hydrogenases: N-Functionalized 2-Aza-1,3-dithiolates Fe2[(SCH2)2NR](CO)x (x = 5, 6) Chem Commun. 2001:1482–1483. [Google Scholar]
- 35.Li H, Rauchfuss TB. Iron Carbonyl Sulfides, Formaldehyde, and Amines Condense To Give the Proposed Azadithiolate Cofactor of the Fe-Only Hydrogenases. J Am Chem Soc. 2002;124:726–727. doi: 10.1021/ja016964n. [DOI] [PubMed] [Google Scholar]
- 36.Berggren G, Adamska A, Lambertz C, Simmons TR, Esselborn J, Atta M, Gambarelli S, Mouesca JM, Reijerse E, Lubitz W, Happe T, Artero V, Fontecave M. Biomimetic Assembly and Activation of [FeFe]-Hydrogenases. Nature. 2013;499:66–69. doi: 10.1038/nature12239. [DOI] [PMC free article] [PubMed] [Google Scholar]; Esselborn J, Lambertz C, Adamska-Venkatesh A, Simmons T, Berggren G, Noth J, Siebel J, Hemschemeier A, Artero V, Reijerse E, Fontecave M, Lubitz W, Happe T. Spontaneous Activation of [FeFe]-Hydrogenases by an Inorganic [2Fe] Active Site Mimic. Nat Chem Biol. 2013;9:607–609. doi: 10.1038/nchembio.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adamska-Venkatesh A, Simmons TR, Siebel JF, Artero V, Fontecave M, Reijerse E, Lubitz W. Artificially Maturated [FeFe] Hydrogenase from Chlamydomonas reinhardtii: a HYSCORE and ENDOR Study of a Non-Natural H-Cluster. Phys Chem Chem Phys. 2015;17:5421–5430. doi: 10.1039/c4cp05426a. [DOI] [PubMed] [Google Scholar]
- 38.Carroll ME, Barton BE, Rauchfuss TB, Carroll PJ. Synthetic Models for the Active Site of the [FeFe]-Hydrogenase: Catalytic Proton Reduction and the Structure of the Doubly Protonated Intermediate. J Am Chem Soc. 2012;134:18843–18852. doi: 10.1021/ja309216v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stanley JL, Heiden ZM, Rauchfuss TB, Wilson SR, De Gioia L, Zampella G. Desymmetrized Diiron Azadithiolato Carbonyls: A Step Toward Modeling the Iron-Only Hydrogenases. Organometallics. 2007;27:119–125. doi: 10.1021/om7009599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eilers G, Schwartz L, Stein M, Zampella G, de Gioia L, Ott S, Lomoth R. Ligand versus Metal Protonation of an Iron Hydrogenase Active Site Mimic. Chem—Eur J. 2007;13:7075–7084. doi: 10.1002/chem.200700019. [DOI] [PubMed] [Google Scholar]
- 41.Barton BE, Olsen MT, Rauchfuss TB. Aza- and Oxadithiolates Are Probable Proton Relays in Functional Models for the [FeFe]-Hydrogenases. J Am Chem Soc. 2008;130:16834–16835. doi: 10.1021/ja8057666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Custelcean R, Jackson JE. Dihydrogen Bonding: Structures, Energetics, and Dynamics. Chem Rev. 2001;101:1963–1980. doi: 10.1021/cr000021b. [DOI] [PubMed] [Google Scholar]
- 43.Liu T, DuBois DL, Bullock RM. An Iron Complex with Pendent Amines as a Molecular Electrocatalyst for Oxidation of Hydrogen. Nat Chem. 2013;5:228–233. doi: 10.1038/nchem.1571. [DOI] [PubMed] [Google Scholar]
- 44.Olsen MT, Rauchfuss TB, Wilson SR. Role of the Azadithiolate Cofactor in Models for [FeFe]-Hydrogenase: Novel Structures and Catalytic Implications. J Am Chem Soc. 2010;132:17733–17740. doi: 10.1021/ja103998v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Justice AK, Zampella G, De Gioia L, Rauchfuss TB, van der Vlugt JI, Wilson SR. Chelate Control of Diiron(I) Dithiolates Relevant to the [Fe–Fe]-Hydrogenase Active Site. Inorg Chem. 2007;46:1655–1664. doi: 10.1021/ic0618706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rakowski DuBois M, DuBois DL. The Roles of the First And Second Coordination Spheres in the Design of Molecular Catalysts for H2 Production and Oxidation. Chem Soc Rev. 2009;38:62–72. doi: 10.1039/b801197b. [DOI] [PubMed] [Google Scholar]
- 47.Huynh MT, Wang W, Rauchfuss TB, Hammes-Schiffer S. Computational Investigation of [FeFe]-Hydrogenase Models: Characterization of Singly and Doubly Protonated Intermediates and Mechanistic Insights. Inorg Chem. 2014;53:10301–10311. doi: 10.1021/ic5013523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fourmond V, Jacques PA, Fontecave M, Artero V. H2 Evolution and Molecular Electrocatalysts: Determination of Overpotentials and Effect of Homoconjugation. Inorg Chem. 2010;49:10338–10347. doi: 10.1021/ic101187v. [DOI] [PubMed] [Google Scholar]
- 49.Thomas CM, Liu T, Hall MB, Darensbourg MY. Series of Mixed Valent Fe(II)Fe(I) Complexes That Model the Hox State of [FeFe]Hydrogenase: Redox Properties, Density-Functional Theory Investigation, and Reactivities with Extrinsic CO. Inorg Chem. 2008;47:7009–7024. doi: 10.1021/ic800654a. [DOI] [PubMed] [Google Scholar]; Justice AK, De Gioia L, Nilges MJ, Rauchfuss TB, Wilson SR, Zampella G. Redox and Structural Properties of Mixed-Valence Models for the Active Site of the [FeFe]-Hydrogenase: Progress and Challenges. Inorg Chem. 2008;47:7405–7414. doi: 10.1021/ic8007552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olsen MT, Barton BE, Rauchfuss TB. Hydrogen Activation by Biomimetic Diiron Dithiolates. Inorg Chem. 2009;48:7507–7509. doi: 10.1021/ic900850u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tilset M, Fjeldahl I, Hamon JR, Hamon P, Toupet L, Saillard JY, Costuas K, Haynes A. Theoretical, Thermodynamic, Spectroscopic, and Structural Studies of the Consequences of One-Electron Oxidation on the Fe-X Bonds in 17- and 18-Electron Cp*Fe(dppe)X Complexes (X = F, Cl, Br, I, H, CH3) J Am Chem Soc. 2001;123:9984–10000. doi: 10.1021/ja0106927. [DOI] [PubMed] [Google Scholar]
- 52.Camara JM, Rauchfuss TB. Mild Redox Complementation Enables H2 Activation by [FeFe]-Hydrogenase Models. J Am Chem Soc. 2011;133:8098–8101. doi: 10.1021/ja201731q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tard C, Liu X, Ibrahim SK, Bruschi M, De Gioia L, Davies SC, Yang X, Wang LS, Sawers G, Pickett CJ. Synthesis of the H-Cluster Framework of Iron-only Hydrogenase. Nature. 2005;433:610–614. doi: 10.1038/nature03298. [DOI] [PubMed] [Google Scholar]
- 54.Camara JM, Rauchfuss TB. Combining Acid–Base, Redox and Substrate Binding Functionalities to Give a Complete Model for the [FeFe]-Hydrogenase. Nat Chem. 2012;4:26–30. doi: 10.1038/nchem.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munery S, Capon JF, De Gioia L, Elleouet C, Greco C, Pétillon FY, Schollhammer P, Talarmin J, Zampella G. New FeI–FeI Complex Featuring a Rotated Conformation Related to [2 Fe]H Subsite of the [Fe–Fe] Hydrogenase. Chem - Eur J. 2013;19:15458–15461. doi: 10.1002/chem.201303316. [DOI] [PubMed] [Google Scholar]; Wang W, Rauchfuss TB, Moore CE, Rheingold AL, De Gioia L, Zampella G. Crystallographic Characterization of a Fully Rotated, Basic Diiron Dithiolate: Model for the Hred State? Chem—Eur J. 2013;19:15476–15479. doi: 10.1002/chem.201303351. [DOI] [PubMed] [Google Scholar]
- 56.Darmon JM, Kumar N, Hulley EB, Weiss CJ, Raugei S, Bullock RM, Helm ML. Increasing the Rate of Hydrogen Oxidation Without Increasing the Overpotential: A Bioinspired Iron Molecular Electrocatalyst with an Outer Coordination Sphere Proton Relay. Chem Sci. 2015;6:2737–2745. doi: 10.1039/c5sc00398a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Crabtree RH. Multifunctional Ligands in Transition Metal Catalysis. New J Chem. 2011;35:18–23. [Google Scholar]
- 58.Lansing JC, Camara JM, Gray DE, Rauchfuss TB. Hydrogen Production Catalyzed by Bidirectional, Biomimetic Models of the [FeFe]-Hydrogenase Active Site. Organometallics. 2014;33:5897–5906. doi: 10.1021/om5004013. [DOI] [PMC free article] [PubMed] [Google Scholar]