

Abstract
The photoenolization/Diels–Alder strategy offers straightforward access to synthetically valuable benzannulated carbocyclic products. This historical light‐triggered process has never before succumbed to efforts to develop an enantioselective catalytic approach. Herein, we demonstrate how asymmetric organocatalysis provides simple yet effective catalytic tools to intercept photochemically generated hydroxy‐o‐quinodimethanes with high stereoselectivity. We used a chiral organic catalyst, derived from natural cinchona alkaloids, to activate maleimides toward highly stereoselective Diels–Alder reactions. An unconventional mechanism of stereocontrol is operative, wherein the organocatalyst is actively involved in both the photochemical pathway, by leveraging the formation of the reactive photoenol, and the stereoselectivity‐defining event.
Keywords: asymmetric catalysis, Diels–Alder reactions, organocatalysis, photochemistry, synthetic methods
The light excitation of 2‐alkyl benzophenones 1 to afford transient hydroxy‐o‐quinodimethanes A (Figure 1 a) is a historical photochemical process established in 1961.1 The unique reactivity of the generated photoenols A,2 which can act as dienes in [4+2] cycloaddition reactions with electron‐poor alkenes 2,3 provides a photochemical alternative to classical Diels–Alder chemistry.4 The resulting chiral benzannulated carbocyclic products 3 are important structural motifs found in a variety of marketed drugs5 and naturally occurring substances6 (see Figure S1 in the Supporting Information for examples). It is thus no surprise that the photoenolization/Diels–Alder (PEDA) strategy, in its non‐enantioselective form, has found widespread application in chemical synthesis.3, 6 However, an enantioselective catalytic variant has remained elusive to date.
Figure 1.

Trapping of photochemically generated hydroxy‐o‐quinodimethanes. a) The classical photoenolization/Diels–Alder (PEDA) strategy and challenges associated with implementing a catalytic enantioselective process; EWG=electron‐withdrawing group. b) The photoenolization mechanism of 2‐methylbenzophenone (1 a).
The mechanism of formation of the key photoenol A (Figure 1 b) has been thoroughly investigated and characterized.7 Light absorption by the carbonyl group in 1 generates a singlet excited state S1‐B, which decays to the triplet state T1‐B by intersystem crossing (ISC). Upon 1,5‐hydrogen transfer, which occurs in T1‐B, the diradical intermediate (Z)‐C undergoes rotation to yield the highly reactive enol (E)‐A. Chemical trapping of A by a dienophile 2 provides straightforward access to stereochemically dense benzocyclohexanol derivatives 3.
Relative stereocontrol over the formation of the PEDA products 3 is secured by the inherent stereospecificity of the Diels–Alder process.4 However, dictating the absolute configuration has proven a more difficult challenge. The only effective asymmetric method reported so far required a stoichiometric amount of a chiral complexing agent to selectively bind a purposely designed 2‐alkyl carbonyl compound, properly adorned with a lactam ring that served as a hydrogen‐bond binding site.8 To date, methods that use substoichiometric stereodifferentiating chiral catalysts remain unprecedented.9 Two fundamental issues have historically frustrated the development of enantioselective catalytic variants: I) the fleeting nature of the hydroxy‐o‐quinodimethane A, which complicates a stereoselective trapping event, and II) the difficulty in controlling racemic background reactions, which occur by direct interception of the highly reactive intermediate A by the dienophile without the assistance of the chiral catalyst.
Herein, we report how organocatalysis offers effective tools for the interception of photogenerated hydroxy‐o‐quinodimethanes A with high stereoselectivity. We used a chiral organic catalyst derived from natural cinchona alkaloids to activate maleimides toward stereoselective Diels–Alder reactions to afford stereochemically dense cyclic products. In the developed methods, simple sources of illumination and readily available substrates and catalysts were used, thus avoiding the need for any tailored or purposely designed reactant.
Our initial explorations focused on the PEDA reaction between N‐tert‐butylmaleimide (2 a) and 2‐methylbenzophenone (1 a; Table 1). Because of its high reactivity, this type of process has found application in the light‐triggered “click” conjugation of polymeric building blocks.10 The experiments were conducted in toluene under irradiation by a single black‐light‐emitting diode (black LED, λ max=365 nm). The rate of the background process confirmed the challenge of making the reaction enantioselective: product 3 a was obtained with complete diastereoselectivity and 80 % yield after 15 h in the absence of a catalyst (Table 1, entry 1). As expected, a control experiment revealed that the process was completely inhibited in the dark (Table 1, entry 2).
Table 1.
Exploratory studies on the feasibility of an organocatalytic asymmetric PEDA process.

| Entry | Catalyst | Solvent | Illumination | Yield [%][a] | ee [%] |
|---|---|---|---|---|---|
| 1 | none | toluene | ON | 80 | 0 |
| 2 | none | toluene | OFF | 0 | – |
| 3 | 4 a | toluene | ON | 18 | 55 |
| 4 | 4 b | toluene | ON | 30 | 80 |
| 5 | 4 c | toluene | ON | 35 | 84 |
| 6 | 4 d | toluene | ON | 18 | 68 |
| 7[b] | 4 c | CyH/toluene (3:1) | ON | 76 | 90 |
[a] Yield of isolated 3 a. [b] The reaction was carried out at −5 °C for 24 h. CyH=cyclohexane. 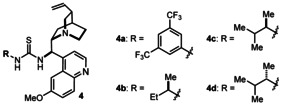
Our approach relied on the use of a chiral organocatalyst that could activate the dienophile 2 a to trap the transient photoenol A stereoselectively. We investigated a variety of chiral catalysts that could use multiple, noncovalent weak attractive interactions to bind 2 a 11 (see the Supporting Information for the full results of an extensive study).
We identified the bifunctional thiourea–amine 4 a, a derivative of natural cinchona alkaloids with well‐known utility in thermal asymmetric processes,12 as a promising catalyst (Table 1, entry 3, 55 % ee). The presence of an additional stereocontrol element on the thiourea moiety was crucial to increasing the enantioselectivity. Derivative 4 c was identified as the catalyst of choice (Table 1, entry 5, 84 % ee). A matched/mismatched combination was observed with catalyst 4 d, which has the opposite configuration at the thiourea stereogenic center (entry 5 vs. entry 6), thus confirming the influence of this chiral moiety on the stereodefining event. A final cycle of optimization with catalyst 4 c revealed that the adduct 3 a could be obtained with excellent results (76 % yield, d.r.>20:1, 90 % ee; Table 1, entry 7) by using a 3:1 mixture of cyclohexane and toluene and stirring the mixture at −5 °C over a 24 h time period.
Aside from providing suitable catalytic conditions for an enantioselective PEDA reaction, the initial studies highlighted that the racemic background process was significantly faster than the stereoselective reaction with the cinchona‐thiourea catalyst 4 c (compare entries 1 and 5 in Table 1; see Figure S3 for more details). Reduction of the rate of the uncatalyzed pathway is crucial to successfully developing any photochemical catalytic asymmetric reaction13 and is generally accomplished through the formation of a chiral catalyst/substrate complex that absorbs light at longer wavelengths or with a higher extinction coefficient than the free substrate. However, optical absorption spectroscopic studies of the individual components of the model reaction and their combination (see Figure S7) confirmed that 2‐methylbenzophenone (1 a) was mainly responsible for the absorption at 365 nm (the operative wavelength in our system), thus excluding the formation of any photoabsorbing substrate/catalyst 4 c aggregation. This puzzling observation prompted us to evaluate other possible pathways available to 4 c for minimizing the background process. We studied the evolution of the formation of product 3 a over time to decipher the effect of the catalyst scaffold on the reactivity of the model reaction (Figure 2 a). Specifically, we investigated the individual behavior of the two main fragments of the organocatalyst, the quinuclidine and the thiourea moieties. A catalytic amount of the achiral thiourea 4 e accelerated the reaction (violet line in Figure 2 a), in consonance with the selective activation of the maleimide 2 a, which facilitates the trapping of the photoenol. In contrast, quinuclidine 4 f greatly inhibited the process (magenta line). The last observation can be reconciled with the established ability of tertiary amines to quench the triplet state of benzophenones,14 the key precursor intermediate in the formation of hydroxy‐o‐quinodimethanes (T1‐B in Figure 1 b). It is also known that geometrically constrained amines, including quinuclidine, use an electron‐transfer quenching mechanism that returns the benzophenone to the ground state along with the unaltered tertiary amine (Figure 2 b), since stereoelectronic effects preclude a hydrogen‐transfer pathway.15 The emerging picture that the quinuclidine core within catalyst 4 c could diminish the formation of the photoenol derived from 1 a was corroborated by laser flash photolysis studies (Figure 2 c), which showed that increasing amounts of 4 c affected both the absorption at 450 nm and the lifetime16 of the transient photoenol generated upon laser excitation of 1 a.17
Figure 2.

Elucidation of the origin of enantiocontrol. a) Evolution of the distribution of product 3 a during the progress of the model reaction in the absence of any catalyst (black line), or in the presence of 20 mol % of cinchona–thiourea 4 c (blue line), N,N′‐dicyclohexylthiourea (4 e; violet line), and quinuclidine (4 f; magenta line). b) Electron‐transfer (ET) quenching mechanism of the triplet state of 1 a, as mediated by geometrically constrained tertiary amines. c) Absorption at 450 nm of the transient E photoenol A (black line) generated upon 355 nm laser excitation of 2‐methylbenzophenone (1 a; [1 a]0=5×10−3 m in benzene). A logarithmic scale is used for time. Absorption decay (red and blue lines) observed in the presence of increasing amounts of the cinchona–thiourea 4 c. Red line: ratio 1 a/4 c mimics the reaction conditions. ΔOD: optical‐density variation. d) Mutualistic relationship between the two chiral fragments in 4 c: The formation of a low amount of the photoenol, as controlled by the tertiary amino moiety, prevents the background reaction to compete with the enantioselective Diels–Alder trap with maleimide 2 a, which is guided by the thiourea moiety. Cy=cyclohexyl.
Although further investigations are needed to better elucidate the mechanism of stereocontrol, these studies indicate that the cinchona–thiourea 4 c plays two opposite yet cooperative roles when promoting the PEDA reaction (Figure 2 d). On the one hand, the quinuclidine moiety interferes with the photoenolization mechanism,16 thus acting as an inhibitor of the PEDA sequence. By a light‐wasting process (Figure 2 b), it lowers the amount of reactive photoenol available, thus decreasing the possibility that the background racemic reaction will take place. On the other hand, 4 c uses the thiourea moiety to act as a chiral catalyst, thus increasing the dienophilic character of the maleimide 2 a upon H‐bonding activation, while channeling the Diels–Alder process toward an enantioselective pathway. Although the two moieties within 4 c exert opposite kinetic effects (Figure 2 a), they are both essential for high stereocontrol.18 In consonance with this mutualistic relationship, when the model reaction was conducted under the same conditions as in entry 5 of Table 1, but in the presence of benzoic acid (20 mol %), it proceeded faster but with greatly lowered enantioselectivity (formation of 3 a in 67 % yield with 4 % ee; see Figure S4). This result was observed because the protonation of the tertiary amine in 4 c disabled the ET‐based inhibition mechanism, thus allowing the background process to compete with the enantioselective pathway.
Adopting the optimized conditions described in Table 1, entry 7, we then demonstrated the generality of the light‐driven method by evaluating a variety of 2‐alkyl benzophenones 1 and maleimides 2. As highlighted in Figure 3, there appears to be significant tolerance for structural and electronic variation of the benzophenone derivative 1 to enable access to a variety of complex tetrahydronaphthalenols 3 a–l, which contain three or four stereogenic centers, with exquisite diastereoselectivity and high enantioselectivity. Different substituents on the enolizable aromatic ring of 1 were well tolerated (products 3 b–d), and a prochiral center at the ortho‐benzylic position of 1 enabled the formation of stereochemically dense products with high fidelity (products 3 e–g). Crystals from compound 3 c and the tetracyclic adduct 3 g were suitable for X‐ray crystal‐structure analysis,19 which established endo selectivity for the Diels–Alder process while securing the absolute configuration of the products. Modifications at the non‐enolizable aryl ring in 1 were also possible: Products 3 h–l with both electron‐withdrawing and electron‐donating groups were obtained in high chemical and optical yields. Also maleimides with different substituents at the nitrogen atom were competent substrates (products 3 m,n). One limitation of the system is that the N‐unprotected maleimide afforded the [4+2] cycloaddition adduct 3 o with poor enantioselectivity (50 % ee).
Figure 3.
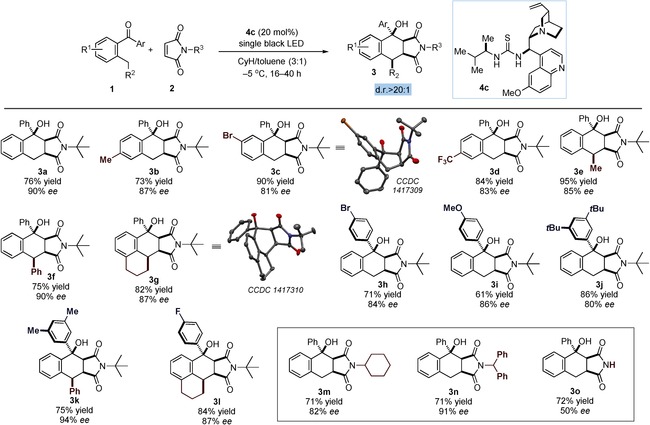
Scope of the Diels–Alder trapping of photochemically generated hydroxy‐o‐quinodimethanes. Reactions were performed on a 0.2 mmol scale; yields and ee values of the isolated products 3 are indicated below each entry. CyH=cyclohexane.
In summary, we have demonstrated that a readily available chiral organic catalyst can activate, under mild conditions, maleimides toward the stereoselective interception of light‐generated hydroxy‐o‐quinodimethanes, thus addressing a longstanding and elusive problem in the realm of photomediated enantioselective catalysis. Our investigations indicate that an unconventional mechanism of stereocontrol is operative, which suggests that this new catalytic blueprint could find application in other enantioselective catalytic photoenol‐trapping processes.
Dedicated to Professor Achille Umani‐Ronchi on the occasion of his 80th birthday
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Financial support was provided by the ICIQ Foundation, MINECO (project CTQ2013‐45938‐P and Severo Ochoa Excellence Accreditation 2014‐2018, SEV‐2013‐0319), the CELLEX Foundation, the AGAUR (2014 SGR 1059), and the European Research Council (ERC 278541—ORGA‐NAUT). L.D. and A.V.‐P. thank the Marie Curie COFUND action (2014‐1‐ICIQ‐IPMP) and the CONACyT (Consejo Nacional de Ciencia y Tecnología, Mexico—Ref. 237346), respectively, for postdoctoral fellowships. We thank Dr. Bryan Matsuura for preliminary investigations and Prof. Emilio Palomares (ICIQ) for assistance with laser flash photolysis experiments.
L. Dell'Amico, A. Vega-Peñaloza, S. Cuadros, P. Melchiorre, Angew. Chem. Int. Ed. 2016, 55, 3313.
References
- 1. Yang N. C., Rivas C., J. Am. Chem. Soc. 1961, 83, 2213–2213. [Google Scholar]
- 2. Sammes P. G., Tetrahedron 1976, 32, 405–422. [Google Scholar]
- 3.“Photoenolization and Its Applications”: Klán P., Wirz J., Gudmundsdottir A. in CRC Handbook of Organic Photochemistry and Photobiology (Ed.: A. Griesbeck), CRC Press, 3rd ed., 2012, chap. 26, pp. 627–651. [Google Scholar]
- 4. Corey E. J., Angew. Chem. Int. Ed. 2002, 41, 1650–1667; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 1724–1741. [Google Scholar]
- 5. Wright P. M., Seiple I. B., Myers A. G., Angew. Chem. Int. Ed. 2014, 53, 8840–8869; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8984–9014. [Google Scholar]
- 6.
- 6a. Nicolaou K. C., Gray D., Tae J., Angew. Chem. Int. Ed. 2001, 40, 3675–3678; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 3787–3790; [Google Scholar]
- 6b. Nicolaou K. C., Gray D., Tae J., Angew. Chem. Int. Ed. 2001, 40, 3679–3683; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 3791–3795; [Google Scholar]
- 6c. Nicolaou K. C., Gray D. L. F., Tae J., J. Am. Chem. Soc. 2004, 126, 613–627; [DOI] [PubMed] [Google Scholar]
- 6d. Charlton J. L., Koh K., J. Org. Chem. 1992, 57, 1514–1516. [Google Scholar]
- 7.
- 7a. Haag R., Wirz J., Wagner P. J., Helv. Chim. Acta 1977, 60, 2595–2607; [Google Scholar]
- 7b. Scaiano J. C., Acc. Chem. Res. 1982, 15, 252–258; [Google Scholar]
- 7c. Wagner P. J., Chen C. P., J. Am. Chem. Soc. 1976, 98, 239–241; [Google Scholar]
- 7d. Das P. K., Scaiano J. C., J. Photochem. 1980, 12, 85–90. [Google Scholar]
- 8. Grosch B., Orlebar C. N., Herdtweck E., Massa W., Bach T., Angew. Chem. Int. Ed. 2003, 42, 3693–3696; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2003, 115, 3822–3824. [Google Scholar]
- 9.Attempts reported in Ref. [6c] to employ a metal-based chiral catalyst proved largely unsuccessful.
- 10. Gruendling T., Oehlenschlaeger K. K., Frick E., Glassner M., Schmid C., Barner-Kowollik C., Macromol. Rapid Commun. 2011, 32, 807–812. [DOI] [PubMed] [Google Scholar]
- 11. Knowles R. R., Jacobsen E. N., Proc. Natl. Acad. Sci. USA 2010, 107, 20678–20685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For a review, see: Connon S. J., Chem. Commun. 2008, 2499–2510. [DOI] [PubMed] [Google Scholar]
- 13.For a review on enantioselective catalytic photochemical processes, see:
- 13a. Brimioulle R., Lenhart D., Maturi M. M., Bach T., Angew. Chem. Int. Ed. 2015, 54, 3872–3890; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3944–3963. For selected examples: [Google Scholar]
- 13b. Brimioulle R., Bach T., Science 2013, 342, 840–843; [DOI] [PubMed] [Google Scholar]
- 13c. Du J., Skubi K. L., Schultz D. M., Yoon T. P., Science 2014, 344, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Cohen S. G., Parola A., Parsons G. H., Chem. Rev. 1973, 73, 141–161; [Google Scholar]
- 14b. von Raumer M., Suppan P., Haselbach E., Chem. Phys. Lett. 1996, 252, 263–266. [Google Scholar]
- 15. Griller D., Howard J. A., Marriott P. R., Scaiano J. C., J. Am. Chem. Soc. 1981, 103, 619–623. [Google Scholar]
- 16.The observation that the lifetime of the photoenol A (half-life: 10 ms) is affected by catalyst 4 c suggests the feasibility of an additional quenching mechanism (e.g. the base-promoted deprotonation of A to reform 1 a). Further photophysical studies are ongoing to better clarify the nature of the interaction between catalyst 4 c and the photoenol and the possible role of this interaction in the stereocontrol of the PEDA reaction.
- 17. Nakayama T., Hamanoue K., Hidaka T., Okamoto M., Teranishi H., J. Photochem. 1984, 24, 71–78. [Google Scholar]
- 18.Although our studies indicate that the mutualistic relationship between the two chiral fragments within the bifunctional organocatalyst 4 c lies at the origin of the stereocontrol, other factors are probably involved. The mechanism of stereoinduction could be more complicated in that 4 c may use multiple weak interactions to simultaneously bind and activate both the transient photoenol A (see Ref. [16]) and the maleimide 2. Investigations are ongoing to experimentally and theoretically evaluate such possibilities.
- 19. CCDC 1417309 (3 c) and 1417310 (3 g) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary