

Abstract
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disease and a worldwide health challenge. Different therapeutic approaches are being developed to reverse or slow the loss of affected neurons. Another plausible therapeutic way that may complement the studies is to increase the survival of existing neurons by mobilizing the existing neural stem/progenitor cells (NSPCs) — i.e. “induce their plasticity” — to regenerate lost neurons despite the existing pathology and unfavorable environment. However, there is controversy about how NSPCs are affected by the unfavorable toxic environment during AD. In this review, we will discuss the use of stem cells in neurodegenerative diseases and in particular how NSPCs affect the AD pathology and how neurodegeneration affects NSPCs. In the end of this review, we will discuss how zebrafish as a useful model organism with extensive regenerative ability in the brain might help to address the molecular programs needed for NSPCs to respond to neurodegeneration by enhanced neurogenesis.
Keywords: Alzheimer’s disease, neural stem/progenitor cell, induced plasticity, Amyloid-beta-42, zebrafish, mouse, human
Introduction
Alzheimer’s disease (AD) is characterized by the chronic loss of neurons and synapses in the cerebral cortex and by a significant loss of brain mass in a progressive manner [1]. AD is the most-common form of dementia [2]. World Alzheimer Report estimates about 46.8 million people worldwide were living with dementia in 2015, and that figure is expected to double over the course of the next two decades. As there is no cure, there is an urgency to better understand the causes of AD in order to carry out prevention strategies. Equally, and maybe more importantly, is to design novel and unconventional therapeutic approaches that not only target the affected neurons, but also the stem cell pool of an adult brain.
Cellular therapies for neurodegenerative diseases are one of the most promising alternatives, along with drug treatments. Cellular replacement implicates the substitution of specific neuronal subtypes lost in disease and successive grafting into affected areas. The newly transplanted cells should incorporate and recapitulate a neural network similar to the healthy brain. Stem cells could provide an environmental support to residing neurons by producing neurotrophic factors and creating additional neural networks in affected areas. Environmental enrichment of stem cells with growth factors, such as glial-derived neurotrophic factor (GDNF), nerve growth factor (NGF), and brain-derived neurotrophic factor (BDNF) would provide a support at the main site of the disease [3-6].
Various types of stem cells, including embryonic stem cells (ESC), mesenchymal stem cells (MSC), induced pluripotent stem cells (iPSCs), and neural stem/progenitor cells (NSPC), have been studied as a cellular therapy in neurodegenerative diseases. Bone marrow-derived MSC transplantation into AD mouse model rescued AD-like pathology via microglial activation [7]. Restorative therapy with ESC-derived neural progenitor cell implantation into AD rat model improved the cognitive function and implanted ESC cells preserved neuronal phenotype. However there are multiple concerns, including immune rejection and/or tumor formation [8,9]. Programming of somatic cells or fibroblasts into iPSCs and iPSC-derived NSCs are another option for stem cell-based therapy for neurodegenerative diseases. iPSCs do not bear immunological complications, but similar to ESCs, have a risk of tumorigenesis in in vivo transplantation. Therefore, to use iPSCs in treatments, safety is an important issue [10]. The development of patient-derived iPSCs gives an exclusive basis to understand the molecular mechanisms in neurodegenerative diseases by providing platforms to perform drug screens, which could otherwise not be possible in vivo [11,12].
One particular way for providing stem cell-based input into the nervous system is to mobilize the endogenous NSPCs. In a healthy brain, the NSPCs are the multipotent stem cells that are capable of proliferation, self-renewal and generation of new neurons, astrocytes, and oligodendrocytes. Enhancing their proliferation rate and differentiation capacity combined with approaches aiming to increase the survival and integration of neurons into circuitry, elevated levels of newly born neurons might provide a regenerative input in a highly unfavorable neurodegenerative environment. Therefore, it is important to understand the behavior of NSPCs during neurodegeneration. In this review, we will elaborate on the current knowledge of how NSPCs are affected by AD and how they affect the AD pathology. In the last section, we will give an outlook on potential uses of model organisms that are capable of regeneration toward understanding the molecular basis of NSPC plasticity and regenerative activity.
The Pathology of AD
AD develops as a result of multiple factors rather than a single cause. Advanced age and certain genetic polymorphisms are the predominant risk factors, yet diabetes, cardiovascular diseases, traumatic brain injury, hypertension, fatty diet, gender, endocrine conditions, oxidative stress, inflammation, stroke, smoking, depression, infection, tumors, vitamin deficiencies, immune and metabolic conditions, and chemical exposure also contribute to the likelihood of developing AD dementia [13-16]. The classical neuropathological hallmarks associated with AD are presence of intracellular and extracellular misfolded protein aggregates: senile plaques and the neurofibrillary tangles (NFTs) [17].
Over the past decades, several studies portrayed the evidence of two competing hypotheses that evolved around AD [18]. The amyloid hypothesis suggests that the depositions of Amyloid precursor protein (APP) cleavage products (39 to 42 amino-acid-long Amyloid β peptides) inside or outside the neuron are the fundamental cause of AD. Amyloid Beta (Aβ) was initially thought to be an abnormal peptide, but studies later showed that it is produced constitutively during normal cell metabolism but the imbalance in amyloidogenic cleavage cascade leads to excessive production of Aβ peptides, which are naturally cleared from the brain by either enzyme degradation [19] or by the process of peptide efflux and influx mechanism [20]. Alternatively, the tau hypothesis states that the hyperphosphorylated tau protein forms the NFTs inside neurons, which in turn acts as the stimulus for the disease progression. Though AD pathogenesis is complicated and elucidating the exact mechanism is difficult, genetic and pathological evidence strongly support the amyloid cascade hypothesis of AD, in which the accumulation of Aβ has an early and critical role to trigger a cascade of events leading to synaptic dysfunction, tau pathology, gliosis, and neuronal loss [21,22].
The major etiology of AD is aggregation of Amyloid protein cleavage products — mainly Aβ42 peptide — either extracellularly or intracellularly [23-30]. Senile plaques, also known as amyloid plaques, are composed of Aβ peptides that exist in extracellular β-pleated sheet conformation in the brain parenchyma [31]. Aβ deposits have also been reported to be found as vascular amyloid in the walls of meningeal and cerebral blood vessels, usually referred to as cerebral amyloid angiopathy [32,33]. Lately, presence of Amyloid deposits inside the neurons have gained much attention as various lines of research suggest that intracellular aggregates of amyloid cleavage products might constitute the early toxicity during the course of neurodegeneration [34].
Post-translational processing of APP occurs in two different pathways: amyloidogenic pathway and non-amyloidogenic pathway [35]. The former pathway features the sequential action of two different enzymes, namely β-secretase (β-site APP-cleaving enzyme, BACE) and γ-secretase, showing a proteolytic action on the APP [26]. BACE cleaves at the N-terminus of the Aβ sequence, releasing a soluble fragment sAPPβ, and another 99 amino-acid-long C-terminal fragment (CTF99) attached to the cellular membrane. The CTF99 fragment is then cleaved by γ-secretase at the C-terminus of the Aβ domain to release the full-length Aβ 40-residue peptide (Aβ40). A small proportion of the longer form of Aβ, a 42-residue peptide Aβ42, is also generated depending on the site of γ–secretase cleavage and is considered to be more cytotoxic [23]. The non-amyloidogenic pathway processes APP with physiological proteolytic cleavage by α-secretase. ADAM10, a disintegrin and metalloprotease component of α-secretase, cleaves APP on the C-terminal side of the Aβ sequence [36-38]. This leads to the destruction of amyloidogenic component, thus preventing the formation of cytotoxic peptides.
Despite the substantial knowledge on the pathological features, the mechanism initiating or leading to the development of AD remains poorly understood. Two forms of AD, namely familial AD (FAD) and sporadic AD (SAD), are known to occur [39]. Early onset FAD shows mutation in three genes: APP, presenilin 1 (PSEN1), and presenilin 2 (PSEN2). The mutations in these genes increase the production of Aβ42 peptide [40]. In the case of the more prevalent late-onset SAD, the main risk factor is the interaction between the genetic susceptibility factors and environment leading to the expression of the ε4 allele of the apolipoprotein E gene (APOE) [41]. The association of early onset FAD with mutations in the APP and γ-secretase components provides a potential tool of generating animal models of the disease. Although various aspects of neuropathophysiology of AD were modeled in various animal models of AD, to date no transgenic animal model fully recapitulated the whole spectrum of the human pathology [42,43].
Lately, preclinical and genetic studies have shown the role and the importance of immune system in AD. Inflammation, various inflammatory cascades, and immune cells seem to contribute to the overall pathology of AD [44]. The link between immune alterations in AD was documented with mutations or deficiencies in microglial or myeloid cell-dependent genes: triggering receptor on myeloid cells 2 (TREM2), myeloid surface antigen CD33, and complement receptor 1 in patients. TREM2 deficiency in AD mice model was shown to enhance the hippocampal Aβ accumulation [44]. CD33 expression was upregulated on microglia in postmortem human AD brains. In contrast to this observation, single-nucleotide polymorphism related with the downregulation in CD33 expression lead to a decrease in Aβ levels [45].
Inflammatory response was mainly generated by central nervous system (CNS)-resident cells, microglia, perivascular myeloid cells, and astrocytes, but also by endothelial cells [46]. It has been showed that receptors, which are expressed by microglia, including CD14, CD36, CD48 and Toll-like receptors (TLRs), can detect soluble Aβ oligomers and Aβ fibrils [47-50]. The binding of Aβ to CD36 or TLR4 results in the production of various inflammatory chemokines and cytokines such as interleukin-1β (IL-1β), IL-6, IL-12, IL-23, and tumor necrosis factor-α, which influence the pathology of AD [51-53]. IL-12 and IL-23 were shown to be increased in the cerebrospinal fluid (CSF) of AD patients [54]. Neutralization studies of these cytokines in AD-like mice models resulted in the reduction of AD-like pathology [55,56]. Regulatory cytokine transforming growth factor β (TGFβ) found to be increased in the plasma, CSF, and brain in AD [57]. Blocking of TGFβ in genetic AD mouse model resulted in reduction in pathology [58].
Mutations in the inflammasome complex NLRP3 or NLRP3-related gene, caspase 1, reduced AD-like pathology in AD mouse model of AD, along with an alteration in microglial phenotype [59]. CD36, the upstream regulator of NLRP3 involved in the inflammation, could enhance the clearance of Aβ in AD, but more in vivo experiments should be conducted to understand the function of CD36 in AD pathology [60]. A deficiency in monocyte-related CC chemokine receptor type 2 leads to Aβ deposition in transgenic mouse model of AD [61]. Another chemokine receptor, CX3CR1, had a positive effect on amyloid deposition, but drastically worsens tau pathology in AD mice [62]. As a potential therapeutic target for AD, roles of IL-12, IL-23, IL-10, and TGFβ cytokines, NLRP3-related molecules (caspase-1, CD36, etc.), and specific chemokine receptors should be more extensively studied. Like myeloid cells, astrocytes are one of the key players in AD pathology. Astrocytes also lead to Aβ plaque-related astrogliosis and possibly contribute to the cognitive impairment in mouse models of AD. The expression of Aβ-degrading enzymes in these cells were upregulated upon the exposure of Aβ peptides ex vivo [63]. Endothelial cells, oligodendrocytes, and neurons have a role in the pathogenesis of AD in neuroinflammatory manner. Complement components that are expressed by oligodendrocytes may contribute to the neuroinflammation by enhanced expression levels in AD brain [64]. Neurons ameliorate the pathology of AD by reducing the expression of anti-inflammatory proteins such as CD59 and CD200 [65,66]. Pro-inflammatory cytokines, IL-1β, IL-6, and CCL2 chemokine were produced by endothelial cells in human AD brains via JNK-AP1 signaling pathway, which is one of the Aβ-induced neuroinflammatory pathways [67]. Since immune-related components are among the main contributors to the pathology of AD, combination therapy of drugs targeting Aβ and/or tau, and modulation of inflammation may be an ultimate way to offset the progression of the disease.
Characteristics of NSPCS
NSPCs are multipotent cells that generate the cell types of the nervous system: neurons, glia, and oligodendrocytes [68]. In vertebrate development, multipotent neuroepithelial cells progressively differentiate into cell types of the nervous system, while sparing undifferentiated cells that maintain glial identity and act as resident stem/progenitor cells of the adult nervous system [69-72]. Stem cell niches in vertebrates show diverse localizations. In adult mammalian brain, neurogenic stem cell niches are restricted to the telencephalon [70], where neural stem cells are found in distinct neurogenic niches: the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone of the dentate gyrus (DG) in the hippocampus (SGZ) [69,73-75]. Recently, the SGZ and NSPCs are multipotent cells that generate the cell types of the nervous system: neurons, glia, and oligodendrocytes [68]. In vertebrate development, multipotent neuroepithelial cells progressively differentiate into cell types of the nervous system, while sparing undifferentiated cells that maintain glial identity and act as resident stem/progenitor cells of the adult nervous system [69-72]. Stem cell niches in vertebrates show diverse localizations. In adult mammalian brain, neurogenic stem cell niches are restricted to the telencephalon [70], where neural stem cells are found in distinct neurogenic niches: the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone of the dentate gyrus (DG) in the hippocampus (SGZ) [69,73-75]. Recently, the SGZ and the striatum were suggested to be stem cell niches in the human brain [76,77]. In non-mammalian vertebrates, the proliferative and germinal zones in the brain are more widespread [78-82]. Thus, there is a fundamental difference in plasticity responses of vertebrate neural stem cells. This is evident in the regenerative capacity of adult vertebrate brains. While lower vertebrates such as teleost fish, frogs, and salamanders can regenerate their CNS [83-89] using an inducible set of molecular programs [90-92], mammalian brains are poorly regenerative [87]. For instance, our aging brains are prone to neurodegeneration, but we are unable to counteract neuronal loss by regenerating lost cells. Patients with neurodegenerative conditions progressively lose neurons yet cannot form new neurons that would replace the lost ones — namely, we humans lack the proper “plasticity response.” Even though the neuropathological outcome in neurons could be hampered, we would still need a neurogenic input from stem cells to replenish the lost neurons. However, as we will elaborate in subsequent sections, most of the neurodegenerative pathology (e.g. Aβ42 deposition) has a negative effect on stem cell proliferation, and even if newborn neurons could be generated, they cease to survive in such an unfavorable environment. Therefore, we might consider neurodegenerative diseases of humans to some degree as “stem cell diseases.” Thus, either providing input through exogenous NSPCs or mobilizing the endogenous stem cells in mammalian brains to proliferate and generate more neurons using “intrinsic” molecular programs of regenerating vertebrates could serve as an alternative (though challenging) therapy option.
NSPC-based therapies for AD
Multipotent NSPCs have been on attention for a considerable time as a cell replacement therapy to prevent the loss of learning and memory function in AD [93-95]. A large portion of the studies using NSPCs undertook transplantation of exogenous mammalian NSPCs. Several studies showed that NSPCs could be used as a potential cell-based treatment in AD mice models. In the mouse model of nucleus basalis of Meynert (NBM) lesion, which manifests as a significant disruption of the working memory, the injection of mouse ESC-derived neural stem cells (NSC) showed improvement in working memory in concordance with the formation of choline acetyltransferase-positive neurons and migration to the cortical cortex [96]. Transplantation of adult mouse NSCs into the hippocampus of the inducible transgenic model with neuronal ablation improved survival, migration, and differentiation of NSCs into neurons, astrocytes, and oligodendrocytes, as well as a significant recovery in memory skills [97].
Transgenic animal models of AD, which recapitulates many of the significant features of the disease, were used to examine the therapeutic effects of mammalian NSPCs (Table 1). Transplantation of postnatal NSPCs to the hippocampi of 3xTg-AD mice recovered behavioral tasks, context-dependent novel object recognition, as well as enhancement in hippocampal synaptic density due to the positive effect of BDNF secreted by NSPCs without changing Aβ or tau pathology [4]. Another mouse model of AD, containing P301S mutation in the Tau gene displays overt Tau pathology, progressive neuronal loss with associated astrogliosis in the cerebral cortex. When fetal NSPCs were injected, astrocytes resulted in improved neuroprotective effect of cortical neurons by the increase in neurotrophins, in particular the GDNF and activity-dependent neuroprotective protein (ADNP). Although the underlying mechanisms are unknown, the differentiation of NSPCs to astrocytes or transplantation of exogenous astrocytes could be the reason for such a neuroprotective effect [98]. Recent studies showed that transplantation of embryonic NSPCs into APP/PS1 double transgenic mice rescued impaired memory and learning ability, along with enhanced long-term potentiation, regeneration of neurons, new synapses, and elevation of neurotrophic factors (NR2B, Trkb/BDNF, SYP and PKCζ), which potentially protect neural function [99]. However, Aβ plaques were not cleared in either of the studies.
Table 1. Effects of various factors on NSPCs and AD pathology.
| Factor | Effect on NSPCs | Effect on AD pathology | Mice model(s) | NSPC type(s) | Reference |
| BDNF, GDNF, ADNP | *Increased NSPC proliferation | *Repair of cognitive impairment | 3x Tg-AD | Fetal | Blurton-Jones et al. 2009, Hampton et al., 2010 |
| *Synaptic remodeling | P301S | ||||
| TLR4 and TLR4-related pathways | *Decrease glial activation | *Deteriorate the course of the disease | Fetal | APP/PS1 | Zhang et al., 2015a |
| Mitochondria-related | *Enhanced differentiation into neurons, astrocytes, oligodendrocytes | *Restoration of spatial learning and memory | APP/PS1 | Fetal | Zhang et al., 2015b |
| VEGF | *Increased NSPC proliferation | *Improvement on cognitive defects | Tg2576 | Feta | Kim et al., 2015 |
| *Reduction of phosphorylated tau levels and Aβ plaques | |||||
| NGF | *Differentiation into functional neurons and astrocytes | *Improvement on learning abilities | Cognitive dysfunction model | Fetal | Lee et al., 2012 |
| Akt/GSK3β pathway | *Differentiation into neuronal and glial cells | *Inhibit tau phosphorylation | NSE/APPsw | Fetal | Lee et al., 2015 |
| IL-1RA | *Microglial proliferation | *Reduction in Aβ plaque formations | Tg2576 | Postnatal | Ben-Menachem-Zidon et al., 2014 |
| *Recovery of cognitive impairment | |||||
| Neprilysin | *Enhancement of synaptic connectivity | *Reduction in Aβ-induced toxicity | 3xTg-AD | Postnatal | Blurton-Jones et al., 2014 |
| *Enhanced NSPC survival |
A potential reason underlying the improvement in learning and memory in APP/PS1 mice at 10 weeks post-transplantation of NSPCs could be the downregulation of inflammation-related pathways TLR4, MyD88, TRIF, p-P38, MAPK, and NFκB because NSPC transplantation also leads to reduced microglial activation [100]. NSC inoculation into AD mice was shown to significantly improve the number of mitochondria and the amount of mitochondria-related proteins (mitochondrial fission factors) [101]. Since a defect in mitochondrial biogenesis is one of the early and prominent features of AD, the level of restoration of the impaired spatial learning and memory could be due to enhanced mitochondrial biogenesis. Fetal NSPCs were also transplanted into the brains of adult Tg2576 mice and showed improvements on cognitive defects, reduction of phosphorylated tau levels, and amyloid plaque levels in the cortex [102]. These outcomes could be due to elevated levels of vascular endothelial growth factor (VEGF) and postsynaptic density protein 95 (PSD-95) at the early stages (12-month-old) of Tg2576 AD model. Similarly, significant improvement in spatial learning and memory were observed in 3xTg-AD mice implanted with NSPCs [103].
NSPC derived from human tissues also showed ameliorating effects on the cognitive decline in various AD mouse models. hNSCs genetically modified to express hNGF differentiated into functional neurons and astrocytes after transplantation helped to improve the learning abilities of a mouse model of cognitive dysfunction [104]. BDNF-producing human CNS-derived NSPCs were injected into hippocampi of 3xTg-AD mice and efficaciously rescued the cognitive defects and upregulated the expression of synaptic and growth-related markers, but did not alter the Aβ or tau pathology [105], similar to other studies [100,101]. Consistent with these reports, promising therapeutic studies were also performed with fetal hNSCs and hNSC cell lines in AD mouse models. When fetal NSCs were introduced into the cerebral lateral ventricles of an APP mouse model that displays Aβ deposits but not plaques, increased levels of neurotrophic factors led to the activation of Akt/GSK3β pathway, which inhibits tau phosphorylation, while implanted cells migrated into the SVZ and differentiated into various types of neuronal and glial cells, improving spatial memory without any adverse effects [106]. Treating Tg2576 mice with neurotrophic drugs combined with injected hNSC cells improved endogenous neurogenesis by the increase in early neurons expressing doublecortin (DCX), inhibit further cognitive impairment and decreased the Aβ levels [107].
Potential NSC-based therapies for AD aim to provide a convenient microenvironment to suppress neurodegeneration and to sustain the survival of mature neurons by supplying neurotrophic factors. For instance, infusions of NGF in aged murine models have been shown to improve cognitive function [108-110]. Phase 1 clinical trials of NGF gene therapy were also performed in AD patients and resulted in an improvement in cognitive behavior and activation of neuronal responses with no adverse effects [5,111]. Delivery of Aβ-degrading enzyme endopeptidase Neprilysin (NEP) or NEP-derived Neuropeptide Y into APP transgenic resulted in neuroprotective activity and led to a reduction of Aβ deposition and inflammation [112,113]. BDNF gene delivery into mice, rat, and nonhuman primate models of AD resulted in reversing synapse loss, cognitive decline, and neuronal atrophy by normalizing the cell survival pathways [114]. Intraventricular BDNF infusion into APP/PS1 mice showed decreased Aβ peptide and enhanced N-acetylaspartate, which is the precursor of the most abundant neuropeptide N-acetylaspartylglutamate in mouse brain [115]. Infusion of bone morphogenic protein 9 (BMP9) or Insulin-like growth factor 2 (IGF2) in APP/PS1 mice models resulted in reduced amyloid plaques and the enhancement of neurotrophic factors such as NGF and BDNF [116,117]. Injection of an adeno-associated virus into the hippocampal region of APP and APP/PS1 mice to express anti-inflammatory glycoprotein CD200 restored the number of differentiated neurons in DG, improved neurogenesis in the SGZ area, and reduced the neuroinflammation and soluble Aβ42 levels [118].
Inflammasome complex NLRP3 is involved in the immunomodulation of AD pathogenesis. NLRP3 regulates the activity of caspase-1, which is involved in the cleavage of proinflammatory Type-1 cytokines such as IL-1 and IL-18. Elevated levels of active caspase-1 and IL-1 have been detected in AD patients [59,119]. IL-1 can affect AD pathology in both detrimental and beneficial ways, such as by stimulating the expression of APP [120,121] or by leading to a reduction in amyloid pathology by microglia-dependent plaque degradation [122-124]. In a study where Neural Progenitor Cells (NPCs) from IL-1 receptor antagonist transgenic mice were transplanted into Tg2576 mouse model, there was a significant increase in hippocampal cells producing BDNF, microglial proliferation, and alleviation of cognitive decline even one month after the transplantation [125]. This study is the first example for the use of genetically manipulated NSPCs in the treatment of AD, and also shows that usage of anti-inflammatory agents can improve the beneficial effects of NSPCs.
Proteolytic enzyme NEP is one of the most potent Aβ degradation enzyme, shown to be found in low levels in AD brains [126,127]. In order to deliver NEP, murine NSCs that overexpress secreted NEP (sNEP) were generated. Modified NSCs were implanted into 3xTg-AD mice and sNEP expressing NSCs drastically decreased Aβ-induced toxicity, enhanced synaptic connectivity, NSCs survived, and continually produced sNEP after the transplantation [128]. Combining NSC implantation with systemic treatment of Cerebrolysin™ (CBL) — a peptide mixture having neurotrophic-like properties — into hAPP transgenic mice showing defects in neurogenesis, high levels of Aβ production, and behavioral deficits significantly improved NSC survival and increased BDNF levels. However, the exact underlying mechanism for this improvement is not clear [129].
Despite the promising results, some limitations need to be clarified before hNSCs and hNSC cell lines can be used for AD treatment primarily because of the immune rejection [130]. Generation of iPS-derived NSPCs from patients is one possible way to overcome graft rejection. Generation of NSPCs from mouse fibroblasts has been reported using stromal feeder co-culture, lentiviral, or retroviral transduction with neural lineage-specific transcription factors Sox2, Klf4, Myc, Pou3f4, and E47/Tcf3; treatment with retinoic acid, or culturing with the mitogens fibroblast growth factor 2 (FGF2) and EGF2. These methods are able to convert stem cells into all three lineages of the CNS in vitro: neurons, astrocytes, and oligodendrocytes [131-136]. Studies showed restricted graft survival but improved functional recovery following implantation of mouse and/or human-derived iNSCs into an animal model of spinal cord injury without tumor formation, revealing the therapeutic potential of this approach in neurodegenerative diseases [137,138]. Although iPS-derived NSPC transplantations generate a quick response on neurodegenerative disease models, they are invasive methods and the long-term effects are still unknown [139]. Therefore, NSPCs can enable major functional improvements in AD animal models both endogenously and exogenously, nevertheless the exact mechanism remains tentative.
Effect of Amyloid Deposition on Endogenous NSPCs
In the past, many studies conducted on various model organisms of AD suggested that the disease modulates the neurogenesis. However, the results still remain contradictory. A considerable portion of the published literature suggests that AD and amyloid deposition has a negative effect on stem cells [99-104], while opposing findings do exist [140-143] (Figure 1).
Figure 1: Effect of Alzheimer’s disease on NSPCs.
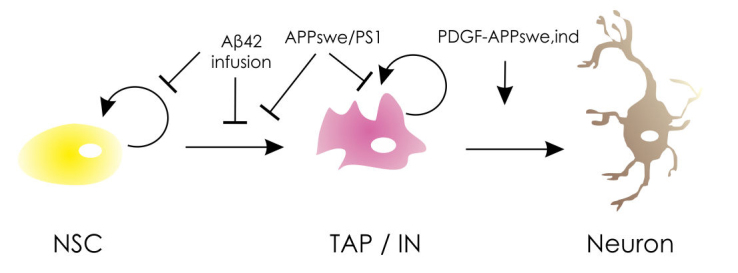
A simplified sketch showing the effects of Aβ deposition in mouse brain on stem cell proliferation, transient amplifying progenitor proliferation, neuronal differentiation and maturation. Aβ42 through infusion or transgenic APPswe/PS1 suppressed NSPC proliferation, while PDGF-driven APPswe,Ind increases the differentiation of progenitors to neurons. NSC: neural stem cell; TAP: transiently amplifying cells, IN: immature neuron. See text for details.
According to a group of studies, the proliferation of NSPCs and neurogenesis in general are enhanced in the presence of Aβ peptide. One key study showed increased neurogenesis in the hippocampus of postmortem brains of AD patients. The markers for early neurons (TUC-4 and DCX) were overexpressed in the SGZ as well as the Grandular Zone (GZ) of the hippocampus in AD patients compared to healthy controls [140], suggesting enhanced neurogenesis. Further studies on transgenic platelet-derived growth factor (PDGF)-APPsw,Ind mouse model of AD supported the previous findings (Figure 1). They were able to show a significantly increased number of BrdU-immunopositive cells in the SGZ of the DG of 3-month-old, as well as year-old transgenic mice. The same hypothesis stood true also for the number of proliferating stem cells in the SVZ of the year-old, but not 3-month-old mice. The different rate of neurogenesis at the two neurogenic zones of murine brain could be explained by earlier pathology in hippocampus compared to SVZ [141]. Similar results of enhanced neurogenesis were obtained from in vitro studies with cultured NSCs from striatum and hippocampus of rat and mouse [142]. The cells were treated with different concentrations of Aβ peptide at different time points. An average of a threefold increase of the number of neurons was observed. However, the increase was noticed to be time-point and dose-dependent. Moreover, the rate of proliferating NSCs in culture was unchanged, thus suggesting that the effect of Aβ is on neurogenic precursors rather than NSCs [144]. These results suggest that Aβ deposition might force the NSPCs to differentiate rather than proliferate, which in the end would deplete the stem cell pool.
An opposing view favors detrimental effects of Aβ peptides on NSPC plasticity. Impaired neurogenesis in the DG and significant reduction in proliferation, survival, and migration of NSPCs in SVZ of adult transgenic mice for mutant APP or in mice infused with Aβ25-35 or Aβ42 in the lateral ventricles were shown [145,146] (Figure 1). The results were consistent with the negative effects of amyloid deposition in NSPCs neurosphere cultures of human embryonic cerebral cortex, possibly due to dysregulated cellular calcium homeostasis [100,109]. Studies on 8- and 9-month-old double transgenic mouse model for APPSwe/PS-1 also suggest defective neurogenesis [147] (Figure 1). However, they did not observe any significant reduction of either NSPCs (MCM2-positive) or neuroblasts (DCX-positive) in an APPSwe knock-in line alone. The reduction of both cell types was slightly higher for PS-1 knock-in and much higher for the double mutant mouse models. This can be explained by the fact that presenilins are expressed in NSPCs and are crucial during both developmental and adult neurogenesis [148-151]. The adverse effect of amyloid deposition was considered long-lasting and persistent up to age 18 months in mice [147]. Interestingly, one study reports a different observation: In this study, the mutant APP is overexpressed exclusively in the mature neurons, thus ensuring the release of Aβ by mature granule neurons into the neurogenic niche. Surprisingly, neither a positive nor a negative effect was seen in terms of adult hippocampal neurogenesis [152]. However, the comparison with previous mouse models is not valid since in former studies, the APP overexpression was driven under NSPCs’ specific promoters. This may indicate that the effects of Aβ deposition on stem cell proliferation might not be due to neuronal Aβ accumulation, but at the stem cell level, possibly at an earlier stage of the disease. Alternatively, AD neuropathology in mature neurons could follow distinct molecular programs and etiology compared to the effects of Aβ42 on neurogenic potential. However, not much is known about the effect of amyloid deposition prior to the onset of the disease phenotype. One study reports hampered neurogenesis in a 2-month-old transgenic mouse co-expressing a chimeric mouse-human APPswe and mutant human PS1dE9. These animals were shown to express decreased numbers of proliferating NSPCs in the SVZ, as well as the hippocampus together with a lower number of newborn neurons [153]. This observation might indicate that the decreased neurogenesis itself contributes to the progression of AD. In addition to the neuronal loss, production of neurons into the circuitry by reduced NSPC proliferation could also be an effect on cognitive decline and other pathologies of AD.
In addition to NSPCs and neurons, another cell type that is affected by Aβ pathology is parenchymal astrocytes. These are the cells distributed in the parenchymal mass, and have been shown to bear neurogenic potential under certain circumstances in vitro [154-158]. Additionally, in vivo, these cells can be converted to become neurogenic in various injury and disease conditions [157,159-162]. Therefore, as discussed in the previous sections, these cells can also be potential targets of therapies to bring back neurons and relevant studies are awaited.
Zebrafish as a Model Organism to Study Neurodegeneration
In the last two decades, different animal models of AD have been generated with an aim to dissect the pathology, dynamics, and molecular mechanisms of the disease [42,43]. More recently, the iPSC technology complemented the existing vertebrate models and provided the community with an excellent in vitro model to test the key questions directly on AD patient-derived cultures. However, in order to elucidate the regenerative capacities of CNS, such model organisms as zebrafish, salamander, or frog came into play [87]. While anuran amphibians (e.g. Xenopus) lose their ability to regenerate the CNS after the larval stage and the urodele amphibians (salamanders) regenerate only some parts of the brain [87-89], zebrafish keeps this widespread neurogenic and regenerative ability to replenish the lost neurons in the CNS throughout adult life [81,82,163-169]. Moreover, the extensive use of zebrafish for various studies led to a deep understanding of zebrafish genetics and a development of reverse genetic tools.
With the advance of such genome editing tools as TALENs, Zinc-finger nucleases, and CRISPR/Cas9, it became plausible to model a wide spectrum of neurodegenerative diseases in zebrafish. Recently, several studies aimed to understand the molecular pathways underlying the regenerative response in adult zebrafish brain suggest that zebrafish might use “induced molecular programs” to endow its NSPCs with a regenerative ability [163,164,170-173]. Attempts to model neurodegeneration in zebrafish have established tools to examine the pathology [169,174-185]. Nevertheless, most of these studies are performed in embryonic or larval stages of early development or ceased to generate a progressing neurodegeneration model that could be assessed in adult stages. Thus, the programs in zebrafish brain might underlie the disparity between the neurogenic abilities of NSPCs and, in turn, the regenerative capacities of zebrafish brains and mammalian brains. This property of zebrafish brain offers enormous opportunities to understand how vertebrates could efficiently form neurons after neuronal loss (e.g. neurodegeneration), and what we might learn from fish could be applied to humans for imposing a regenerative capacity to mammalian NSPCs, which would be useful for designing regenerative therapies. Thus, the modulation of adult zebrafish NSPCs to produce more neurons to compensate the damaged neuronal cells by AD-like mechanisms might open up new avenues in regenerative medicine. Also, high-throughput drug screening opportunities [186] make zebrafish an excellent model to study the neurodegenerative mechanisms as well as the regenerative potential for future therapeutic purposes in AD patients.
This goal definitely demands currently nonexistent tools for efficient analysis of gene function in adult zebrafish brain. However, although zebrafish brain has a highly conserved phylogenetic similarity to humans in terms of development, neuronal types, and brain structure [187,188], it does not reflect the exact same physiological and neurochemical complexity of the human brain (just as rodent brains do not). Thus, there is a definite need to combine all possible lines of experimental approaches and findings (in zebrafish, mouse, iPSCs, and human) to reach a consolidated molecular understanding of stem cell plasticity upon neurodegenerative conditions. These molecular programs will be extremely useful and informative because they could be the direct clinical targets to turn on in human brains to treat many neurodegenerative diseases, including AD.
Conclusion
Neurodegenerative diseases are complex disorders where various cell types are involved in the overall pathology. Regeneration in such diseases, the causes of which are not fully elucidated, may seem a far dream; however, findings in model organisms may herald a promise for advancement toward cellular therapies. The field requires novel approaches and new model organisms to tackle the hurdles of reverting neuronal death, preventing synaptic degeneration, ameliorating cognitive decline, and inducing the plasticity of neural stem/progenitor cells.
Abbreviations
- AD
Alzheimer’s disease
- ADAM10
A disintegrin and metalloproteinase domain-containing protein 10
- ADNP
activity-dependent neuroprotective protein
- APOE
Apolipoprotein E
- APP
Amyloid precursor protein
- Aβ
Amyloid beta
- AB40
amyloid beta 40
- BACE
beta-site APP cleaving enzyme
- BDNF
Brain-a derived neurotrophic factor
- BMP
Bone morphogenic protein
- CBL
Cerebrolysin
- CNS
central nervous system
- CSF
cerebrospinal fluid
- CTF
C-terminal fragment of amyloid precursor protein
- DCX
doublecortin
- DG
Dentate gyrus of the hippocamps
- ESC
embryonic stem cells
- FAD
familial Alzheimer’s disease
- GDNF
glial cell-derived neurotrophic factor
- GZ
granular zone
- IGF2
insulin growth factor 2
- IL
Interleukin
- IL-1RA
Interleukin-1 receptor antagonist
- iPSC
induced Pluripotent stem cells
- MSC
mesenchymal stem cells
- NEP
Neprilysin
- NFT
neurofibrillary tangle
- NGF
neural growth factor
- NLRP3:
NACHT, LRR, and PYD domains-containing protein 3
- NPC
neural progenitor cell
- NSPC
neural stem/progenitor cell
- PDGF
Platelet-derived growth factor
- PS-1 and PS-2
Presenilin 1 and 2, respectively
- SAD
sporadix Alzheimer’s Diease
- SGZ
subgranular zone
- sNEP
soluble neprilysin
- SVZ
subventricular zone of the lateral ventricle
- TGFBN
transforming growth factor beta N
- NSC
nueral stem cell
- TLR
toll-like receptor
- TREM
triggering receptor expressed on myeloid cells
- VEGF
Vascular endothelial growth factor
Author Contributions
Gizem Tincer and Violeta Mashkaryan contributed equally.
References
- Wenk GL. Neuropathologic changes in Alzheimer’s disease: potential targets for treatment. J Clin Psychol. 2006;67 Suppl 3:3–7. quiz 23. [PubMed] [Google Scholar]
- Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007;3(3):186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- Behrstock S, Ebert AD, Klein S, Schmitt M, Moore JM, Svendsen CN. Lesion-induced increase in survival and migration of human neural progenitor cells releasing GDNF. Cell Transplant. 2008;17(7):753–762. doi: 10.3727/096368908786516819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Müller F-J, Loring JF. et al. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A. 2009;106(32):13594–13599. doi: 10.1073/pnas.0901402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuszynski MH, Thal L, Pay M, Salmon DP, U HS, Bakay R. et al. A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med. 2005;11(5):551–555. doi: 10.1038/nm1239. [DOI] [PubMed] [Google Scholar]
- Ferreira D, Westman E, Eyjolfsdottir H, Almqvist P, Lind G, Linderoth B. et al. Brain changes in Alzheimer’s disease patients with implanted encapsulated cells releasing nerve growth factor. J Alzheimers Dis. 2015;43(3):1059–1072. doi: 10.3233/JAD-141068. [DOI] [PubMed] [Google Scholar]
- Huang B, Tabata Y, Gao J-G. Mesenchymal stem cells as therapeutic agents and potential targeted gene delivery vehicle for brain diseases. J Control Release. 2012;162(2):464–473. doi: 10.1016/j.jconrel.2012.07.034. [DOI] [PubMed] [Google Scholar]
- Dantuma E, Merchant S, Sugaya K. Stem cells for the treatment of neurodegenerative diseases. Stem Cell Res Ther. 2010;1(5):37. doi: 10.1186/scrt37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghadam FH, Alaie H, Karbalaie K, Tanhaei S, Nasr Esfahani MH, Baharvand H. Transplantation of primed or unprimed mouse embryonic stem cell-derived neural precursor cells improves cognitive function in Alzheimerian rats. Differentiation. 2009;78(2-3):59–68. doi: 10.1016/j.diff.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Jung YW, Hysolli E, Kim K-Y, Tanaka Y, Park I-H. Human induced pluripotent stem cells and neurodegenerative disease: prospects for novel therapies. Curr Opin Neurol. 2012;25(2):125–130. doi: 10.1097/WCO.0b013e3283518226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T. et al. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20(23):4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C. et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482(7384):216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RC, Lockwood AH, Sonawane BR. Neurodegenerative diseases: an overview of environmental risk factors. Environ Health Perspect. 2005;113(9):1250–1256. doi: 10.1289/ehp.7567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. Alzheimer’s disease pathogenesis: role of aging. Ann N Y Acad Sci. 2006;1067:454–460. doi: 10.1196/annals.1354.065. [DOI] [PubMed] [Google Scholar]
- Sastre M, Klockgether T, Heneka MT. Contribution of inflammatory processes to Alzheimer’s disease: molecular mechanisms. Int J Dev Neurosci. 2006;24(2-3):167–176. doi: 10.1016/j.ijdevneu.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Yaffe Y. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819–828. doi: 10.1016/S1474-4422(11)70072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368(9533):387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- Mudher A, Lovestone S. Alzheimer’s disease--do tauists and baptists finally shake hands? Trends Neurosci. 2002;25(1):22–26. doi: 10.1016/s0166-2236(00)02031-2. [DOI] [PubMed] [Google Scholar]
- Carson JA, Turner AJ. Beta-amyloid catabolism: roles for neprilysin (NEP) and other metallopeptidases? J Neurochem. 2002;81(1):1–8. doi: 10.1046/j.1471-4159.2002.00855.x. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Moir RD, Wagner SL. Clearance of Alzheimer’s Abeta peptide: the many roads to perdition. Neuron. 2004;43(5):605–608. doi: 10.1016/j.neuron.2004.08.024. [DOI] [PubMed] [Google Scholar]
- Hardy J. The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J Neurochem. 2009;110(4):1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Esler WP, Wolfe MS. A portrait of Alzheimer secretases -- new features and familiar faces. Science. 2001;293(5534):1449–1454. doi: 10.1126/science.1064638. [DOI] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S. et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012;488(7409):96–99. doi: 10.1038/nature11283. [DOI] [PubMed] [Google Scholar]
- Nagele RG, D’Andrea MR, Anderson WJ, Wang HY. Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience. 2002;110(2):199–211. doi: 10.1016/s0306-4522(01)00460-2. [DOI] [PubMed] [Google Scholar]
- Nalbantoglu J, Tirado-Santiago G, Lahsaini A, Poirier J, Goncalves O, Verge G. et al. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature. 1997;387(6632):500–505. doi: 10.1038/387500a0. [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J. et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26(40):10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AJ, Knauer M, Burdick DA, Glabe C. Intracellular A beta 1-42 aggregates stimulate the accumulation of stable, insoluble amyloidogenic fragments of the amyloid precursor protein in transfected cells. J Biol Chem. 1995;270(24):14786–14792. doi: 10.1074/jbc.270.24.14786. [DOI] [PubMed] [Google Scholar]
- Wild-Bode C, Yamazaki T, Capell A, Leimer U, Steiner H, Ihara Y. et al. Intracellular generation and accumulation of amyloid beta-peptide terminating at amino acid 42. J Biol Chem. 1997;272(26):16085–16088. doi: 10.1074/jbc.272.26.16085. [DOI] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120(3):885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Beher D, Graham SL. Protease inhibitors as potential disease-modifying therapeutics for Alzheimer’s disease. Expert Opin Investig Drugs. 2005;14(11):1385–1409. doi: 10.1517/13543784.14.11.1385. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Gurol ME, Rosand J, Smith EE. Amyloid angiopathy-related vascular cognitive impairment. Stroke. 2004;35(11 Suppl 1):2616–2619. doi: 10.1161/01.STR.0000143224.36527.44. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci. 2007;8(7):499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- Younkin SG. The role of A beta 42 in Alzheimer’s disease. J Physiol Paris. 1998;92(3-4):289–292. doi: 10.1016/s0928-4257(98)80035-1. [DOI] [PubMed] [Google Scholar]
- Haass C. Take five--BACE and the gamma-secretase quartet conduct Alzheimer’s amyloid beta-peptide generation. EMBO J. 2004;23(3):483–488. doi: 10.1038/sj.emboj.7600061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L. et al. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci. 2010;30(14):4833–4844. doi: 10.1523/JNEUROSCI.5221-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW. et al. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010;29(17):3020–3032. doi: 10.1038/emboj.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornsten A, Lieberthal J, Fadia S, Malins R, Ha L, Xu X. et al. APL-1, a Caenorhabditis elegans protein related to the human beta-amyloid precursor protein, is essential for viability. Proc Natl Acad Sci U S A. 2007;104(6):1971–1976. doi: 10.1073/pnas.0603997104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N. et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2(8):864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW. et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Gotz J, Ittner LM. Animal models of Alzheimer’s disease and frontotemporal dementia. Nat Rev Neurosci. 2008;9(7):532–544. doi: 10.1038/nrn2420. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Green KN. Animal models of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(11) doi: 10.1101/cshperspect.a006320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudduth TL, Schmitt FA, Nelson PT, Wilcock DM. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol Aging. 2013;34(4):1051–1059. doi: 10.1016/j.neurobiolaging.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K. et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78(4):631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16(6):358–372. doi: 10.1038/nrn3880. [DOI] [PubMed] [Google Scholar]
- Du Yan S, Zhu H, Fu J, Yan SF, Roher A, Tourtellotte WW. et al. Amyloid-beta peptide-receptor for advanced glycation endproduct interaction elicits neuronal expression of macrophage-colony stimulating factor: a proinflammatory pathway in Alzheimer disease. Proc Natl Acad Sci U S A. 1997;94(10):5296–5301. doi: 10.1073/pnas.94.10.5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature. 1996;382(6593):716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- Dzamba D, Harantova L, Butenko O, Anderova M. Glial cells -- the key elements of Alzheimer’s disease. Curr Alzheimer Res. 2016 doi: 10.2174/1567205013666160129095924. [DOI] [PubMed] [Google Scholar]
- Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17(3):553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A. et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11(2):155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel NS, Paris D, Mathura V, Quadros AN, Crawford FC, Mullan MJ. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J Neuroinflammation. 2005;2(1):9. doi: 10.1186/1742-2094-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, Lawlor B. et al. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci Lett. 1991;129(2):318–320. doi: 10.1016/0304-3940(91)90490-k. [DOI] [PubMed] [Google Scholar]
- Griffin WST. Neuroinflammatory cytokine signaling and Alzheime’s disease. N Engl J Med. 2013;368(8):770–771. doi: 10.1056/NEJMcibr1214546. [DOI] [PubMed] [Google Scholar]
- Vom Berg J, Prokop S, Miller KR, Obst J, Kälin RE, Lopategui-Cabezas I. et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat Med. 2012;18(12):1812–1819. doi: 10.1038/nm.2965. [DOI] [PubMed] [Google Scholar]
- Tan M-S, Yu J-T, Jiang T, Zhu X-C, Guan H-S, Tan L. IL12/23 p40 inhibition ameliorates Alzheimer’s disease-associated neuropathology and spatial memory in SAMP8 mice. J Alzheimers Dis. 2014;38(3):633–646. doi: 10.3233/JAD-131148. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L. et al. TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7(5):612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J. et al. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14(6):681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A. et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB. et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. 2013;14(8):812–820. doi: 10.1038/ni.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C. et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13(4):432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CKE, Burgold S, Page RM, Mitteregger G. et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer’s disease. Nat Neurosci. 2010;13(4):411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros R, LaFerla FM. Astrocytes: conductors of the Alzheimer disease neuroinflammatory symphony. Exp Neurol. 2013;239:133–138. doi: 10.1016/j.expneurol.2012.10.007. [DOI] [PubMed] [Google Scholar]
- Hosokawa M, Klegeris A, Maguire J, McGeer PL. Expression of complement messenger RNAs and proteins by human oligodendroglial cells. Glia. 2003;42(4):417–423. doi: 10.1002/glia.10234. [DOI] [PubMed] [Google Scholar]
- Yang YB, Li R, Meri S, Rogers J, Shen Y. Deficiency of complement defense protein CD59 may contribute to neurodegeneration in Alzheimer’s disease. J Neurosci. 2000;20(20):7505–7509. doi: 10.1523/JNEUROSCI.20-20-07505.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DG, Dalsing-Hernandez JE, Campbell NA, Lue L-F. Decreased expression of CD200 and CD200 receptor in Alzheime’s disease: a potential mechanism leading to chronic inflammation. Exp Neurol. 2009;215(1):5–19. doi: 10.1016/j.expneurol.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vukic V, Callaghan D, Walker D, Lue L-F, Liu QY, Couraud P-O. et al. Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells andin Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol Dis. 2009;34(1):95–106. doi: 10.1016/j.nbd.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke DL, Johansson CB, Wilbertz J, Veress B, Nilsson E, Karlstrom H. et al. Generalized potential of adult neural stem cells. Science. 2000;288(5471):1660–1663. doi: 10.1126/science.288.5471.1660. [DOI] [PubMed] [Google Scholar]
- Altman J, Das GD. Post-natal origin of microneurones in the rat brain. Nature. 1965;207(5000):953–956. doi: 10.1038/207953a0. [DOI] [PubMed] [Google Scholar]
- Doetsch F, Garcia-Verdugo JM, Alvarez-Buylla A. Cellular composition and three-dimensional organization of the subventricular germinal zone in the adult mammalian brain. J Neurosci. 1997;17(13):5046–5061. doi: 10.1523/JNEUROSCI.17-13-05046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch F, Scharff C. Challenges for brain repair: insights from adult neurogenesis in birds and mammals. Brain Behav Evol. 2001;58(5):306–322. doi: 10.1159/000057572. [DOI] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman J. Autoradiographic and histological studies of postnatal neurogenesis. IV. Cell proliferation and migration in the anterior forebrain, with special reference to persisting neurogenesis in the olfactory bulb. J Comp Neurol. 1969;137(4):433–457. doi: 10.1002/cne.901370404. [DOI] [PubMed] [Google Scholar]
- Kempermann G, van Praag H, Gage FH. Activity-dependent regulation of neuronal plasticity and self repair. Prog Brain Res. 2000;127:35–48. doi: 10.1016/s0079-6123(00)27004-0. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Gast D, Kronenberg G, Yamaguchi M, Gage FH. Early determination and long-term persistence of adult-generated new neurons in the hippocampus of mice. Development. 2003;130(2):391–399. doi: 10.1242/dev.00203. [DOI] [PubMed] [Google Scholar]
- Ernst A, Alkass K, Bernard S, Salehpour M, Perl S, Tisdale J. et al. Neurogenesis in the striatum of the adult human brain. Cell. 2014;156(5):1072–1083. doi: 10.1016/j.cell.2014.01.044. [DOI] [PubMed] [Google Scholar]
- Magnusson JP, Goritz C, Tatarishvili J, Dias DO, Smith EM, Lindvall O. et al. A latent neurogenic program in astrocytes regulated by Notch signaling in the mouse. Science. 2014;346(6206):237–241. doi: 10.1126/science.346.6206.237. [DOI] [PubMed] [Google Scholar]
- Alvarez-Buylla A, Seri B, Doetsch F. Identification of neural stem cells in the adult vertebrate brain. Brain Res Bull. 2002;57(6):751–758. doi: 10.1016/s0361-9230(01)00770-5. [DOI] [PubMed] [Google Scholar]
- Chapouton P, Jagasia R, Bally-Cuif L. Adult neurogenesis in non-mammalian vertebrates. Bioessays. 2007;29(8):745–757. doi: 10.1002/bies.20615. [DOI] [PubMed] [Google Scholar]
- Kaslin J, Ganz J, Brand M. Proliferation, neurogenesis and regeneration in the non-mammalian vertebrate brain. Philos Trans R Soc Lond B Biol Sci. 2008;363(1489):101–122. doi: 10.1098/rstb.2006.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zupanc GK. Adult neurogenesis and neuronal regeneration in the brain of teleost fish. J Physiol Paris. 2008;102(4-6):357–373. doi: 10.1016/j.jphysparis.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Kizil C, Kaslin J, Kroehne V, Brand M. Adult neurogenesis and brain regeneration in zebrafish. Dev Neurobiol. 2012;72(3):429–461. doi: 10.1002/dneu.20918. [DOI] [PubMed] [Google Scholar]
- Baumgart EV, Barbosa JS, Bally-Cuif L, Gotz M, Ninkovic J. Stab wound injury of the zebrafish telencephalon: a model for comparative analysis of reactive gliosis. Glia. 2012;60(3):343–357. doi: 10.1002/glia.22269. [DOI] [PubMed] [Google Scholar]
- Kishimoto N, Shimizu K, Sawamoto K. Neuronal regeneration in a zebrafish model of adult brain injury. Dis Model Mech. 2012;5(2):200–209. doi: 10.1242/dmm.007336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroehne V, Freudenreich D, Hans S, Kaslin J, Brand M. Regeneration of the adult zebrafish brain from neurogenic radial glia-type progenitors. Development. 2011;138(22):4831–4841. doi: 10.1242/dev.072587. [DOI] [PubMed] [Google Scholar]
- Marz M, Schmidt R, Rastegar S, Strahle U. Regenerative response following stab injury in the adult zebrafish telencephalon. Dev Dyn. 2012;240(9):2221–2231. doi: 10.1002/dvdy.22710. [DOI] [PubMed] [Google Scholar]
- Tanaka EM, Ferretti P. Considering the evolution of regeneration in the central nervous system. Nat Rev Neurosci. 2009;10(10):713–723. doi: 10.1038/nrn2707. [DOI] [PubMed] [Google Scholar]
- Berg DA, Kirkham M, Beljajeva A, Knapp D, Habermann B, Ryge J. et al. Efficient regeneration by activation of neurogenesis in homeostatically quiescent regions of the adult vertebrate brain. Development. 2010;137(24):4127–4134. doi: 10.1242/dev.055541. [DOI] [PubMed] [Google Scholar]
- Endo T, Yoshino J, Kado K, Tochinai S. Brain regeneration in anuran amphibians. Dev Growth Differ. 2007;49(2):121–129. doi: 10.1111/j.1440-169X.2007.00914.x. [DOI] [PubMed] [Google Scholar]
- Kizil C, Dudczig S, Kyritsis N, Machate A, Blaesche J, Kroehne V. et al. The chemokine receptor cxcr5 regulates the regenerative neurogenesis response in the adult zebrafish brain. Neural Dev. 2012;7:27. doi: 10.1186/1749-8104-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizil C, Kyritsis N, Dudczig S, Kroehne V, Freudenreich D, Kaslin J. et al. Regenerative neurogenesis from neural progenitor cells requires injury-induced expression of Gata3. Dev Cell. 2012;23(6):1230–1237. doi: 10.1016/j.devcel.2012.10.014. [DOI] [PubMed] [Google Scholar]
- Kyritsis N, Kizil C, Zocher S, Kroehne V, Kaslin J, Freudenreich D. et al. Acute inflammation initiates the regenerative response in the adult zebrafish brain. Science. 2012;338(6112):1353–1356. doi: 10.1126/science.1228773. [DOI] [PubMed] [Google Scholar]
- Wang Q-H, Xu R-X, Nagao S. Transplantation of cholinergic neural stem cells in a mouse model of Alzheimer’s disease. Chin Med J. 2005;118(6):508–511. [PubMed] [Google Scholar]
- Oliveira AA, Hodges HM. Alzheimer’s disease and neural transplantation as prospective cell therapy. Curr Alzheimer Res. 2005;2(1):79–95. doi: 10.2174/1567205052772759. [DOI] [PubMed] [Google Scholar]
- Taupin P. Adult neurogenesis, neural stem cells and Alzheimer’s disease: developments, limitations, problems and promises. Curr Alzheimer Res. 2009;6(6):461–470. doi: 10.2174/156720509790147151. [DOI] [PubMed] [Google Scholar]
- Wang Q, Matsumoto Y, Shindo T, Miyake K, Shindo A, Kawanishi M. et al. Neural stem cells transplantation in cortex in a mouse model of Alzheimer’s disease. J Med Invest. 2006;53(1-2):61–69. doi: 10.2152/jmi.53.61. [DOI] [PubMed] [Google Scholar]
- Yamasaki TR, Blurton-Jones M, Morrissette DA, Kitazawa M, Oddo S, LaFerla FM. Neural stem cells improve memory in an inducible mouse model of neuronal loss. J Neurosci. 2007;27(44):11925–11933. doi: 10.1523/JNEUROSCI.1627-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton DW, Webber DJ, Bilican B, Goedert M, Spillantini MG, Chandran S. Cell-mediated neuroprotection in a mouse model of human tauopathy. Neurosci J. 2010;30(30):9973–9983. doi: 10.1523/JNEUROSCI.0834-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang P-J, Sha H-Y, Ni J, Li M-H, Gu G-J. Neural stem cell transplants improve cognitive function without altering amyloid pathology in an APP/PS1 double transgenic model of Alzheimer’s disease. Mol Neurobiol. 2014;50(2):423–437. doi: 10.1007/s12035-014-8640-x. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Wu H-H, Wang Y, Gu G-J, Zhang W, Xia R. Neural stem cell transplantation decreases neuroinflammation in a transgenic mouse model of Alzheimer’s disease. J Neurochem. 2015 doi: 10.1111/jnc.13413. [DOI] [PubMed] [Google Scholar]
- Zhang W, Gu G-J, Shen X, Zhang Q, Wang G-M, Wang P-J. Neural stem cell transplantation enhances mitochondrial biogenesis in a transgenic mouse model of Alzheimer’s disease-like pathology. Neurobiol Aging. 2015;36(3):1282–1292. doi: 10.1016/j.neurobiolaging.2014.10.040. [DOI] [PubMed] [Google Scholar]
- Kim JA, Ha S, Shin KY, Kim S, Lee KJ, Chong YH. et al. Neural stem cell transplantation at critical period improves learning and memory through restoring synaptic impairment in Alzheimer’s disease mouse model. Cell Death Dis. 2015;6:e1789. doi: 10.1038/cddis.2015.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S-Q, Cai Q, Shen Y-Y, Wang P-Y, Li M-H, Teng G-Y. Neural stem cell transplantation improves spatial learning and memory via neuronal regeneration in amyloid-β precursor protein/presenilin 1/tau triple transgenic mice. Am J Alzheimers Dis Other Demen. 2014;29(2):142–149. doi: 10.1177/1533317513506776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MJ, Kim MY, Heo SC, Kwon YW, Kim YM, Do EK. et al. Macrophages regulate smooth muscle differentiation of mesenchymal stem cells via a prostaglandin F(2)alpha-mediated paracrine mechanism. Arterioscler Thromb Vasc Biol. 2012;32(11):2733–2740. doi: 10.1161/ATVBAHA.112.300230. [DOI] [PubMed] [Google Scholar]
- Ager RR, Davis JL, Agazaryan A, Benavente F, Poon WW, LaFerla FM. et al. Human neural stem cells improve cognition and promote synaptic growth in two complementary transgenic models of Alzheimer’s disease and neuronal loss. Hippocampus. 2015;25(7):813–826. doi: 10.1002/hipo.22405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I-S, Jung K, Kim I-S, Lee H, Kim M, Yun S. et al. Human neural stem cells alleviate Alzheimer-like pathology in a mouse model. Mol Neurodegener. 2015;10:38. doi: 10.1186/s13024-015-0035-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilja AM, Malmsten L, Röjdner J, Voytenko L, Verkhratsky A, Ögren SO. et al. Neural Stem Cell Transplant-Induced Effect on Neurogenesis and Cognition in Alzheimer Tg2576 Mice Is Inhibited by Concomitant Treatment with Amyloid-Lowering or Cholinergic α7 Nicotinic Receptor Drugs. Neural Plast. 2015;2015:370432. doi: 10.1155/2015/370432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuszynski MH, U HS, Amaral DG, Gage FH. Nerve growth factor infusion in the primate brain reduces lesion-induced cholinergic neuronal degeneration. J Neurosci. 1990;10(11):3604–3614. doi: 10.1523/JNEUROSCI.10-11-03604.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W, Wictorin K, Björklund A, Williams LR, Varon S, Gage FH. Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature. 1987;329(6134):65–68. doi: 10.1038/329065a0. [DOI] [PubMed] [Google Scholar]
- Birch AM, McGarry NB, Kelly AM. Short-term environmental enrichment, in the absence of exercise, improves memory, and increases NGF concentration, early neuronal survival, and synaptogenesis in the dentate gyrus in a time-dependent manner. Hippocampus. 2013;23(6):437–450. doi: 10.1002/hipo.22103. [DOI] [PubMed] [Google Scholar]
- Tuszynski MH, Yang JH, Barba D, U H-S, Bakay RAE, Pay MM. et al. Nerve Growth Factor Gene Therapy: Activation of Neuronal Responses in Alzheimer Disease. JAMA Neurol. 2015;72(10):1139–1147. doi: 10.1001/jamaneurol.2015.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Amouri SS, Zhu H, Yu J, Marr R, Verma IM, Kindy MS. Neprilysin: an enzyme candidate to slow the progression of Alzheimer’s disease. Am J Pathol. 2008;172(5):1342–1354. doi: 10.2353/ajpath.2008.070620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JB, Crews L, Rockenstein E, Adame A, Mante M, Hersh LB. et al. Neuropeptide Y fragments derived from neprilysin processing are neuroprotective in a transgenic model of Alzheimer’s disease. J Neurosci. 2009;29(4):1115–1125. doi: 10.1523/JNEUROSCI.4220-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM. et al. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer’s disease. Nat Med. 2009;15(3):331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang P-J, Li M-H, Wang G-L, Li P, Gao X-L. 1H-MRS assessment of the therapeutic effect of bilateral intraventricular BDNF infusion into APP/PS1 double transgenic mice. J Mol Neurosci. 2013;50(3):434–442. doi: 10.1007/s12031-013-9951-5. [DOI] [PubMed] [Google Scholar]
- Burke RM, Norman TA, Haydar TF, Slack BE, Leeman SE, Blusztajn JK. et al. BMP9 ameliorates amyloidosis and the cholinergic defect in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2013;110(48):19567–19572. doi: 10.1073/pnas.1319297110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellott TJ, Pender SM, Burke RM, Langley EA, Blusztajn JK. IGF2 ameliorates amyloidosis, increases cholinergic marker expression and raises BMP9 and neurotrophin levels in the hippocampus of the APPswePS1dE9 Alzheimer’s disease model mice. PloS One. 2014;9(4):e94287. doi: 10.1371/journal.pone.0094287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum MM, Kiyota T, Ingraham KL, Ikezu S, Ikezu T. The anti-inflammatory glycoprotein, CD200, restores neurogenesis and enhances amyloid phagocytosis in a mouse model of Alzheimer’s disease. Neurobiol Aging. 2015;36(11):2995–3007. doi: 10.1016/j.neurobiolaging.2015.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T. et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9(8):857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgaber D, Harris HW, Hla T, Maciag T, Donnelly RJ, Jacobsen JS. et al. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc Natl Acad Sci U S A. 1989;86(19):7606–7610. doi: 10.1073/pnas.86.19.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Ito K, Skinner RD, Mrak RE, Rovnaghi CR, Van Eldik LJ. et al. In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. Neurobiol Aging. 1996;17(5):761–766. doi: 10.1016/0197-4580(96)00104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaftel SS, Kyrkanides S, Olschowka JA, Miller J-NH, Johnson RE, O’Banion MK. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest. 2007;117(6):1595–1604. doi: 10.1172/JCI31450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachida Y, Nakagawa K, Saito T, Saido TC, Honda T, Saito Y. et al. Interleukin-1 beta up-regulates TACE to enhance alpha-cleavage of APP in neurons: resulting decrease in Abeta production. J Neurochem. 2008;104(5):1387–1393. doi: 10.1111/j.1471-4159.2007.05127.x. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA. et al. Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci. 2013;33(11):5053–5064. doi: 10.1523/JNEUROSCI.4361-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Menachem-Zidon O, Ben-Menachem-Zidon O, Ben-Menachem Y, Ben-Hur T, Yirmiya R. Intra-hippocampal transplantation of neural precursor cells with transgenic over-expression of IL-1 receptor antagonist rescues memory and neurogenesis impairments in an Alzheimer’s disease model. Neuropsychopharmacology. 2014;39(2):401–414. doi: 10.1038/npp.2013.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Wang R, Chen L, Bennett DA, Dickson DW, Wang D-S. et al. Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer’s brain. J Neurochem. 2010;115(1):47–57. doi: 10.1111/j.1471-4159.2010.06899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X. et al. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40(6):1087–1093. doi: 10.1016/s0896-6273(03)00787-6. [DOI] [PubMed] [Google Scholar]
- Blurton-Jones M, Spencer B, Michael S, Castello NA, Agazaryan AA, Davis JL. et al. Neural stem cells genetically-modified to express neprilysin reduce pathology in Alzheimer transgenic models. Stem Cell Res Ther. 2014;5(2):46. doi: 10.1186/scrt440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E, Desplats P, Ubhi K, Mante M, Florio J, Adame A. et al. Neuro-peptide treatment with Cerebrolysin improves the survival of neural stem cell grafts in an APP transgenic model of Alzheimer disease. Stem Cell Res. 2015;15(1):54–67. doi: 10.1016/j.scr.2015.04.008. [DOI] [PubMed] [Google Scholar]
- Xu L, Xu CJ, Lu HZ, Wang YX, Li Y, Lu PH. Long-term fate of allogeneic neural stem cells following transplantation into injured spinal cord. Stem Cell Rev. 2010;6(1):121–136. doi: 10.1007/s12015-009-9104-y. [DOI] [PubMed] [Google Scholar]
- Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132(4):661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Kim J, Efe JA, Zhu S, Talantova M, Yuan X, Wang S. et al. Direct reprogramming of mouse fibroblasts to neural progenitors. Proc Natl Acad Sci U S A. 2011;108(19):7838–7843. doi: 10.1073/pnas.1103113108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Flasskamp H, Hermann A, Arauzo-Bravo MJ, Lee SC, Lee SH. et al. Direct conversion of mouse fibroblasts into induced neural stem cells. Nat Protoc. 2014;9(4):871–881. doi: 10.1038/nprot.2014.056. [DOI] [PubMed] [Google Scholar]
- Lujan E, Chanda S, Ahlenius H, Sudhof TC, Wernig M. Direct conversion of mouse fibroblasts to self-renewing, tripotent neural precursor cells. Proc Natl Acad Sci U S A. 2012;109(7):2527–2532. doi: 10.1073/pnas.1121003109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thier M, Worsdorfer P, Lakes YB, Gorris R, Herms S, Opitz T. et al. Direct conversion of fibroblasts into stably expandable neural stem cells. Cell Stem Cell. 2012;10(4):473–479. doi: 10.1016/j.stem.2012.03.003. [DOI] [PubMed] [Google Scholar]
- Kawabata S, Takano M, Numasawa-Kuroiwa Y, Itakura G, Kobayashi Y, Nishiyama Y. et al. Grafted Human iPS Cell-Derived Oligodendrocyte Precursor Cells Contribute to Robust Remyelination of Demyelinated Axons after Spinal Cord Injury. Stem Cell Reports. 2016;6(1):1–8. doi: 10.1016/j.stemcr.2015.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y, Okada Y, Itakura G, Iwai H, Nishimura S, Yasuda A. et al. Pre-evaluated safe human iPSC-derived neural stem cells promote functional recovery after spinal cord injury in common marmoset without tumorigenicity. PLoS One. 2012;7(12):e52787. doi: 10.1371/journal.pone.0052787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JY, Lee SH, Lee SC, Kim JW, Kim KP, Kim SM. et al. Therapeutic potential of induced neural stem cells for spinal cord injury. J Biol Chem. 2014;289(47):32512–32525. doi: 10.1074/jbc.M114.588871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong LM, Fong H, Huang Y. Stem cell therapy for Alzheimer’s disease and related disorders: current status and future perspectives. Exp Mol Med. 2015;47:e151. doi: 10.1038/emm.2014.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Galvan V, Xie L, Mao XO, Gorostiza OF, Bredesen DE. et al. Enhanced neurogenesis in Alzheimer’s disease transgenic (PDGF-APPSw,Ind) mice. Proc Natl Acad Sci U S A. 2004;101(36):13363–13367. doi: 10.1073/pnas.0403678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC. et al. Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101:343–347. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Toledano MA, Shelanski ML. Neurogenic effect of beta-amyloid peptide in the development of neural stem cells. J Neurosci. 2004;24(33):5439–5444. doi: 10.1523/JNEUROSCI.0974-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Toledano MA, Shelanski ML. Increased neurogenesis in young transgenic mice overexpressing human APP(Sw, Ind) J Alzheimers Dis. 2007;12(3):229–240. doi: 10.3233/jad-2007-12304. [DOI] [PubMed] [Google Scholar]
- Lopez-Toledano MA. Neurogenic Effect of -Amyloid Peptide in the Development of Neural Stem Cells. J Neurosci. 2004;24:5439–5444. doi: 10.1523/JNEUROSCI.0974-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP. Disruption of neurogenesis by amyloid b -peptide, and perturbed neural progenitor cell homeostasis , in models of Alzheimer’s disease. J Neurochem. 2002;83:1509–1524. doi: 10.1046/j.1471-4159.2002.01267.x. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Liu D, Nath A, Borchard AC, Mattson MP. Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid beta-peptide: implications for the pathogenesis of Alzheimer’s disease. Neuromolecular Med. 2002;1:125–135. doi: 10.1385/NMM:1:2:125. [DOI] [PubMed] [Google Scholar]
- Zhang C, Mcneil E, Dressler L, Siman R. Long-lasting impairment in hippocampal neurogenesis associated with amyloid deposition in a knock-in mouse model of familial Alzheimer’s disease. Exp Neurol. Author manuscript. 2007;204(1):77–87. doi: 10.1016/j.expneurol.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadadhar A, Marr R, Lazarov O. Presenilin-1 regulates neural progenitor cell differentiation in the adult brain. J Neurosci. 2011;31(7):2615–2623. doi: 10.1523/JNEUROSCI.4767-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Handler M, Yang X, Shen J. Presenilin-1 regulates neuronal differentiation during neurogenesis. Development. 2000;127(12):2593–2606. doi: 10.1242/dev.127.12.2593. [DOI] [PubMed] [Google Scholar]
- Shen J, Bronsono RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89(4):629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- Yetman MJ, Jankowsky JL. Wild-type neural progenitors divide and differentiate normally in an amyloid-rich environment. J Neurosci. 2013;33:17335–17341. doi: 10.1523/JNEUROSCI.1917-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demars M, Hu YS, Gadadhar A, Lazarov O. Impaired neurogenesis is an early event in the etiology of familial Alzheimer’s disease in transgenic mice. J Neurosci Res. 2010;88(10):2103–2117. doi: 10.1002/jnr.22387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffo A, Rite I, Tripathi P, Lepier A, Colak D, Horn AP. et al. Origin and progeny of reactive gliosis: A source of multipotent cells in the injured brain. Proc Natl Acad Sci U S A. 2008;105(9):3581–3586. doi: 10.1073/pnas.0709002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffo A, Vosko MR, Erturk D, Hamann GF, Jucker M, Rowitch D. et al. Expression pattern of the transcription factor Olig2 in response to brain injuries: implications for neuronal repair. Proc Natl Acad Sci U S A. 2005;102(50):18183–18188. doi: 10.1073/pnas.0506535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa MR, Gotz M, Berninger B. What determines neurogenic competence in glia? Brain Res Rev. 2010;63(1-2):47–59. doi: 10.1016/j.brainresrev.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Heinrich C, Blum R, Gascon S, Masserdotti G, Tripathi P, Sanchez R. et al. Directing astroglia from the cerebral cortex into subtype specific functional neurons. PLoS Biol. 2010;8(5):e1000373. doi: 10.1371/journal.pbio.1000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robel S, Berninger B, Gotz M. The stem cell potential of glia: lessons from reactive gliosis. Nat Rev Neurosci. 2011;12(2):88–104. doi: 10.1038/nrn2978. [DOI] [PubMed] [Google Scholar]
- Karow M, Sanchez R, Schichor C, Masserdotti G, Ortega F, Heinrich C. et al. Reprogramming of pericyte-derived cells of the adult human brain into induced neuronal cells. Cell Stem Cell. 2012;11(4):471–476. doi: 10.1016/j.stem.2012.07.007. [DOI] [PubMed] [Google Scholar]
- Brill MS, Ninkovic J, Winpenny E, Hodge RD, Ozen I, Yang R. et al. Adult generation of glutamatergic olfactory bulb interneurons. Nat Neurosci. 2009;12(12):1524–1533. doi: 10.1038/nn.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med. 2002;8(9):963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- Thored P, Heldmann U, Gomes-Leal W, Gisler R, Darsalia V, Taneera J. et al. Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia. 2009;57(8):835–849. doi: 10.1002/glia.20810. [DOI] [PubMed] [Google Scholar]
- Becker T, Bernhardt RR, Reinhard E, Wullimann MF, Tongiorgi E, Schachner M. Readiness of zebrafish brain neurons to regenerate a spinal axon correlates with differential expression of specific cell recognition molecules. J Neurosci. 1998;18(15):5789–5803. doi: 10.1523/JNEUROSCI.18-15-05789.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosacak MI, Papadimitriou C, Kizil C. Regeneration, Plasticity, and Induced Molecular Programs in Adult Zebrafish Brain. Biomed Res Int. 2015 doi: 10.1155/2015/769763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisch VC, Fraser B, Allison WT. Investigating regeneration and functional integration of CNS neurons: lessons from zebrafish genetics and other fish species. Biochim Biophys Acta. 2010;1812(3):364–380. doi: 10.1016/j.bbadis.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Poss KD, Keating MT, Nechiporuk A. Tales of regeneration in zebrafish. Dev Dyn. 2003;226(2):202–210. doi: 10.1002/dvdy.10220. [DOI] [PubMed] [Google Scholar]
- Poss KD. Advances in understanding tissue regenerative capacity and mechanisms in animals. Nat Rev Genet. 2010;11(10):710–722. doi: 10.1038/nrg2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana S, Rico EP, Burgos JS. Can zebrafish be used as animal model to study Alzheimer’s disease? Am J Neurodegener Dis. 2012;1(1):32–48. [PMC free article] [PubMed] [Google Scholar]
- Newman M, Ebrahimie E, Lardelli M. Using the zebrafish model for Alzheimer’s disease research. Front Genet. 2014;5:189. doi: 10.3389/fgene.2014.00189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyritsis N, Kizil C, Brand M. Neuroinflammation and central nervous system regeneration in vertebrates. Trends Cell Biol. 2014;24(2):128–135. doi: 10.1016/j.tcb.2013.08.004. [DOI] [PubMed] [Google Scholar]
- Kizil C, Kyritsis N, Brand M. Effects of inflammation on stem cells: together they strive? EMBO Rep. 2015;16(4):416–426. doi: 10.15252/embr.201439702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diotel N, Vaillant C, Gueguen MM, Mironov S, Anglade I, Servili A. et al. Cxcr4 and Cxcl12 expression in radial glial cells of the brain of adult zebrafish. J Comp Neurol. 2010;518(24):4855–4876. doi: 10.1002/cne.22492. [DOI] [PubMed] [Google Scholar]
- Sallinen V, Kolehmainen J, Priyadarshini M, Toleikyte G, Chen YC, Panula P. Dopaminergic cell damage and vulnerability to MPTP in Pink1 knockdown zebrafish. Neurobiol Dis. 2011;40(1):93–101. doi: 10.1016/j.nbd.2010.06.001. [DOI] [PubMed] [Google Scholar]
- Newman M, Tucker B, Nornes S, Ward A, Lardelli M. Altering presenilin gene activity in zebrafish embryos causes changes in expression of genes with potential involvement in Alzheimer's disease pathogenesis. J Alzheimers Dis. 2009;16(1):133–147. doi: 10.3233/JAD-2009-0945. [DOI] [PubMed] [Google Scholar]
- Newman M, Verdile G, Martins RN, Lardelli M. Zebrafish as a tool in Alzheimer’s disease research. Biochim Biophys Acta. 2010;1812(3):346–352. doi: 10.1016/j.bbadis.2010.09.012. [DOI] [PubMed] [Google Scholar]
- Paquet D, Bhat R, Sydow A, Mandelkow EM, Berg S, Hellberg S. et al. A zebrafish model of tauopathy allows in vivo imaging of neuronal cell death and drug evaluationn. J Clin Invest. 2009;119(5):1382–1395. doi: 10.1172/JCI37537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquet D, Schmid B, Haass C. Transgenic zebrafish as a novel animal model to study tauopathies and other neurodegenerative disorders in vivo. Neurodegener Dis. 2010;7(1-3):99–102. doi: 10.1159/000285515. [DOI] [PubMed] [Google Scholar]
- Tomasiewicz HG, Flaherty DB, Soria JP, Wood JG. Transgenic zebrafish model of neurodegeneration. J Neurosci Res. 2002;70(6):734–745. doi: 10.1002/jnr.10451. [DOI] [PubMed] [Google Scholar]
- Anichtchik O, Diekmann H, Fleming A, Roach A, Goldsmith P, Rubinsztein DC. Loss of PINK1 function affects development and results in neurodegeneration in zebrafish. J Neurosci. 2008;28(33):8199–8207. doi: 10.1523/JNEUROSCI.0979-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Q, Garver JA, Hukriede NA, Burton EA. Generation of a transgenic zebrafish model of Tauopathy using a novel promoter element derived from the zebrafish eno2 gene. Nucleic Acids Res. 2007;35(19):6501–6516. doi: 10.1093/nar/gkm608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gama Sosa MA, De Gasperi R, Elder GA. Modeling human neurodegenerative diseases in transgenic systems. Hum Genet. 2011;131(4):535–563. doi: 10.1007/s00439-011-1119-1. [DOI] [PubMed] [Google Scholar]
- Malaga-Trillo E, Salta E, Figueras A, Panagiotidis C, Sklaviadis T. Fish models in prion biology: underwater issues. Biochim Biophys Acta. 2011;1812(3):402–414. doi: 10.1016/j.bbadis.2010.09.013. [DOI] [PubMed] [Google Scholar]
- O’Donnell KC, Lulla A, Stahl MC, Wheat ND, Bronstein JM, Sagasti A. Axon degeneration and PGC-1alpha-mediated protection in a zebrafish model of alpha-synuclein toxicity. Dis Model Mech. 2014;7(5):571–582. doi: 10.1242/dmm.013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y, Noble S, Ekker M. Modeling neurodegeneration in zebrafish. Curr Neurol Neurosci Rep. 2011;11(3):274–282. doi: 10.1007/s11910-011-0182-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W. Exploring Alzheimer’s disease in zebrafish. J Alzheimers Dis. 2010;20(4):981–990. doi: 10.3233/JAD-2010-1412. [DOI] [PubMed] [Google Scholar]
- Musa A, Lehrach H, Russo VA. Distinct expression patterns of two zebrafish homologues of the human APP gene during embryonic development. Dev Genes Evol. 2001;211(11):563–567. doi: 10.1007/s00427-001-0189-9. [DOI] [PubMed] [Google Scholar]
- Mueller T, Vernier P, Wullimann MF. The adult central nervous cholinergic system of a neurogenetic model animal, the zebrafish Danio rerio. Brain Res. 2004;1011(2):156–169. doi: 10.1016/j.brainres.2004.02.073. [DOI] [PubMed] [Google Scholar]
- Guo S. Linking genes to brain, behavior and neurological diseases: what can we learn from zebrafish? Genes Brain Behav. 2004;3(2):63–74. doi: 10.1046/j.1601-183x.2003.00053.x. [DOI] [PubMed] [Google Scholar]