

Abstract
Background
Inflammatory bowel disease (IBD) consists of Crohn's disease (CD) and ulcerative colitis (UC), two widespread diseases of unknown, multifactorial etiology. Colitis pathology involves a pathological angiogenic response where increases in vascular density participate in colitis histopathology. Vascular endothelial growth factor-A (VEGF-A) is a potent angiogenesis stimulator known to be involved in pathological angiogenesis in several diseases including colitis. However, the pathogenic importance of specific VEGF-A isoforms during T-cell-mediated experimental colitis remains largely unknown.
Methods
The CD4+CD45RBhigh T-cell transfer model of experimental colitis was used for these studies. The VEGF lac-Z trans-genic reporter mouse was used to examine specific cellular sources of VEGF-A production. The VEGF164 aptamer (Macu-gen), adenoviral VEGF164, and the VEGF Trap were used to evaluate pathological importance.
Results
VEGF lac-Z reporter mice experiments showed that both infiltrating T cells and local tissue cells produce VEGF-A in the colon during disease. Inhibition of VEGF164 using a highly selective RNA aptamer significantly attenuated CD4+CD45RBhigh T-cell-dependent experimental colitis by reducing pathological angiogenesis and inflammatory pathology. Conversely, broad-spectrum VEGF inhibition with VEGF Trap did not attenuate disease, nor did adenoviral VEGF164 overexpression significantly alter colitis pathology.
Conclusions
VEGF164 is actively produced by multiple cell types during T-cell-mediated colitis. Importantly, specific inhibition of the VEGF164 isoform during T-cell-mediated colitis dose-dependently attenuated disease progression, while broad-scale inhibition of all VEGF-A isoforms was not therapeutically beneficial.
Keywords: IBD, colitis, VEGF, inflammation, angiogenesis, T cell
Inflammatory bowel disease (IBD) occurs in people of all ages and is a life-long debilitating condition. It consists of two diseases, Crohn's disease (CD) and ulcerative colitis (UC), both of which have unknown, multifactorial etiologies. Dietary, immune, and genetic factors are all implicated in the onset of these diseases; however, the contribution of each remains unknown. Clinical data has long shown increased vascular density to be a hallmark characteristic of IBD.1–4 Recently, we and others have examined and quantified this angiogenic response.5,6 There is now considerable evidence that angiogenesis contributes to the establishment and maintenance of this chronic disease.4–7 Angiogenesis is a critical component of many chronic inflammatory diseases, including IBD, rheumatoid arthritis, atherosclerosis, and age-related macular degeneration (AMD), making modulation of pathological angiogenesis therapeutically relevant for these diseases. Recent studies from our laboratory and others provide strong evidence that a pathological angiogenic response is necessary for establishing chronic inflammation in IBD and experimental colitis, and that attenuation of this angiogenic response ameliorates disease in animal models.5,8,9 Our data shows that, not only do increases in vascular density correlate highly with tissue pathology, but inhibition of angiogenesis reduces disease activity in the CD4+CD45RBhigh and dextran sulfate sodium (DSS) mouse models of experimental colitis.5 Specifically, the use of antiangiogenic agents such as thalidomide have been shown to induce remission in patients with active disease and protect against experimental colitis.5,10,11
Vascular endothelial growth factor-A (VEGF-A) production is significantly increased during disease in human IBD and in models of experimental colitis.5,12–14 VEGF-A is a potent angiogenic growth factor that stimulates endothelial cell growth, multiplication, and motility. VEGF-A also activates endothelial signaling pathways, resulting in transient microvasculature dilation and permeability increases, and upregulation of endothelial adhesion molecule expression, factors that all contribute to pathological angiogenic responses and when not properly regulated potentially contribute to chronic inflammatory states. Production of VEGF-A is known to occur in many cell types including endothelial cells, leukocytes, epithelial cells, and others.15–18 The production of VEGF by different tissues in the body has been elegantly documented during development and postnatally with use of VEGF lac-Z transgenic mice.19–21 However, the cellular sources of VEGF-A production during T-cell-mediated experimental colitis have not been examined.
VEGF-A involvement in pathological processes of both chronic inflammatory and cancer settings has been documented, and new therapeutic approaches targeting it are being developed and used for the treatment of these and other diseases.22–28 The overproduction of VEGF-A and other proangiogenic mediators during pathological states can cause a dysregulated angiogenic response characterized by the development of immature, tortuous, and leaky neovascular structures that aid edema formation and ease leukocyte entry into inflamed tissues.5,29 This occurrence allows inflammation to continue unchecked and likely is key to establishing a chronic inflammatory state. Importantly, VEGF-A has four major isoform splice variants of the VEGF gene: VEGF121, VEGF165, VEGF189, and VEGF205, each of which exist as a one amino acid shorter protein in mice. These isoforms play various roles in developmental, wound healing, and pathological angiogenesis as dictated by their size, solubility, and binding properties. It has been proposed that VEGF164 may be responsible for many of the pathological VEGF-A responses seen during disease, due to its intermediate biological characteristics and strong affinity for VEGFR2 and neuropilin-1 (Nrp1), a coreceptor that amplifies VEGFR2 signaling in endothelium.
Here we examine the hypothesis that the VEGF164 isoform plays a key role in the linking of angiogenesis and chronic inflammation during T-cell-mediated colitis, and is therefore the pathogenic isoform necessary for the establishment of chronic disease. Herein we show, using VEGF lac-Z transgenic mice, T cell and local tissue production of VEGF-A during colitis, verifying that VEGF-A is produced in large quantities by multiple cell types during colitis. We also demonstrate that broad inhibition of multiple VEGF-A isoforms with the VEGF Trap did not attenuate disease, while VEGF164 isoform neutralization significantly decreased T-cell colitis pathology and associated angiogenesis. Importantly, these data suggest that other VEGF family members may play protective or regulatory roles during T-cell-dependent colitis, indicating that selective targeting of specific VEGF isoforms may be more beneficial than wide-scale inhibition of multiple isoforms across different protein families. These findings contribute new insight into how chronic inflammation during IBD is regulated by VEGF-A, and delineates VEGF164 as a specific target for the treatment of colitis through unlinking angiogenesis and inflammation to prevent the development of a chronic inflammatory state.
Materials and Methods
Mice
Animals used in these studies were bred and housed at the Association for Assessment and Accreditation of Laboratory Animal Care, international-accredited Louisiana State University Health Sciences Center-Shreveport (LSUHSC) animal resource facility and maintained according to the National Research Council's Guide for Care and Use of Laboratory Animals. C57B6/J and Rag-1−/−(tm1Mom) mice used in these experiments were ordered from Jax Mice and housed at LSUHSC. VEGF Lac-Z mice were bred in-house and backcrossed onto the Rag-1−/− background for eight generations.
CD4+CD45RBHigh T-cell Transfer Model of Colitis
T-cell-dependent CD4+CD45RBHigh experimental colitis was induced in 10-12-week-old male Rag-1−/− and VEGF lac-Z Rag-1−/− mice as previously reported.5 Animals were monitored throughout the study for weight loss as a determinant of disease progression.
VEGF Antagonist Dosing Regimens
One day after T-cell transfer, animals were randomized to either vehicle or experimental treatment groups. VEGF Trap or human Fc placebo was administered at a dose of 20 mg/kg by intraperitoneal (i.p.) injection three times a week (M,W,F) throughout the 6-week study period. VEGF164 aptamer was administered at a range of doses including 40 mg/kg, 80 mg/kg, and 160 mg/kg by i.p. injection every 2 days throughout the 6-week study period.
Histopathological Scoring of the CD4+CD45RBHigh Model of Colitis
Histopathological scoring of the colons from mice used in these experiments was done on paraffin-embedded hematoxylin and eosin (H&E)-stained sections in a double-blinded manner as previously reported.5 Briefly, eight parameters were examined, including: 1) the degree of inflammation in the lamina propria (scored 1–3); 2) goblet cell lost indicative of mucin depletion (scored 0–2); 3) reactive epithelial hyperplasia/atypia with nuclear changes (scored 0–3); 4) number of intraepithelial lymphocytes in the epithelial crypt (scored 0–3); 5) abnormal crypt architecture (scored 0–3); 6) number of crypt abscesses (scored 0–2); 7) mucosal erosion to frank ulceration (scored 0–2); and 8) submucosal spread to transmural involvement (scored 0–2). Final histopathological scores for each mouse were based on the sum of these scores for a maximal score of 20.
Histological Staining for Lac-Z
Staining for the lac-Z reporter gene was done using an anti-β-gal primary antibody (AbCam, Cambridge, MA) at a concentration of 1:200, incubated overnight at 4°C in conjunction with the Vector ABC Elite Kit (Vector Laboratories, Burlingame, CA), Nova Red (Vector Laboratories), and hematoxylin nuclear counterstain (Vector Laboratories).
Tissue Angiogenic Index
Cross-sections of colon tissue from each animal were analyzed as previously reported.5 Slides were fixed and stained for platelet-endothelial cellular adhesion molecule-1 (PECAM-1) (CD31) and a Nikon Eclipse TE2000-S epifluorescent scope was used to capture images using Simple PCI software (C-Imaging Systems; Compix, Lake Oswego, OR) as previously described.5 Total pixel area stained for PECAM-1 was divided by DAPI pixel area staining to calculate an angiogenic index (AI).
Enzyme-linked Immunosorbent Assays (ELISAs)
VEGF164 ELISAs were obtained from Calbiochem (La Jolla, CA) and used according to their protocols with tissue lysates from the colons of control and experimental mice.
In Vitro Transwell Assay of VEGF164 Chemotaxis
Transwell assays were performed as previously described.30 Briefly, cells were seeded at 400,000 cells per insert and allowed to sit for 2 hours. Fifty ng/mL VEGF164 was then added to the ablumenal chamber and cells were allowed to chemotax for 6 hours, then fixed. Cells were cleared from the top of each insert and fluorescence readings were taken.
Statistical Analyses
Statistical analyses were done using Student's t-test or analysis of variance (ANOVA) with a Bonferroni's posttest among all groups and are presented as mean ± standard error of the mean (SEM). P values <0.05, <0.01, or <0.0001 are significant as indicated in the figures. All experiments were replicated at least twice and experimental P values are reported in the text and N values are reported in the figure legends.
Results
Cellular Production of VEGF-A During CD4+ Colitis
VEGF-A is known to play a role in pathological angiogenic settings where its overexpression has been documented, including rheumatoid arthritis, IBD, atherosclerosis, diabetic retinopathy, and psoriasis.5,29, Here we examine VEGF-A production during CD4+ T-cell-induced experimental colitis in order to determine the cellular sources of VEGF-A during disease. Using CD4+CD45RB Hig T cells isolated from VEGF lac-Z transgenic mice we induced colitis in Rag-1 mice, and T cells isolated from C57BL/6J mice were used to induce colitis in VEGF lac-Z Rag-1−/− mice. These mice were followed for 6 weeks and weight loss was documented to measure disease activity. Figure 1A shows the percent change in weight and colon lengths of the control and experimental groups, these data were not significantly different between groups, and all mice lost significant weight indicative of disease (10%-20% total body weight). Figure 1B shows the survival data for these groups over the 6-week experimental time course. Figure 1C (lac-Z T cells into Rag-1−/− mice) and 1D (wildtype [WT] T cells into VEGF lac-Z Rag-1−/− mice) show sample pictomicrographs of the histopathology from both experimental groups verifying the full development of colitis; for comparison, sample images of normal colon tissue are shown in Figure 3A and Supporting Information Figure 2. Figure 1E shows β-gal staining of colon tissue from Rag-1−/− mice where colitis was induced with VEGF lac-Z T cells; this sample image shows the nuclei of T cells producing VEGF-A (brown) with a hematoxylin counterstain (blue); black arrows indicate positive staining of T-cell nuclei. Figure 1F is a sample image of β-gal staining in VEGF lac-Z Rag-1−/− mice; here the nuclei of all tissue cells producing VEGF-A stain positive for β-gal (brown) and hematoxylin counterstain was used (blue); black arrows indicate positive staining of nuclei for β-gal. These data clearly show that multiple cell types (e.g., infiltrating leukocytes and mucosal epithelial cells) actively produce VEGF-A during disease. These findings are significant as they clearly demonstrate VEGF-A cytokine expression is involved in multiple tissue niches during the development of chronic disease.
Figure 1.
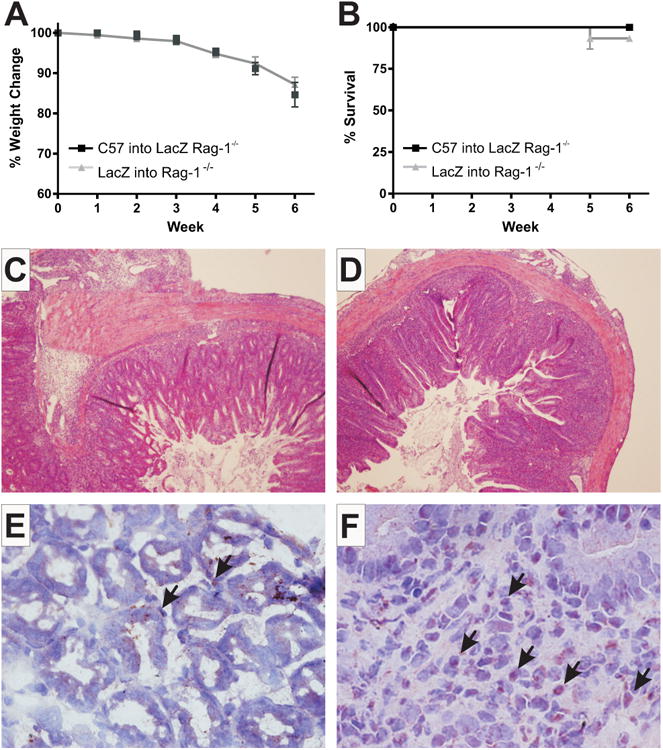
VEGF lac-Z reporter mice reveal VEGF-A production by infiltrating immune and local tissue cells. Weight loss data for VEGF Lac-Z T cells transferred into Rag-1−/− (gray) and C57 T cells transferred into VEGF Lac-Z Rag-1−/− (black) mice is shown in (A). (B) Survival data for the same groups. Sample H&E stained histopathology images are shown in (C,D) for Rag-1−/− and VEGF Lac-Z Rag-1−/− groups, respectively. (E) Lac-Z staining (brown) with hematoxylin counterstain (blue) of T cells producing VEGF-A during colitis. (F) The same for leukocytes other than T cells and local tissue cells. N = 14 for Lac-Z into Rag-1−/−, N = 10 for C57 into Lac-Z Rag-1−/− groups.
Figure 3.
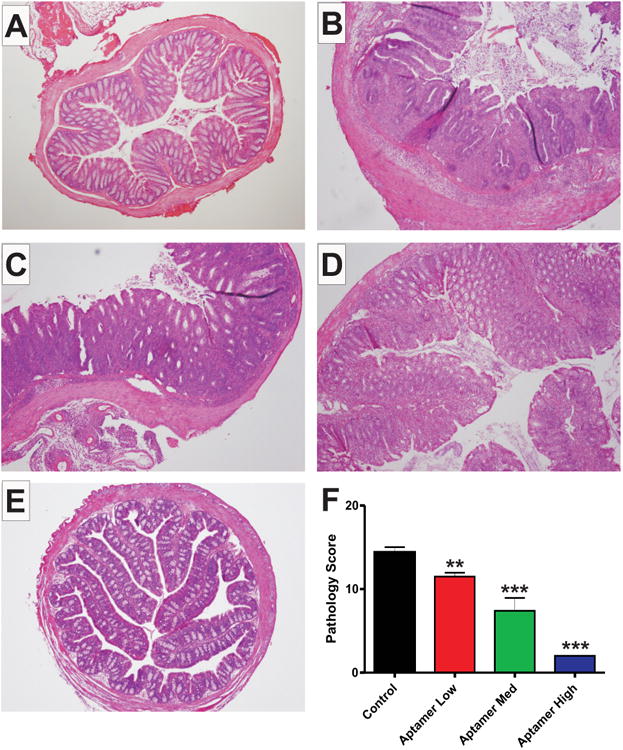
The VEGF164 aptamer dose-dependently inhibits CD4+ experimental colitis. This figure shows sample images of H&E staining of colonic cross-sections from Control noncolitic (A), Control colitic (B), Aptamer Low (C), Aptamer Med (D), and Aptamer High (E) dose groups. (F) The histopathology scores for these groups, Control colitic (black), Aptamer Low (red), Aptamer Med (green), and Aptamer High (blue). N = 28 for Control, N = 14 for Aptamer Low, N = 18 for Aptamer Med, N = 18 for Aptamer High.
Isoform-specific Dose-dependent Attenuation of CD4+ Experimental Colitis by the VEGF164 aptamer
Having shown previously that VEGF-A is upregulated during CD4+ T-cell-mediated colitis we next examined the effect of VEGF164 isoform-specific inhibition in our model using a VEGF164 aptamer. These studies make use of three increasing doses of the aptamer (40 mg/kg/day, low dose; 80 mg/kg/day, med dose; and 160 mg/kg/day, high dose) to examine the utility of VEGF164 inhibition and dose-dependent responses to the same. Table 1 depicts the aptamer doses used, number and frequency of dosing, and total amount of the aptamer given to each group as compared to the dose given to patients for treatment of AMD. Figure 2A shows the weight loss data from all groups revealing that the medium and high dose groups of aptamer significantly attenuated weight loss during the experimental time course beginning at experimental week 4. Survival data for all four groups is reported in Figure 2B. Measurements of colonic shortening as an indication of disease progression are reported in Figure 2C, revealing increased colon lengths with the aptamer treatment (low, 83.50 ± 3.423, P = 0.0160; med, 88.33 ± 1.252, P < 0.0001; high, 87.78 ± 1.553, P < 0.0001) compared to control (73.45 ± 2.033). Figure 2D reports the gross scores for the combined control group (2.375 ± 0.1451), and low (1.625 ± 0.1825, P = 0.0103), med (1.385 ± 0.1804, P = 0.0002), and high (1.100 ± 0.2333, P < 0.0001) dose aptamer treated groups. Here we saw a significant, dose-dependent reduction in gross score for all groups with increasing significant differences for each increase in dose. ELISAs of colon tissue lysates from these groups revealed decreasing concentrations of VEGF164 levels concomitant with increasing doses of the aptamer (control, 1329 ± 57.96 pg/μL; low dose, 1082 ± 156.7 pg/μL, P = 0.0973; med dose, 755.2 ± 53.08 pg/μL, P < 0.0001; and high dose, 605 ± 40.00 pg/μL, P < 0.0001) (Fig. 2E).
Table 1. Various Dosing Regimens for VEGF164 Aptamer during 6 Week T cell Colitis Model.
| Dose Group | Total Amount | Time between injections | Total Amount Given |
|---|---|---|---|
| AMD Patient | 0.3 mg | 6 weeks | 0.3 mg |
| Low | 40 mg/kg | 2 days | 18 mg |
| Medium | 80 mg/kg | 2 days | 36 mg |
| High | 160 mg/kg | 2 days | 72 mg |
The amount of the VEGF164 aptamer (Macugen) given in AMD treatment, and the amount given in our low, medium, and high doses per day, the frequency of dosing, and the total dose given over a 6- week period.
Figure 2.
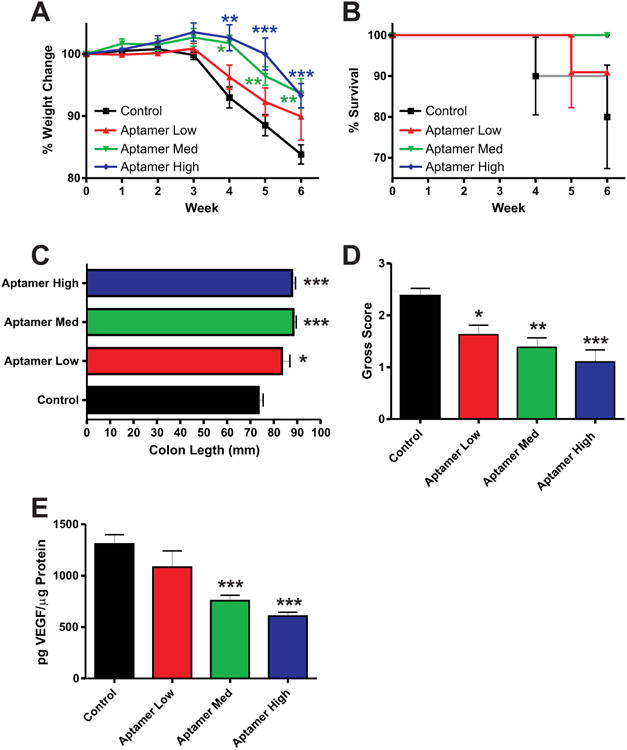
Disease activity and progression in the VEGF164 aptamer treatment groups. (A) Weight loss data for the aptamer-dosed groups; control (black), Aptamer Low (red), Aptamer Med (green), and Aptamer High (blue). (B) Survival data for the same. (C,D) Colon lengths and gross scores, respectively, upon takedown for all groups. (E) VEGF164 ELISA data for the same. N = 28 for Control, N = 14 for Aptamer Low, N = 18 for Aptamer Med, N = 18 for Aptamer High, N = 5 for ELISA data.
We next examined the effects of the aptamer on disease histopathology. Figure 3 contains sample histopathology pictomicrographs of normal (A), control (B), low dose (C), medium dose (D), and high dose (E) aptamer treated colons. Graphical analysis of combined histopathology scores for the same groups is reported in Figure 3F. The dose-dependent response is evident with decreased pathology compared to controls (14.41 ± 0.6138) in the low (11.50 ± 0.4564, P = 0.0019) and medium (7.412 ± 1.536, P < 0.0001) groups, and a near absence of pathological indicators in the high (2.000 ± 0.000, P < 0.0001) dose group. The attenuation of tissue histopathology provides clear evidence in a highly relevant disease model that the targeting of VEGF164 is cytoprotective during T-cell-mediated colitis.
We then examined changes in pathological angiogenesis and vascular density in the aptamer treated groups using the angiogenic index (AI) analysis. Figure 4 shows sample images from untreated (A), low dose (B), medium dose (C), and high dose (D), VEGF164 aptamer treatment cohorts of CD4+ colitis stained for PECAM-1 (red) and DAPI nuclear counterstain. PECAM-1 staining showed a trend for reduced vascular density in the low-dose group (0.2043 ± 0.2080, P = 0.1355), and significantly attenuated vascular density in the medium (0.1700 ± 0.03215, P = 0.0012) and high (0.06067 ± 0.006434, P < 0.0001) dose aptamer-treated groups versus control (0.2445 ± 0.01507) (Fig. 4E). These data clearly demonstrate that the isoform-specific inhibition of VEGF164 results in attenuation of CD4+ experimental colitis, and that high doses of the aptamer largely inhibit development of disease.
Figure 4.
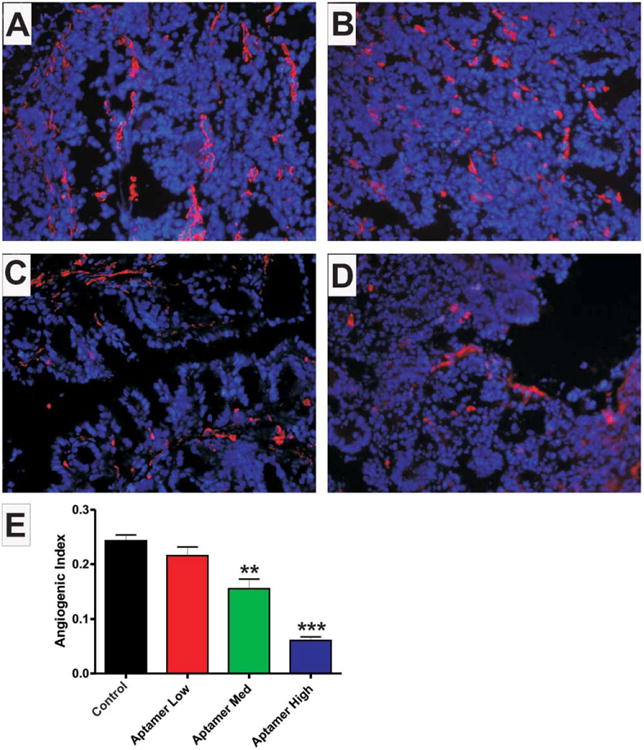
The VEGF164 aptamer dose-dependently prevents increases in vascular density associated with CD4+ experimental colitis. This figure shows sample images of PECAM-1 (red) staining with DAPI nuclear counterstain (blue) of colonic cross-sections from Control colitic (A), Aptamer Low (B), Aptamer Med (C), and Aptamer High (D) dose groups. (E) The angiogenic indexes for these groups, Control colitic (black), Aptamer Low (red), Aptamer Med (green), and Aptamer High (blue). N = 28 for Control, N = 14 for Aptamer Low, N = 18 for Aptamer Med, N = 18 for Aptamer High.
Heat-denatured Aptamer Does Not Inhibit T-cell-mediated Colitis
Having shown that the VEGF164 aptamer is able to attenuate CD4+ colitis in mice, we next heat-denatured the aptamer (HD aptamer) and administered it at the medium dose (80 mg/kg/day) as a control experiment. Figure 5A,B show weight loss and survival data for control and HD aptamer-treated groups, respectively. Histopathology scores (15.30 ± 0.3958, control; 16.12 ± 0.2241, HD aptamer; P = 0.0703) and AI analysis (0.2400 ± 0.01281, control; 0.2287 ± 0.02451, HD aptamer; P = 0.6636), are shown in Figure 5C,D, respectively. Figure 5E shows VEGF164 ELISA data for the control (1366 ± 50.45 pg/μL) and HD aptamer (1307 ± 91.21 pg/μL) treated groups (P = 0.3666). These data confirm the efficacy of the aptamer as a specific VEGF164 inhibitor and effective therapy for colitis.
Figure 5.
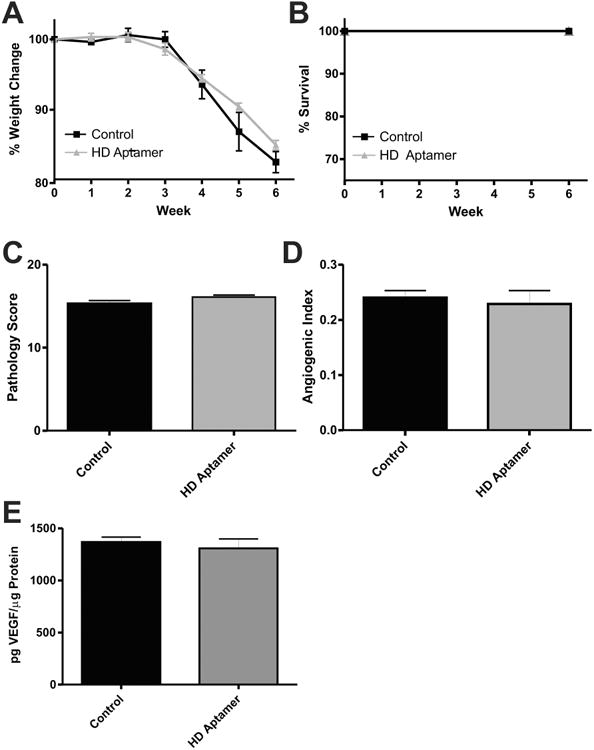
Heat-denatured aptamer fails to prevent disease pathology and increases in vascular density. (A) Weight loss data for control (black) and HD aptamer (gray) treated groups. (B) Survival data for the same. The histopathology scores and angiogenic indexes for these groups are shown in (C,D), respectively. VEGF164 ELISA data is shown in (E). N = 8 for control and HD aptamer groups, N = 5 for ELISA data.
As a further control we performed an in vitro Transwell cell migration assay to verify that the aptamer was efficiently binding and inhibiting VEGF164. Mouse recombinant VEGF164 treatment (50 ng/μL) resulted in significantly increased transmigration of colonic endothelial cells as measured by relative fluorescence intensity (40,024 ± 2708), 6.27-fold increase versus control (nonlabeled) (6379 ± 210.6, P = 0.0005) and 2.61-fold increase versus untreated (CellTracker green labeled, nonstimulated) (15,312 ± 205.5, P = 0.0119) mouse colonic endothelial cells (Supporting Information Fig. 1). Stimulation with VEGF164 and coincubation with the aptamer at doses used in AMD therapy (patient dose 0.3 mM) (26,483 ± 5134, P = 0.1613, 1.5-fold decrease), and our low (1 mM) (24,700 ± 1608; P = 0.0132, 1.6-fold decrease), medium, 2 mM (21,503 ± 1928; P = 0.0051, 1.86-fold decrease), and high, 4 mM (18,179 ± 2406; P = 0.0053, 2.20-fold decrease) doses resulted in a stepwise reduction in endothelial transmigration versus the VEGF164-treated group (ANOVA, P = 0.0003) (Supporting Information Fig. 1). Together, these data verify the activity of the aptamer used in these experiments and highlight that the VEGF164 aptamer dose-dependently attenuates CD4+ colitis in mice.
Overexpression of VEGF-A in CD4+ Colitis
Having shown that pharmacologic inhibition of all VEGF-A isoforms was not beneficial in the previous experiment, we next tested whether or not VEGF164 over-expression had an effect on disease. Here we used VEGF164 adenovirus or β-gal control adenovirus treatment in the CD4+ model of colitis. Figure 6A reports the observed weight change during the experimental period; a transient separation was observed at week 5 with greater weight loss in the Ad.β-gal (86.88 ± 1.945) than the Ad.VEGF-treated group (94.90 ± 1.672) (P = 0.0198), but no difference was present at the end of the experimental period. Survival data for both groups is reported in Figure 6B. Gross scores from the colons of both groups are shown in Figure 6C; no difference was observed between these groups (2.400 ± 0.2449, Ad.β-gal versus 2.400 ± 0.4000, Ad.VEGF; P = 1.000). Supporting Information Figure 2A–C illustrates sample H&E-stained images of normal, Ad.β-gal, and Ad.VEGF-treated colon cross-sections, respectively. Histopathological analysis of colon sections showed no difference in score between groups (13.30 ± 1.100, Ad.β-gal versus 12.70 ± 0.9000, Ad.VEGF; P = 0.7140) (Fig. 6D). We next examined the colons of these mice for differences in vascular density. Supporting Information Figure 2 contains sample images of normal (D), Ad.β-gal (E), and Ad.VEGF (F) treated colons, respectively, stained for PECAM-1 (red) and DAPI nuclear counterstain (blue). AI analysis showed that both groups have increased vascular density compared to normal colons (0.2375 6 0.03259, Ad.β-gal, and 0.2258 ± 0.01877, Ad.VEGF). Importantly, VEGF164 ELISA revealed increased levels of VEGF164 in the Ad.β-gal group (3628 ± 549.8 pg/μL) and an even larger increase in VEGF164 protein in Ad.VEGF-treated colon tissue (8540 6 321.3 pg/μL), indicating the effectiveness of the adenovirus (P = 0.0007) (Fig. 6F). These data suggest that induction of VEGF164 normally observed during the development of CD4+ colitis is sufficient to develop robust tissue damage and vascular density, and that further increasing the VEGF164 concentration has little additional effect on T-cell-mediated colitis.
Figure 6.
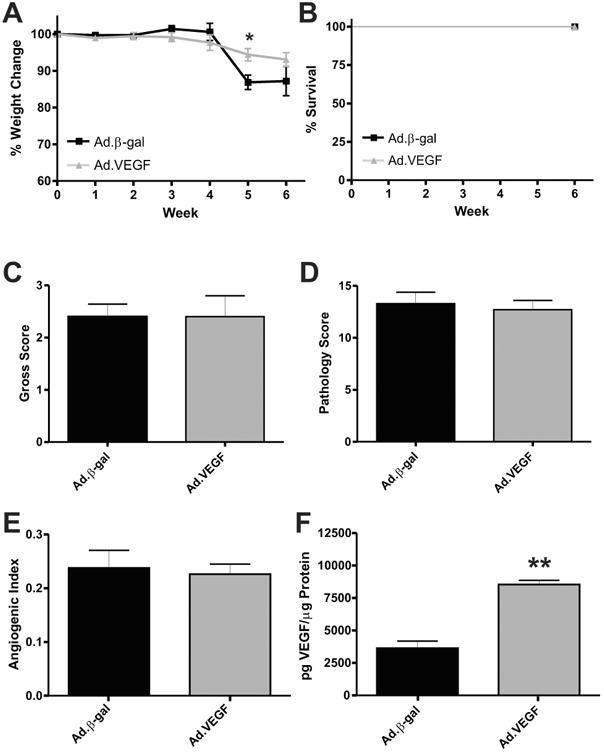
Adenoviral overexpression of VEGF164 results in maximal development of CD4+ experimental colitis. (A) Weight loss data for Ad.β-gal (black) and Ad.VEGF164 (gray) treated mice. (B) Survival data for the same. The gross scores and histopathological scores for these groups are reported in (C,D), respectively. Angiogenic Index data for the same are shown in (E). (F) The VEGF164 ELISA data for these groups. N = 8 for Ad.β-gal, N = 9 for Ad.VEGF, N = 5 for ELISA data.
Inhibition of All VEGF-A Isoforms in CD4+ Colitis
As noted previously, the CD4+ model exhibited rampant upregulation of proangiogenic factors, including VEGF-A, similar to what is seen in human disease, with some downregulation of antiangiogenic mediators.5 Lastly, we examined whether inhibition of all VEGF-A isoforms using the VEGF Trap altered the development of CD4+ T-cell-dependent colitis. VEGF Trap and human Fc (control) treated groups were used in this experiment. Importantly, neither VEGF Trap nor Fc control significantly altered weight loss; however, a significant decrease in survival occurred in the VEGF Trap-treated group (Supporting Information Fig. 3A,B, respectively). Gross scores of the colons from the human Fc (3.000 ± 0.5164) and VEGF Trap (2.429 ± 0.4266) (P = 0.4085) were not significantly different (Supporting Information Fig. 3C). Moreover, histo-pathological scores of diseased colons between the human Fc (11.76 6 2.240) and VEGF Trap (15.94 6 2.399) treated groups showed no significant difference (P = 0.2114), with both groups receiving high scores (Supporting Information Fig. 3D). Sample images of H&E-stained colon cross-sections from human Fc and VEGF Trap-treated experimental groups revealing significant pathology in both groups are shown in Supporting Information Figure 3E,F, respectively. Surprisingly, angiogenic index analysis of vascular density showed that both groups have similar vascular density levels (0.2325 ± 0.04502, human Fc versus 0.1988 ± 0.01794, VEGF Trap; P = 0.7593) (Supporting Information Fig. 4A). Sample images of human Fc and VEGF Trap-treated colons, stained for PECAM-1 (red) and DAPI nuclear counterstain (blue), are shown in Supporting Information Figure 4B,C, respectively. Conversely, VEGF-A ELISA data revealed a reduced level of VEGF-A protein in tissues from VEGF Trap-treated mice (704.0 ± 152.0 pg/μL) versus human Fc treated mice (2874 ± 539.8 pg/μL), verifying the potency of the VEGF Trap (P = 0.0083) (Supporting Information Fig. 4D). Both groups in this experiment exhibited significant development of disease, suggesting that inhibition of all VEGF-A isoforms may not be beneficial during experimental colitis. These data combined with the VEGF164 aptamer data support the hypothesis that VEGF120 and 188 may act in a physiological/wound healing manner during disease, while VEGF164 drives pathological angiogenesis.
Discussion
While the etiology of IBD remains largely unknown, the pathological basis of disease progression is widely studied and involves both innate mucosal and acquired immune responses. However, work over the past decade has revealed that the colon microvasculature plays an important role in controlling disease progression and severity through angiogenesis and leukocyte recruitment processes.5,6,29,31 As such, several clinical and experimental studies have shown increased VEGF-A levels in the serum and colon tissue during colitis.12–14,29,32 Increased VEGF-A expression could emanate from immune cells including neutro-phils, monocytes, T cells, and platelets, as well as produced locally by endothelial cells, epithelial cells, and other cell types. Our findings are the first to unambiguously document specific tissue niches of VEGF-A expression during T-cell colitis implicating infiltrating leukocytes as well as local colon tissue production of angiogenic cytokines during disease. These data also provide additional explanation for the abundant amount of VEGF-A protein expression in situ during the T-cell transfer model,5 indicating that VEGF-A expression must occur from multiple cell sources to result in high tissue levels seen during T-cell-mediated colitis.
Our current findings clearly show that VEGF164 plays an important role in the process of pathological angiogenesis occurring during colitis. We have previously shown that this isoform drives inflammatory responses of colonic endothelium that contribute to colitis.33 Importantly, VEGF164 has been repeatedly indicated as the pathological isoform of VEGF-A due to its role in multiple disease pathologies.34 Moreover, inhibition of angiogenesis attenuates inflammation and disease development in different models of colitis, and the reduced inflammatory capacity of CD18−/− mice results in decreased vascular density and attenuated disease.5 Gene array analysis of both the CD4+ T cell and DSS models of experimental colitis, and data from clinical studies shows that during disease there is a prominent upregulation of multiple proangiogenic factors such as VEGF, bFGF, TGF-β, TNF-α, and others which could outweigh the biological effects of antiangiogenic factors which are found at lower levels during disease.5 However, the fact that selective inhibition of VEGF164 conferred protection against disease in a dose-dependent manner surprisingly demonstrates that this angiogenic factor serves as an important intermediary between angiogenesis and inflammation despite upregulation of other cytokines that could engage in similar processes. This relationship between angiogenesis and inflammation suggests that the ideal targets for therapy may be those factors that are closely tied to both events, such as VEGF164.35,36
In the VEGF164 adenovirus experiments, we showed that overexpression of VEGF164 did not affect the disease process. This is important because were VEGF164 to act in a protective and wound healing manner, its overexpression should protect against disease; however, this was not the case. Moreover, these data suggest that during T-cell-mediated colitis, VEGF164 is produced at elevated levels that are above maximal pathologic VEGF164-dependent effects, as tissue histopathology does not go beyond previously documented severity levels, even with a greater overexpression of VEGF-A above disease levels. Another possible explanation for our findings could be that such significant overexpression of VEGF164 downregulates VEGFRs or VEGF coreceptors (Nrp-1), which would diminish further induction of angiogenesis and inflammation. Whichever the explanation may be, our data demonstrate that VEGF164 overexpression does not significantly alter chronic T-cell-mediated colitis.
However, pharmacological inhibition of the VEGF164 isoform using a specific RNA aptamer (Macugen) did confer significant protection against T-cell-mediated colitis in a dose-dependent manner. We found that a low dose (40 mg/kg/day) of the aptamer was unable to drastically alter the disease phenotype. This was not entirely unexpected, since Macugen is used for the treatment of AMD, where it is injected and retained in a localized intraocular environment, thereby necessitating lesser amounts of the drug for efficacy. However, when treating a large and less confined organ with greater surface area and ability to produce VEGF164 (e.g., the colon), it seems logical that larger drug doses may be necessary to effectively neutralize elevated tissue VEGF164 protein levels. As we increased the dose of the aptamer we observed a clear, dose-dependent therapeutic response, resulting in near total attenuation of disease and significant reduction of tissue VEGF164 protein levels at the highest dose. These data support the hypothesis that VEGF164 is the dominant pathological isoform of VEGF-A during T-cell-mediated colitis.34 It is likely that the loss of VEGF164 affects the progression of inflammation through altered regulation of immune cell recruitment, as previously reported.33 Moreover, the known roles of the different VEGF family members (i.e., VEGF-B) during physiological angiogenesis and other functions of VEGF-A isoforms (120 and 188) are consistent with the hypothesis that specific inhibition of VEGF164 could be very beneficial during disease.
Data from our VEGF Trap (Aflibercept) studies, which binds all isoforms of VEGF-A, VEGF-B, and PlGF, showed no protection against disease, with a surprising increase in mortality. While at first this may seem contrary to potential pathological roles of VEGF-A in disease, upon further consideration this result is not unexpected. VEGF-B, has been shown to have a “decoy effect” when interacting with VEGFR1 to diminish VEGF-A binding to and signaling by VEGFR2, the major ligand for VEGF164, which may partly explain the effects of VEGF Trap therapy in the T-cell colitis model.17 If, as the literature suggests, various VEGF-A isoforms are important for tissue healing, then broad-scale isoform inhibition would not be protective, which agrees with our data.37,38 It has also recently been reported that genetic deficiency of PlGF expression exacerbates DSS-induced mucosal hypoxia and aggravates colon injury, which could be due to a possible protective role in regulating vascular growth which could also occur in the T-cell colitis model.39
Results from our current study significantly advance our understanding of VEGF-A during experimental colitis as initially reported by Scaldaferri et al36 showing that VEGF-A inhibition with soluble Flt-1 (VEGFR1) adenovirus attenuates DSS-mediated colitis. Several important differences between our studies include: 1) soluble Flt-1 confers limited VEGF protein binding (i.e., all VEGF-A isoforms), whereas the VEGF164 aptamer only binds this isoform and the VEGF Trap (containing binding regions of VEGFR1 and VEGFR2) binds VEGF-A, VEGF-B, and PlGF; 2) different disease mechanisms are involved between the DSS and CD4+ T-cell transfer models of colitis that could influence VEGF isoform function and biology; and 3) our study employed approved pharmacological agents to antagonize VEGF function in the Rag-1−/− C57BL/6J mouse strain versus adenovirus-based approaches in C57BL/6N mouse strain which may involve strain-dependent responses. It is important to emphasize that inhibition of multiple VEGF family proteins may not be protective due to the broad range of actions they are able to regulate. Numerous clinical side effects have been noted with broad-scale anti-VEGF therapy including disturbances of cardiovascular (thrombosis, hemorrhage, and hypertension) and renal (proteinuria and renal dysfunction) systems, and in wound healing.40,41 Nonetheless, it is clear that VEGF164 isoform specific inhibition is therapeutically beneficial, whereas VEGF Trap therapy does not confer protection against T-cell-mediated colitis and actually increases disease mortality. These results demonstrate that proper consideration and thought must be given to any potential antiangiogenic therapy for colitis.
The profound effect of VEGF164 inhibition is of critical importance in defining key physiological links between pathological angiogenesis and chronic inflammation during T-cell-mediated colitis.5,33 The complexity of angiogenic regulation during colitis is only beginning to be elucidated and we report that VEGF-A expression occurs from several different cell types and that VEGF164 plays a prominent pathophysiological role during T-cell-mediated colitis. While the VEGF164 aptamer doses required to treat experimental colitis are clearly cost-limiting in human disease, our findings represent a major step forward in understanding potential antiangiogenic targets for novel IBD therapies.
Supplementary Material
Acknowledgments
Supported by NIH grant P01 DK043785-18 Project 4, and Animal models and Histopathology cores.
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Warren S, Sommers SC. Pathology of regional ileitis and ulcerative colitis. JAMA. 1954;154:189–193. doi: 10.1001/jama.1954.02940370001001. [DOI] [PubMed] [Google Scholar]
- 2.Reifferscheid M, Wolfram E. Resection or by-pass anastomosis in regional enteritis? Chirurg. 1962;33:164–172. [PubMed] [Google Scholar]
- 3.Bacaner MB. Quantitative measurement of regional colon blood flow in the normal and pathological human bowel. Gastroenterology. 1966;51:764–777. [PubMed] [Google Scholar]
- 4.Knutson H, Lunderquist A, Lunderquist A. Vascular changes in Crohn's disease. Am J Roentgenol Radium Ther Nucl Med. 1968;103:380–385. doi: 10.2214/ajr.103.2.380. [DOI] [PubMed] [Google Scholar]
- 5.Chidlow JH, Jr, Langston W, Greer JJ, et al. Differential angiogenic regulation of experimental colitis. Am J Pathol. 2006;169:2014–2030. doi: 10.2353/ajpath.2006.051021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danese S, Sans M, de la Motte C, et al. Angiogenesis as a novel component of inflammatory bowel disease pathogenesis. Gastroenterology. 2006;130:2060–2073. doi: 10.1053/j.gastro.2006.03.054. [DOI] [PubMed] [Google Scholar]
- 7.Brahme F, Lindstrom C. A comparative radiographic and pathological study of intestinal vaso-architecture in Crohn's disease and in ulcerative colitis. Gut. 1970;11:928–940. doi: 10.1136/gut.11.11.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hatoum OA, Heidemann J, Binion DG. The intestinal microvascula-ture as a therapeutic target in inflammatory bowel disease. Ann N Y Acad Sci. 2006;1072:78–97. doi: 10.1196/annals.1326.003. [DOI] [PubMed] [Google Scholar]
- 9.Laroux FS, Grisham MB. Immunological basis of inflammatory bowel disease: role of the microcirculation. Microcirculation. 2001;8:283–301. doi: 10.1038/sj/mn/7800095. [DOI] [PubMed] [Google Scholar]
- 10.Fishman SJ, Feins NR, Folkman J. Long-term remission of Crohn's disease treated with thalidomide: a seminal case report. Angiogenesis. 1999;3:201–204. doi: 10.1023/a:1009027315912. [DOI] [PubMed] [Google Scholar]
- 11.Bariol C, Meagher AP, Vickers CR, et al. Early studies on the safety and efficacy of thalidomide for symptomatic inflammatory bowel disease. J Gastroenterol Hepatol. 2002;17:135–139. doi: 10.1046/j.1440-1746.2002.02564.x. [DOI] [PubMed] [Google Scholar]
- 12.Beck PL, Podolsky DK. Growth factors in inflammatory bowel disease. Inflamm Bowel Dis. 1999;5:44–60. doi: 10.1097/00054725-199902000-00007. [DOI] [PubMed] [Google Scholar]
- 13.Kanazawa S, Tsunoda T, Onuma E, et al. VEGF, basic-FGF, and TGF-beta in Crohn's disease and ulcerative colitis: a novel mechanism of chronic intestinal inflammation. Am J Gastroenterol. 2001;96:822–828. doi: 10.1111/j.1572-0241.2001.03527.x. [DOI] [PubMed] [Google Scholar]
- 14.Kapsoritakis A, Sfiridaki A, Maltezos E, et al. Vascular endothelial growth factor in inflammatory bowel disease. Int J Colorectal Dis. 2003;18:418–422. doi: 10.1007/s00384-003-0495-y. [DOI] [PubMed] [Google Scholar]
- 15.Sunderkotter C, Steinbrink K, Goebeler M, et al. Macrophages and angiogenesis. J Leukoc Biol. 1994;55:410–422. doi: 10.1002/jlb.55.3.410. [DOI] [PubMed] [Google Scholar]
- 16.Griga T, Werner S, Koller M, et al. Vascular endothelial growth factor (VEGF) in Crohn's disease: increased production by peripheral blood mononuclear cells and decreased VEGF165 labeling of peripheral CD14+ monocytes. Dig Dis Sci. 1999;44:1196–1201. doi: 10.1023/a:1026640610621. [DOI] [PubMed] [Google Scholar]
- 17.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 18.Kusumanto YH, Dam WA, Hospers GA, et al. Platelets and granulocytes, in particular the neutrophils, form important compartments for circulating vascular endothelial growth factor. Angiogenesis. 2003;6:283–287. doi: 10.1023/B:AGEN.0000029415.62384.ba. [DOI] [PubMed] [Google Scholar]
- 19.Ng YS, Rohan R, Sunday ME, et al. Differential expression of VEGF isoforms in mouse during development and in the adult. Dev Dyn. 2001;220:112–121. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1093>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 20.Breier G, Albrecht U, Sterrer S, et al. Expression of vascular endothe-lial growth factor during embryonic angiogenesis and endothelial cell differentiation. Development. 1992;114:521–532. doi: 10.1242/dev.114.2.521. [DOI] [PubMed] [Google Scholar]
- 21.Maharaj AS, Saint-Geniez M, Maldonado AE, et al. Vascular endothelial growth factor localization in the adult. Am J Pathol. 2006;168:639–648. doi: 10.2353/ajpath.2006.050834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 23.Ferrara N, Hillan KJ, Gerber HP, et al. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. doi: 10.1038/nrd1381. [DOI] [PubMed] [Google Scholar]
- 24.Fukasawa M, Korc M. Vascular endothelial growth factor-trap suppresses tumorigenicity of multiple pancreatic cancer cell lines. Clin Cancer Res. 2004;10:3327–3332. doi: 10.1158/1078-0432.CCR-03-0820. [DOI] [PubMed] [Google Scholar]
- 25.Gordon MS, Margolin K, Talpaz M, et al. Phase I safety and pharma-cokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. J Clin Oncol. 2001;19:843–850. doi: 10.1200/JCO.2001.19.3.843. [DOI] [PubMed] [Google Scholar]
- 26.Gragoudas ES, Adamis AP, Cunningham ET, Jr, et al. Pegaptanib for neovascular age-related macular degeneration. N Engl J Med. 2004;351:2805–2816. doi: 10.1056/NEJMoa042760. [DOI] [PubMed] [Google Scholar]
- 27.Lee JH, Canny MD, De Erkenez A, et al. A therapeutic aptamer inhibits angiogenesis by specifically targeting the heparin binding domain of VEGF165. Proc Natl Acad Sci U S A. 2005;102:18902–18907. doi: 10.1073/pnas.0509069102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Presta LG, Chen H, O'Connor SJ, et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593–4599. [PubMed] [Google Scholar]
- 29.Chidlow JH, Jr, Shukla D, Grisham MB, et al. Pathogenic angiogenesis in IBD and experimental colitis: new ideas and therapeutic avenues. Am J Physiol Gastrointest Liver Physiol. 2007;293:G5–G18. doi: 10.1152/ajpgi.00107.2007. [DOI] [PubMed] [Google Scholar]
- 30.Langston W, Chidlow JH, Jr, Booth BA, et al. Regulation of endothe-lial glutathione by ICAM-1 governs VEGF-A-mediated eNOS activity and angiogenesis. Free Radic Biol Med. 2007;42:720–729. doi: 10.1016/j.freeradbiomed.2006.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chidlow JH, Jr, Greer JJ, Anthoni C, et al. Endothelial caveolin-1 regulates pathologic angiogenesis in a mouse model of colitis. Gastroenterology. 2009;136:575–584 e2. doi: 10.1053/j.gastro.2008.10.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsiolakidou G, Koutroubakis IE, Tzardi M, et al. Increased expression of VEGF and CD146 in patients with inflammatory bowel disease. Dig Liver Dis. 2008;40:673–679. doi: 10.1016/j.dld.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 33.Goebel S, Huang M, Davis WC, et al. VEGF-A stimulation of leukocyte adhesion to colonic microvascular endothelium: implications for inflammatory bowel disease. Am J Physiol Gastrointest Liver Physiol. 2006;290:G648–654. doi: 10.1152/ajpgi.00466.2005. [DOI] [PubMed] [Google Scholar]
- 34.Usui T, Ishida S, Yamashiro K, et al. VEGF164(165) as the pathological isoform: differential leukocyte and endothelial responses through VEGFR1 and VEGFR2. Invest Ophthalmol Vis Sci. 2004;45:368–374. doi: 10.1167/iovs.03-0106. [DOI] [PubMed] [Google Scholar]
- 35.Mor F, Quintana FJ, Cohen IR. Angiogenesis-inflammation cross-talk: vascular endothelial growth factor is secreted by activated T cells and induces Th1 polarization. J Immunol. 2004;172:4618–4623. doi: 10.4049/jimmunol.172.7.4618. [DOI] [PubMed] [Google Scholar]
- 36.Scaldaferri F, Vetrano S, Sans M, et al. VEGF-A links angiogenesis and inflammation in inflammatory bowel disease pathogenesis. Gastroenterology. 2009;136:585–595 e5. doi: 10.1053/j.gastro.2008.09.064. [DOI] [PubMed] [Google Scholar]
- 37.Sandor Z, Deng XM, Khomenko T, et al. Altered angiogenic balance in ulcerative colitis: a key to impaired healing? Biochem Biophys Res Commun. 2006;350:147–150. doi: 10.1016/j.bbrc.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 38.Jones MK, Tomikawa M, Mohajer B, et al. Gastrointestinal mucosal regeneration: role of growth factors. Front Biosci. 1999;4:D303–309. doi: 10.2741/a428. [DOI] [PubMed] [Google Scholar]
- 39.Hindryckx P, Waeytens A, Laukens D, et al. Absence of placental growth factor blocks dextran sodium sulfate-induced colonic mucosal angiogenesis, increases mucosal hypoxia and aggrevates acute colonic injury. Lab Invest. 2010;90:566–576. doi: 10.1038/labinvest.2010.37. [DOI] [PubMed] [Google Scholar]
- 40.Chen HX, Cleck JN. Adverse effects of anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol. 2009;6:465–477. doi: 10.1038/nrclinonc.2009.94. [DOI] [PubMed] [Google Scholar]
- 41.Tunon J, Ruiz-Moreno JM, Martin-Ventura JL, et al. Cardiovascular risk and antiangiogenic therapy for age-related macular degeneration. Surv Ophthalmol. 2009;54:339–348. doi: 10.1016/j.survophthal.2009.02.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.