

Abstract
p38 mitogen-activated protein kinase (MAPK) stabilises mRNAs of pro-inflammatory mediators by inhibiting AU-rich element-mediated decay. We show that in bone marrow-derived macrophages (BMDM) tumour necrosis factor (TNF) mRNA is stabilised by inactivation of the AU-rich element-binding protein tristetraprolin (TTP) by the p38 MAPK pathway. We show that this mechanism is not limited to TNF mRNA and that p38 MAPK also inhibits TTP-directed decay of interleukin (IL)-10 mRNA. Lipopolysaccharide-induced IL-10 mRNA and protein expression was increased in TTP−/− compared with wild-type BMDM. We also provide evidence that TTP directly targets IL-10 mRNA by binding to its 3’-untranslated region. We found that p38 MAPK activation also prevents TTP-directed decay of cyclooxygenase-2, IL-6, and IL-1α mRNAs. However, despite destabilisation of IL-6 mRNA by TTP, IL-6 mRNA and protein expression was reduced in TTP−/− BMDM and IL-12p40 mRNA and protein expression was also reduced in TTP−/− BMDM. Neutralisation of IL-10 in lipopolysaccharide-treated BMDM with an anti-IL-10 antibody rescued the inhibition of IL-6 and IL-12p40 protein in TTP−/− cells. Thus the p38 MAPK pathway acting in conjunction with TTP regulates the stability and expression of not only pro-inflammatory mRNAs but also the mRNA of the anti-inflammatory cytokine, IL-10.
INTRODUCTION
It is now recognised that in addition to transcriptional control, post-transcriptional mechanisms play a key role in regulating gene expression. The control of mRNA stability is of particular importance for the expression of proteins of the inflammatory response. A large number of mRNAs of these proteins contain AU-rich elements (ARE) in their 3’-untranslated regions (UTR). These elements target mRNAs for rapid decay. The decay of ARE-containing reporter mRNAs is blocked following the activation of p38 mitogen-activated protein kinase (MAPK) (1,2). Many ARE-containing endogenous mRNAs of proteins of the inflammatory response are destabilised upon inhibition of this protein kinase (3–9). Indeed, in a microarray study in THP-1 cells, the stability of 42 different ARE-containing mRNAs was found to be regulated by p38 MAPK (10).
Tristetraprolin (TTP) has long been known to regulate the expression of tumor necrosis factor (TNF) by binding to the ARE in the 3’UTR of TNF mRNA and targeting it for degradation (11). TTP knockout mice develop a complex inflammatory phenotype and display an inflammatory arthritis, cachexia, conjunctivitis and myeloid hyperplasia (12). Treatment of TTP−/− mice with anti-TNF antibody prevented almost all aspects of the phenotype (12) suggesting that TTP might specifically target TNF. Initial studies of the role of TTP in TNF production were performed in macrophages. Since then, TTP deficiency has been found to affect granulocyte-macrophage colony-stimulating factor (13), interleukin (IL)-2 (14) and immediate-early response gene 3 (15) mRNA stability in bone marrow stromal cells, T cells and embryonic fibroblasts respectively. The minimal ARE sequence required for TTP binding on the RNA has been previously shown to be only nine (16,17) or seven (18) nucleotides long, suggesting that there are additional potential TTP targets that remain to be identified.
The stabilisation of inflammatory mediator ARE-containing mRNAs by p38 MAPK involves the downstream kinase, MAPK-activated protein kinase (MK2) (1,2,19). TTP is phosphorylated by MK2 (20) on Ser 52 and Ser 178 (21). Mutation of these two phosphorylation sites to alanine prevented MK2-mediated stabilisation of a TNF reporter mRNA (22). Previous work has shown that lipopolysaccharide (LPS)-induced TNF protein production by bone marrow-derived macrophages (BMDM) from TTP−/− mice was insensitive to p38 MAPK inhibition (23) although others failed to observe such an effect (24). A more recent study in mice deficient in both TTP and MK2 showed that TNF mRNA is stable in TTP−/−MK2−/− macrophages consistent with the involvement of TTP in p38 MAPK/MK2-mediated mRNA stabilisation (25). Definitive evidence in support of this model has been lacking, although, TTP does destabilise ARE-containing mRNAs by promoting their deadenylation (13,26,27), and p38 MAPK stabilises these transcripts by inhibiting their deadenylation (28). To date, only TNF mRNA has been shown to be regulated by a mechanism that involves both TTP and p38 MAPK, and it is unclear how p38 MAPK stabilises other mRNAs.
Blockade of p38 MAPK activity in human monocytes (29) and murine BMDM (30) inhibits IL-10 expression. p38 MAPK induces IL-10 by activating its transcription (31). The human IL-10 promoter contains a binding site for the transcription factor, Sp1 and p38 MAPK has been suggested to induce IL-10 by activating Sp1-dependent transcription (31). To our knowledge, post-transcriptional regulation of IL-10 mRNA by p38 MAPK and TTP has not been examined previously.
We now provide definitive evidence that p38 MAPK inhibits TTP-directed decay of TNF mRNA in BMDM. p38 MAPK, TTP and MK2 represent important targets for therapies aimed at treating chronic inflammatory diseases such as rheumatoid arthritis. Ideally such therapies would specifically block the expression of pro-inflammatory mediators, such as TNF, whilst sparing anti-inflammatory IL-10. We have investigated whether the same post-transcriptional mechanism that regulates TNF mRNA also controls the expression of other mRNAs of the inflammatory response, including that for the anti-inflammatory cytokine IL-10.
EXPERIMENTAL PROCEDURES
Materials
4-(4-Fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole (SB 202190) was from Calbiochem-Novabiochem. We found that, in contrast to when added after LPS treatment, pre-treatment of BMDM with inhibitor prior to LPS addition resulted in strong suppression of MK2 activity only at concentrations ≥5 μM (Supplementary Fig. S1). When added 4h post-LPS (as in the case of actinomycin D chases) a concentration of 1 μM of SB202190 was sufficient to completely block LPS-induced MK2 activity (Supplementary Fig. S1). Therefore for pre-treatment experiments it was decided to use SB 202190 at a concentration of 5 μM. LPS from Salmonella typhimurium was from Sigma-Aldrich. [α-32P]-UTP was purchased from GE Healthcare. A hamster monoclonal antibody against murine TNF (TN3-19.12) (32) was used to neutralise TNF at a final concentration of 10 μg ml−1 (generous gift from Robert Schreiber, Washington University School of Medicine, St. Louis, MO). Anti-IL-10 neutralising antibody (R & D Systems) was used at 20 μg ml−1. Anti-COX-2 antibody was from Alexis and anti-α-tubulin antibody was from Sigma-Aldrich. The rabbit antiserum against the C-terminus of murine TTP has been described previously (20).
Plasmids
Antisense murine TNF 3’UTR riboprobe template plasmid was generated by PCR amplification of a 250-bp sequence spanning nt 1359–1609 of the 3’UTR of TNF which was cloned into pCR-Blunt (Invitrogen). Antisense murine COX-2 riboprobe template plasmid, was prepared by PCR of cDNA prepared by reverse transcription of RNA from LPS-treated BMDM. A 317-bp COX-2 fragment (nt 1522-1838) was then amplified by PCR. The 317-bp COX-2 PCR product was then cloned in a pCR®II-TOPO® vector (Invitrogen) using a “TOPO TA Cloning ® Kit” (Invitrogen) according to the manufacturer’s protocol. A plasmid for in vitro transcription of nucleotides 613-1295 (from the translation stop codon to the polyadenylation signal) of the murine IL-10 3’UTR was prepared in a similar fashion. Primer sequences are available upon request.
Mice
TTP−/− mice were originally generated as described previously (12) by insertion of a neomycin-resistance cassette into the TTP coding region of the exon 2 of the TTP gene. Gene disruption results in the expression of a TTP-neo fusion mRNA without the subsequent expression of TTP protein. TTP−/− mice were of mixed 129 and C57BL/6 background as originally obtained from Perry Blackshear. All animal experiments were performed according to ethical procedures.
Bone marrow-derived macrophage (BMDM) preparation
Mice (four to eight weeks old) were humanely culled and bone marrow was flushed from femurs and tibiae. Bone marrow cells were resuspended in 15 % DMSO in foetal calf serum (FCS) (v/v) and stored on dry ice for 24 to 48h. Cells were thawed at 37 °C and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 100 units ml−1 penicillin, 100 pg ml−1 streptomycin, 10% (v/v) FCS and 10% (v/v) L929-cell conditioned medium (as a source of macrophage colony stimulating factor) in bacteriological Petri dishes in 5% CO2 at 37 °C for 6 days. Cells were then trypsinised, scraped and seeded in 6 well plates at a density of 3 × 106 cells/well in DMEM supplemented with 100 units ml−1 penicillin, 100 pg ml−1 streptomycin, 10% (v/v) FCS and 10 ng ml−1 macrophage colony stimulating factor (PeproTech). Graphs in figures 1–3 show mean data from one experiment using BMDM from TTP−/− mice and wild-type littermates and two experiments using C57BL/6 mice as controls. Similar data were obtained using littermate or C57BL/6 controls. All other experiments were performed using wild-type littermates as controls.
FIGURE 1. Regulation of TNF mRNA stability by p38 MAPK requires TTP.

Wild-type (A) or TTP−/− (B) BMDM were left untreated or treated with LPS (10 ng ml−1) for 4h and then actinomycin D (10 μg ml−1) was added together with SB 202190 (final concentration 1 μM) or vehicle (0.1% DMSO). Cells were harvested at the times shown and RNA was extracted and northern blotted with an antisense riboprobe to TNF 3’UTR. Graphs show mean TNF mRNA in wild-type (C) or TTP−/− cells (D) normalised to GAPDH mRNA expressed as a percentage of t=0 actinomycin D treatment ± S.E.M. from three independent experiments. Where not shown error bars are smaller than the symbols. Significance of the effect of SB 202190 was measured by paired Student’s t-test *p<0.01.
FIGURE 3. IL-10 mRNA and protein expression is increased in TTP−/− BMDM.

A) Wild-type and TTP−/− BMDM were left untreated or pretreated for 30 min with anti-TNF neutralising antibody TN3-19.12 (10 μg ml−1) and then treated with 5 μM SB 202190 or vehicle control (0.1% DMSO) for 1h, followed by LPS treatment for a further 4h. Cells were then harvested, lysed and RNA was isolated. IL-10 and GAPDH mRNAs were analysed by quantitative-real-time PCR. Graph shows amount of IL-10 mRNA normalised to GAPDH calculated using the ΔΔCt method of analysis and expressed relative to that for untreated wild-type cells. Similar results were obtained in two separate experiments. B) IL-10 protein in culture medium from the cells (described in A) was measured by ELISA. Similar results were found in two independent experiments.
RAW 264.7 cell culture
RAW 264.7 cells were cultured in DMEM supplemented with 10% (v/v) FCS in a humidified atmosphere of at 37 °C. 5% CO2
RNA analysis
Total RNA was isolated from BMDM using a QIAamp RNA Blood Mini kit (Qiagen) according to the manufacturer’s instructions. TNF, COX-2 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNAs were analysed by northern blotting. Typically 5 μg of RNA was electrophoresed on denaturing formaldehyde/ 2% agarose gels. RNA was capillary-transferred onto Hybond XL membranes (GE Healthcare) and fixed by UV cross-linking. Membranes were prehybridised for 2h and hybridised overnight with [α-32P]-UTP-labeled riboprobes at 65 °C in Ultrahyb (Ambion). Blots were then washed 3 times for 30 min at 65 °C with 5 × SSC/0.1% SDS, 1 × SSC/0.1% SDS, and 0.1 × SSC/0.1% SDS. Signals were visualised and quantified using a phosphorimager (Fuji FLA-2000). [α-32P]-UTP-labeled murine TNF, COX-2 and GAPDH riboprobes used for northern blotting were prepared from template plasmids by in vitro transcription as described previously (28).
Some mRNAs were quantified using the Riboquant multiprobe RPA kit (BD Biosciences) according to the manufacturer’s instructions. Probes were synthesised using the mCK-2b template set by in vitro transcription (as described in (28) except that the concentration of unlabelled UTP was 2.4 μM). Typically 400,000 cpm was used per point. Protected RNA fragments were resolved by electrophoresis on denaturing 4% polyacrylamide gels and visualised and quantified by phosphorimaging.
IL-10 and GAPDH mRNAs were measured by quantitative RT-PCR. For this cDNA was generated using the Reverse Transcription System kit (Promega) and random hexamers (Promega) and 0.5 μg of total RNA per reaction. 4 % of this cDNA was measured by quantitative RT-PCR using TaqMan technology and prevalidated primer-probe sets for IL-10 (Mm 00439616_m1) and GAPDH (Mm 9999915_g1) purchased from Applied Biosystems. A Rotor-Gene 6000 thermal cycler (Corbett Research) was used. The ΔΔCt method of relative quantitation and the Rotor-Gene 6000 software (Corbett Research) was used to calculate relative mRNA amounts for each of the transcripts examined.
Preparation of RAW 264.7 cell extracts
Confluent RAW 264.7 cells were rinsed and harvested in ice-cold PBS. Cells were pelleted by centrifugation at 500 × g for 5 min, resuspended in 1 ml of lysis buffer (10 mM HEPES pH 7.6, 3mM MgCl2, 40 mM KCl, 2mM DTT, 5% glycerol, 0.5% IGEPAL CA-630, 2mM NaF, 1mM sodium orthovanadate and protease inhibitors [1mM phenylmethylsulfonyl fluoride, 3 μg ml−1 aprotinin, 10 μM E64, 1 μg ml−1 pepstatin A and 10 μM mycrocystin]) and lysed for 10 min on ice. The lysate was centrifuged at 1500 x g for 5 min and the supernatant (cytoplasmic fraction) was collected, aliquoted and stored at −70 °C. Protein concentrations of lysates were estimated by Bradford assay.
Electrophoretic mobility shift assays (EMSA) and antibody supershifts
50 μg of RAW 264.7 cell extracts were incubated with a rabbit antiserum against TTP or a non-immune serum (rabbit anti-HSP27) for 30 min on ice followed by incubation with 500,000 cpm of [α-32P]UTP-labeled RNA probe and bandshift buffer (10 mM HEPES pH 7.6, 3 mM MgCl2, 20 mM KCl, 1mM DTT, 5 % glycerol) for 15 min on ice. RNase T1 and heparin sulfate were added to final concentrations of 5000 U ml−1 and 5 mg ml−1, respectively, together with 3 μl loading buffer (80 % glycerol and 0.1 % bromophenol blue) and the reaction mixture was incubated for 15 min. Samples were electrophoresed (150 V for 3h at 4 °C) on 4 % non-denaturing polyacrylamide gels containing 0.5× Tris-borate EDTA and 2.5 % glycerol (which had been pre-run for 1h at 150 V). The gels were dried on 3MM blotting paper and visualised by phosphorimaging.
Enzyme-linked immunosorbent assay (ELISA) and western blotting
ELISAs for murine cytokines were performed using reagents from BD Biosciences according to the manufacturer’s instructions. Western blotting was performed as described previously (33).
RESULTS
TTP is required for destabilisation of TNF mRNA upon p38 MAPK inhibition
To investigate whether TTP is required for p38 MAPK-dependent regulation of TNF mRNA stability, actinomycin D chases were carried out in BMDM from wild-type and TTP−/− mice in the presence or absence of SB 202190. BMDM were left untreated or treated with LPS for 4h and then actinomycin D was added together with SB 202190 (1 μM) or vehicle (0.1% DMSO). Cells were harvested at the times shown, lysed and RNA was prepared. TNF mRNA was detected by northern blotting (Fig. 1A and 1B). In wild-type BMDM, the addition of SB 202190 destabilised TNF mRNA (t1/2 = 18 min compared to t1/2 = 46 min for vehicle control; Fig. 1A and 1C). In TTP−/− cells, the stability of TNF mRNA was about 2-fold greater in the absence of SB 202190 compared to wild-type cells (t1/2 = 89 min for TTP−/− compared to t1/2 = 46 min for wild-type BMDM) (Fig 1B and 1D). The destabilising effect of SB 202190 on TNF mRNA was largely ablated in TTP−/− BMDM (Fig. 1B and 1D). There was a small statistically significant effect of SB 202190 in these cells (Fig. 1D). However, overall, TTP appeared to be responsible for most of p38 MAPK-mediated regulation of TNF mRNA stability in LPS-treated BMDM.
Chronic inhibition of p38 MAPK inhibits the induction of TTP mRNA and protein in RAW264.7 cells (20). Pretreatment with SB 202190 also inhibited the induction of TTP mRNA by LPS in BMDM (Supplementary Fig. S2). Addition of SB 202190 4h post-LPS resulted in rapid decay of TNF mRNA in wild-type BMDM (Fig. 1 and Supplementary Fig. S2), but addition of the inhibitor 1h before LPS treatment did not destabilise the mRNA (Supplementary Fig. S2). This is consistent with the requirement of TTP for TNF mRNA decay and shows that p38 MAPK siRNA or dominant negative mutants cannot be used for such experiments.
p38 MAPK inhibits TTP-directed decay of IL-10 mRNA
To date, p38 MAPK has only been shown to regulate the stability of pro-inflammatory mRNAs (34). p38 MAPK-mediated post-transcriptional regulation has only been shown to involve TTP in the case of TNF mRNA (22,23,25). IL-10 is an important anti-inflammatory cytokine and the induction of IL-10 protein is regulated by p38 MAPK (29,30). To investigate whether IL-10 is regulated post-transcriptionally by p38 MAPK and TTP, the stability of IL-10 mRNA was examined in TTP−/− BMDM in the presence or absence of SB 202190 by actinomycin D chase and quantitative RT-PCR. In wild-type cells, IL-10 mRNA was relatively stable but in the presence of SB 202190 it decayed rapidly (Fig. 2A). In TTP−/− cells, the decay of IL-10 mRNA induced by SB 202190 was blocked (Fig. 2B). In the absence of the p38 MAPK inhibitor, IL-10 mRNA stability was similar in wild-type and TTP−/− cells (Fig. 2A and 2B). Overall, these results show that IL-10 is a target for both TTP and p38 MAPK and that p38 MAPK inhibits TTP-directed decay of IL-10 mRNA.
FIGURE 2. p38 MAPK inhibits TTP-directed decay of IL-10 mRNA.

Wild-type (A) and TTP−/− (B) BMDM were treated with LPS for 4h and then actinomycin D was added together with 1 μM SB 202190 or vehicle (0.1% DMSO). Cells were harvested at the times shown and RNA was isolated. IL-10 and GAPDH mRNAs were measured by quantitative real-time PCR using the ΔΔCt method of analysis. Graphs show IL-10 mRNA normalised to GAPDH mRNA expressed as a percentage of t=0 actinomycin D treatment from one experiment. Qualitatively similar data were obtained by RPA in two additional independent experiments.
IL-10 expression is increased in LPS-treated TTP−/− BMDM
To investigate whether TTP regulates IL-10 expression, IL-10 mRNA was analysed by quantitative RT-PCR in BMDM from wild-type and TTP−/− P mice treated with LPS for 4h in the presence or absence of 5 μM SB 202190 (Fig. 3A). Endogenous TNF induced by LPS can up-regulate IL-10 expression (29). Therefore a neutralising anti-TNF antibody was also added to certain BMDM cultures 30 min before addition of LPS. IL-10 mRNA was induced by LPS treatment in wild-type and TTP−/− BMDM (Fig. 3A).
In three separate experiments TTP−/− BMDM expressed 6-fold more LPS-induced IL-10 mRNA than wild-type cells (Fig. 3A). In resting wild-type cells, a small amount of IL-10 mRNA was detected with higher levels found in TTP−/− P cells (Fig. 3A). LPS-induced IL-10 mRNA was similarly inhibited by SB 202190 in wild-type cells and TTP−/− BMDM (Fig. 3A) in two separate experiments. TNF neutralisation resulted in little effect on IL-10 mRNA and even in the presence of the anti-TNF neutralising antibody, IL-10 mRNA was increased ≥5-fold in TTP−/− BMDM compared to wild-type cells (Fig. 3A). This suggests that TTP negatively regulates IL-10 mRNA expression in LPS-treated BMDM.
IL-10 protein secreted into the culture medium in the above experiments was also measured by ELISA (Fig. 3B). As seen for IL-10 mRNA, LPS-induced IL-10 protein was upregulated in TTP−/− BMDM albeit to a slightly lesser extent, inhibited to a similar degree by SB202190 in wild-type and TTP−/− cells and there was little effect of anti-TNF (Fig. 3B). Therefore TTP regulates the expression of both IL-10 mRNA and IL-10 protein and this is not an indirect effect caused by TNF overproduction in TTP−/− cells and subsequent paracrine signalling.
TTP binds to the IL-10 3’UTR in EMSA
The IL-10 3’UTR contains several copies of the core heptamer UAUUUAU which is a putative TTP binding site. To examine whether TTP regulates IL-10 mRNA directly by binding to it EMSAs were performed using cytoplasmic extracts from RAW 264.7 macrophage-like cells that were left untreated or treated with LPS for 2h. Extracts were analysed by EMSA using a 32P labelled probe spanning the IL-10 3’UTR. To supershift any TTP-containing complexes an antiserum against TTP or a non-immune serum was included in binding reactions. RNA-protein complexes were resolved by electrophoresis on a non-denaturing acrylamide gel. A constitutive (complex 1) and an LPS-inducible complex (complex 2) were detected (Fig. 4). Incubation of the extracts with the anti-TTP antiserum reduced the intensity of the LPS-inducible complex and resulted in a supershifted band (Fig. 4). No such supershifted complex was seen using a non-immune serum (Fig. 4). Therefore TTP targets IL-10 mRNA for degradation by binding to it directly.
FIGURE 4. TTP binds an IL-10 mRNA 3’UTR probe in EMSA.
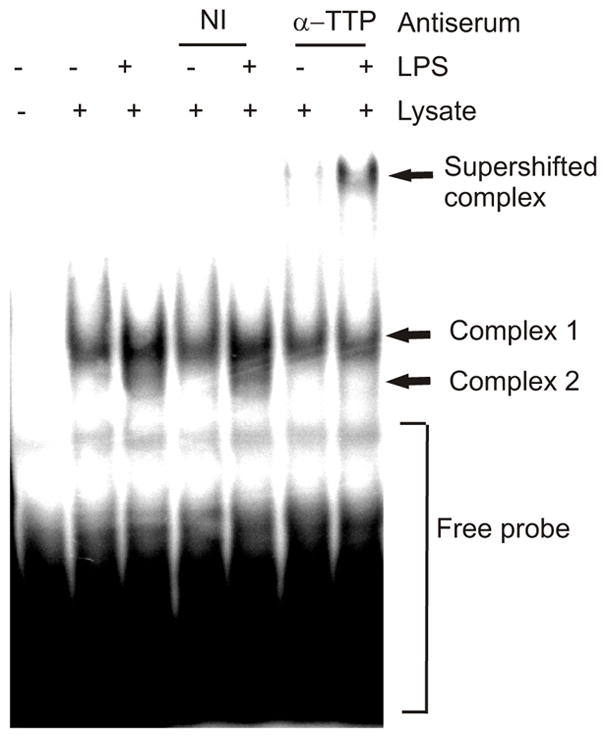
RAW 264.7 cells either left untreated or treated with LPS for 2h. EMSA was performed using 50 μg of cytoplasmic extract and 20 fmol of 32P labelled IL-10 3’UTR RNA probe per lane. Some samples were pre-incubated with either a rabbit antiserum against HSP27 (non-immune, NI) or an antiserum against TTP. Samples were electrophoresed on a non-denaturing acrylamide gel at 4°C and analysed by phosphorimaging. Similar results were obtained in two separate experiments.
TTP is required for destabilisation of COX-2 mRNA upon p38 MAPK inhibition
To investigate whether TTP regulates the stability of other mRNAs that are also targeted by the p38 MAPK pathway or whether this effect is specific to TNF and IL-10, the stability of COX-2 mRNA was examined in TTP−/− BMDM in the presence or absence of SB 202190 by northern blotting. In wild-type cells there was little decay of COX-2 mRNA in the absence of SB 202190 (Fig. 5A and 5C). Addition of the inhibitor resulted in an initial rapid decrease in COX-2 mRNA (t1/2 = 29 min), after which there was no further decay (Fig. 5A and 5C). The 4.6-kb and the 2.8-kb COX-2 transcripts that arise owing to the presence of an internal polyadenylation site appeared to decay at a similar rate (Fig. 5A). Both COX-2 transcripts include conserved region 1 (35) which contains the p38 MAPK-regulated ARE (2) and this is consistent with both being regulated by p38 MAPK. In the absence of p38 MAPK blockade, the stability of COX-2 mRNA was similar in wild-type and TTP−/− BMDM (Fig 5C and 5D). However, in the presence of SB 202190 COX-2 mRNA was not destabilised in TTP−/− BMDM (Fig. 5B and 5D). Thus, as for TNF and IL-10 mRNAs, the destabilisation of COX-2 mRNA consequent upon p38 MAPK inhibition is also mediated by TTP.
FIGURE 5. Regulation of COX-2 mRNA stability by p38 MAPK requires TTP.

Actinomycin D chases were performed in wild-type (A) or TTP−/− (B) BMDM as in Figure 1. Cells were harvested at the times shown and RNA was extracted and northern blotted with an antisense COX-2 riboprobe. Graphs show mean COX-2 mRNA in wild-type cells (C) or TTP−/− (D) cells normalised to GAPDH expressed as a percentage of t = 0 actinomycin D treatment for three independent experiments. Error bars show the S.E.M. Error bars are not shown where smaller that the symbol. Significance of the inhibition compared that for DMSO only was measured by paired Student’s t-test *p<0.01.
TTP is also required for destabilisation of IL-6 and IL-1α mRNAs upon SB 202190 addition
To examine whether co-regulation by TTP and p38 MAPK represents a general mechanism, multi-probe ribonuclease protection assays (RPA) were performed to examine the stabilities of different cytokine mRNAs upon p38 MAPK blockade in wild-type and TTP−/− BMDM. Actinomycin D chases were performed as before, and RNA was isolated and analysed by RPA (Fig. 6A and 6B).
FIGURE 6. Regulation of IL-1α and IL-6 mRNA stability by p38 MAPK requires TTP.

Actinomycin D chases were performed in wild-type (A) or TTP−/− (B) BMDM as in Figure 1. Cells were harvested at the times shown and RNA was extracted and subjected to ribonuclease protection assay (RPA) with the RiboQuant multi-probe template set mCK-2b. Protected RNA fragments were detected and quantified by phosphorimaging. Graphs show mean IL-6 (C) and IL-1α mRNA (D) mRNA ± S.E.M. from four experiments in wild-type cells and mean IL-6 (E) and IL-1α mRNA (F) mRNA ± S.E.M. from three experiments in TTP−/− cells. Significance of the effect of SB 202190 on mRNA levels was measured by paired Student’s t-test *p<0.01. Where smaller than the symbols error bars are not shown.
In wild-type cells, in the absence of SB 202190, all of the mRNAs detected (IL-12p40, IL-1α, IL-1β, IL-1 receptor antagonist (IL-1Ra), IL-6, macrophage inhibitory factor, L32 and GAPDH) appeared stable (Fig. 6A). IL-6 mRNA was destabilised upon addition of SB 202190 (t1/2 = 50 min for decay over the initial 1 h of the actinomycin D chase compared to t1/2 = 203 min for vehicle control) (Fig. 6A and 6C). As for IL-10 (Fig. 2A) and COX-2 (Fig. 5A) mRNAs, most of the decay occurred in the first 30 min following actinomycin D addition after which IL-6 mRNA appeared stable (Fig. 6C). Similarly, IL-1α mRNA was stable in the absence of SB 202190 (t1/2 = 365 min) but was destabilised following addition of the inhibitor (t1/2 = 61 min for initial decay) (Fig. 6A and 6D).
In TTP−/− BMDM, in the absence of SB 202190, IL-6 mRNA appeared slightly more stable compared to wild-type cells (Fig. 6B and 6E). IL-1α mRNA was stable in TTP−/− cells in the absence of p38 MAPK inhibitor (Fig. 6B and 6F) as seen in wild-type cells (Fig. 6A and 6D). However, in contrast to that in wild-type cells, in TTP−/− BMDM the decay of IL-6 (Fig. 6E) and IL-1α mRNAs (Fig. 6F) was insensitive to p38 MAPK inhibition. Therefore, treatment of macrophages with SB 202190 results in rapid decay of IL-6 and IL-1α mRNAs and TTP is required for the destabilisation of these mRNAs upon SB 202190 addition. Taken together these data show that p38 MAPK inhibits TTP-directed decay of TNF, IL-10, COX-2, IL-6 and IL-1α mRNAs.
Expression of inflammatory mediator mRNAs in wild-type and TTP−/− BMDM
For most of the mRNAs examined, the effect of TTP deficiency on mRNA stability was only detected in cells treated with p38 MAPK inhibitor. To find out whether, in addition to regulating mRNA stability, TTP also regulates the expression of these mRNAs their abundance was measured in BMDM derived from wild-type and TTP−/− mice. The effect of SB 202190 on LPS-induced mRNA expression was also tested.
In broad agreement with previous work (11), TNF mRNA induced by LPS was increased in TTP−/− compared with wild-type BMDM (Fig. 7A). SB202190 did not inhibit LPS-induced TNF mRNA in wild-type (consistent with a previous report (36)) or TTPP −/− cells (Fig. 7A). COX-2 mRNA expression was only slightly increased in TTP−/− BMDM compared with wild-type cells and it was weakly inhibited by SB 202190 in both (Fig. 7A).
FIGURE 7. Abundance of inflammatory mediator mRNAs in LPS-treated wild-type and TTP−/- BMDM.

A) BMDM derived from wild-type and TTP−/− littermate mice were left untreated or pretreated for 1h with 5 μM SB 202190. Some cells were treated with LPS for a further 4h. Cells were harvested, lysed and RNA was extracted and analysed by northern blotting for TNF and COX-2 mRNAs. B) Cells were left untreated or pretreated for 30 min with an anti-TNF neutralising antibody and then treated with 5 μM SB 202190 or vehicle control (0.1% DMSO) for 1h, followed by LPS treatment for a further 4h. Cells were harvested, lysed and RNA was extracted and analysed by RPA as before. Similar results were obtained in two independent experiments. C) Plot of mean cytokine mRNA in TTP−/− BMDM normalised to GAPDH mRNA expressed as fold change of that in wild-type cells treated with LPS for 4h ± S.E.M. from three independent experiments.
Since we found that TTP also targets IL-6 and IL-1α mRNAs for decay (Fig. 6), it was expected that the expression of these mRNAs in TTP−/− cells would also be increased. BMDM from TTP−/− mice are known to display increased TNF production (Fig. 7A) and this might result in an upregulation of inflammatory response genes, therefore a neutralising anti-TNF antibody was used to block paracrine stimulation of the cells by TNF. BMDM from wild-type and TTPP −/− were left untreated or treated with a TNF neutralising antibody for 30 min and then either treated with SB 202190 or vehicle (0.1% DMSO) for a further 1h, followed by LPS for a further 4h. Cells were harvested, lysed and RNA was isolated and analysed by RPA.
Despite TTP-dependent regulation of IL-1α mRNA stability (Fig. 6), IL-1α mRNA expression was similar in LPS-treated TTP−/− BMDM compared to wild-type cells (Fig. 7B and 7C). Surprisingly, although IL-6 mRNA stability was regulated by TTP IL-6 mRNA was reduced in TTP−/− cells compared with wild-type (Fig. 7B and 7C). IL-12p40 mRNA expression was also suppressed in TTP−/− BMDM compared to wild-type cells (Fig. 7B and 7C). Of the other mRNAs detected, IL-1β mRNA expression was similar in wild-type and TTP−/− BMDM (Fig. 7B and 7C) and IL-1Ra mRNA was weakly upregulated in TTP−/− BMDM in some experiments (Fig. 7C).
At first sight, these data appear confusing. However, a possible explanation is provided by the fact that the anti-inflammatory cytokine, IL-10, negatively regulates the expression of IL-1, IL-6 and IL-12p40 and positively regulates IL-1Ra (37).
COX-2 (Fig 7A), IL-1α, IL-1β, and IL-6, (Fig. XB) were all inhibited by SB 202190 not only in wild-type but also in TTP−/− BMDM, suggesting that in addition to acting on TTP, p38 MAPK also regulates their expression in a TTP-independent fashion. The inclusion of neutralising anti-TNF antibody in cultures had no effect on the expression at 4h post-LPS of any of the inflammatory mediator mRNAs measured (Fig. 7B) although it did inhibit IL-1α mRNA at 16h post-LPS (data not shown).
Expression of pro-inflammatory mediator proteins in wild-type and TTP−/− BMDM
It was of interest to examine whether in addition to regulation of mRNA levels, the expression of pro-inflammatory proteins was also regulated in TTP−/− BMDM. In agreement with the data for TNF mRNA, TNF protein was upregulated approximately 2-fold in TTP−/− BMDM (Fig. 8A). As previously reported (23,25) the effect of p38 MAPK inhibition on TNF protein was reduced in TTP−/− cells. COX-2 protein was weakly upregulated in TTP−/− BMDM as found by others (38) and was inhibited by SB202190 in both wild-type and TTP−/− cells (Fig. 8B). As seen for IL-6 mRNA, IL-6 protein was expressed at a lower level in TTP−/− compared with wild-type cells and it was inhibited by SB202190 in both cell types in three separate experiments (Fig. 8C).
FIGURE 8. Effect of TTP deficiency on the expression of pro-inflammatory mediator proteins.

A) Cells were treated as described in Fig. 7A and TNF protein secreted into the culture medium was measured by ELISA. Similar results were obtained in two separate experiments. The error shown is the standard deviation of ELISA measurements made in triplicate. B) Western blot for COX-2 protein expressed in lysates from cells treated as described above. C) Graph of IL-6 protein measured by ELISA as for (A). Results are representative of three independent experiments.
IL-10 blockade rescues IL-6 and IL-12p40 production in TTP−/− BMDM
It was possible that the reduction in expression of IL-6, IL-1α and IL-12p40 mRNAs and the increase in IL-1Ra in TTP−/− BMDM was due to increased production of IL-10 in these cells. To test this possibility, BMDM were either left untreated, or pre-treated with an anti-IL-10 antibody for 1h and then incubated for a further 4h in the presence of LPS. LPS-induced IL-6 expression was lower in TTP−/− cells than in wild-type cells (Fig. 9A). Pre-treatment with anti-IL-10 antibody had little effect on IL-6 production by wild-type cells, but restored IL-6 expression in TTP−/− BMDM (Fig. 9A). Similar results were obtained for IL-12p40 (Fig. 9B). Thus the reduced expression of IL-6 and IL-12p40 in TTP−/− P BMDM can be attributed to the increased production of IL-10.
FIGURE 9. IL-10 neutralisation rescues IL-6 and IL-12p40 protein expression in TTP-/- BMDM.
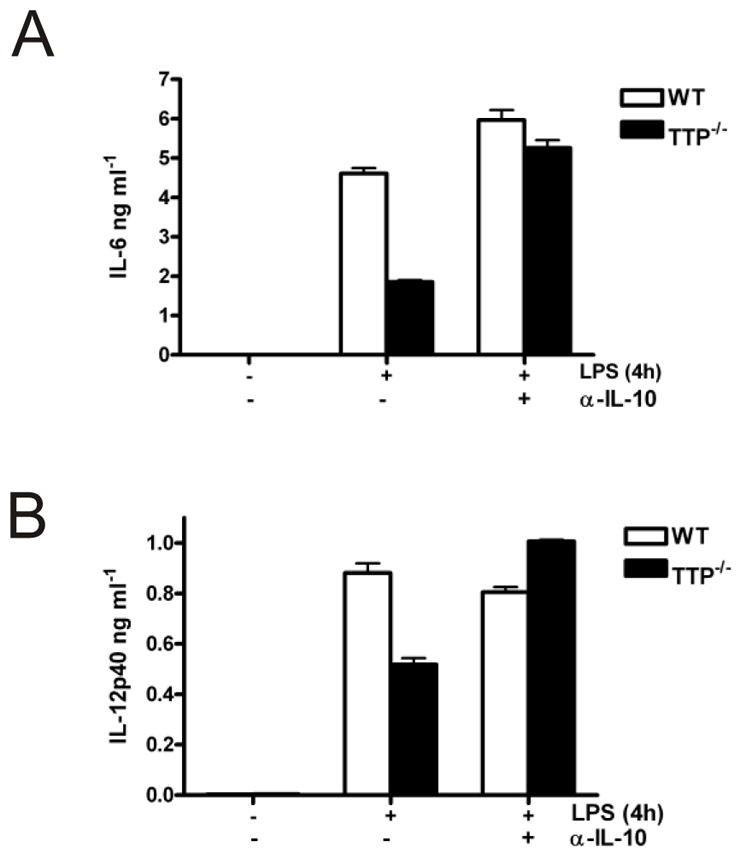
Wild-type and TTP−/− BMDM were treated with anti-IL-10 antibody for 1h, or left untreated. Some cultures were then treated with LPS for a further 4h. IL-6 (A) and IL-12p40 protein (B) in culture medium was measured by ELISA.
DISCUSSION
p38 MAPK has been previously shown to regulate many different pro-inflammatory mRNAs (34). TTP has also been shown to play a pivotal role in the expression of the pro-inflammatory cytokine, TNF. We have shown that p38 MAPK and TTP not only regulate the stability of pro-inflammatory mRNAs, but also play a crucial role in the post-transcriptional regulation of anti-inflammatory IL-10.
p38 MAPK regulates IL-10 mRNA stability by inhibiting TTP-directed mRNA decay. This is demonstrated by the lack of an effect of SB 202190 on IL-10 mRNA stability in TTP−/− BMDM. IL-10 appears to be a direct target of TTP since an LPS-inducible complex containing TTP protein bound an IL-10 3’UTR probe in EMSA. The murine IL-10 3’UTR does not contain the originally proposed (17) nonameric TTP binding site. However, we note that it does contain four heptameric UAUUUAU motifs, two of which are separated by 26 nucleotides. TTP has been found to bind with high affinity to such sites (18). All of the mRNAs that we found were co-regulated by p38 MAPK and TTP (except IL-6 mRNA) contain at least two heptamers separated by an intervening sequence of similar length. Murine TNF, COX-2 and IL-1α contain heptamers separated by 28, 26 and 23 nucleotides respectively. IL-6 mRNA contains only one heptamer, however, it also has a pentamer in a U-rich context with intervening sequence present as in the other regulated mRNAs. Of the mRNAs that we found were not regulated, IL-1β has two heptamers which are separated by only 5 nt; IL-12p40 has only one heptamer; IL-1Ra has none. This is consistent with the presence of two heptamers separated by 23–28 nt in the majority of p38-MAPK- and TTP-regulated mRNAs.
TTP plays an important role in regulating IL-10 since steady-state IL-10 mRNA and IL-10 protein was found to be upregulated in LPS-treated BMDM. In the absence of LPS stimulation, IL-10 mRNA was also found to be somewhat increased in TTP−/− compared with wild-type BMDM suggesting that circulating levels of IL-10 protein may be increased in TTP−/− mice. Whilst TTP−/− mice develop a complex inflammatory phenotype, largely arising from increased TNF production (12), unlike mice with a targeted deletion of the TNF ARE (36), they do not develop a Crohn’s-like inflammatory bowel disease (IBD). The anti-inflammatory function of IL-10 plays a key protective role in protecting against IBD. This has been clearly demonstrated by the finding that IL-10−/− mice spontaneously develop ulcerative colitis (39). An interesting possibility is that the increased levels of IL-10 in the TTP−/− mice protects them against IBD-like disease that may otherwise have been caused by the elevated TNF production in these animals. It is possible that therapies for the treatment of chronic inflammatory diseases which target p38 MAPK, MK2, or TTP could be complicated by the consequences of inhibition of IL-10.
p38 MAPK has now been shown to stabilise a plethora of different transcripts, many of which are mRNAs of proteins of the inflammatory response (34). However, TNF (11) and granulocyte-macrophage colony-stimulating factor (13) are the only mRNAs that have been shown to be regulated in TTP−/− cells and which are also stabilised by p38 MAPK (6–8). Thus it was unclear whether inactivation of TTP by the p38 MAPK pathway represents a general post-transcriptional mechanism, or whether it is restricted to only a few mRNAs. We have now shown that TTP is required for p38 MAPK-dependent regulation of not only TNF, but also of other long known p38 MAPK-regulated mRNAs such as COX-2 and IL-6. We have also found that IL-1α and IL-10 are relatively stable mRNAs in LPS-treated BMDM, but are destabilised following the addition of p38 MAPK inhibitor to the cells. IL-1 α mRNA has previously been found to be regulated by p38 MAPK in THP-1 cells (10). The decay of these mRNAs in the presence of the inhibitor was completely blocked in TTP−/− macrophages. Thus we have identified five different mRNAs of the inflammatory response which are stabilised by p38 MAPK-mediated inactivation of TTP.
It is noted that the decay of all of the mRNAs co-regulated by p38 MAPK and TTP, except TNF mRNA, all decay rapidly following addition of p38 MAPK inhibitor, after which they appear to be relatively stable. This effect is probably due to the degradation of TTP protein that occurs following p38 MAPK blockade (40). Under physiological circumstances the decline in p38 MAPK activity following stimulation of the cells would likely be more gradual and TTP protein could function for longer.
In a previous study we investigated the involvement of different ARE-binding proteins in the p38 MAPK-mediated stabilisation of a COX-2 ARE reporter mRNA in a HeLa cell line (41). We reported that AUF1, AUF2, FBP1, HuR and TTP were not required for this (41). However, we ruled out a role for TTP largely because of our inability to detect TTP in these cells (41). We have now been able to detect small amounts of TTP protein in HeLa cells (C. Asensio and A. Clark, unpublished). Therefore it is of interest to test the function of TTP in regulating the expression of COX-2 mRNA in these cells, as well as in human fibroblasts. It is unclear which mechanism would predominate in such cells for COX-2 mRNA regulation as other ARE-binding proteins with a stabilising function including HuR (42,43) and CUGBP2 (44) could be involved. However HuR does not appear to be required for the induction of COX-2 by IL-1 in HeLa cells as shown by depletion of HuR by RNAi (J.L.E.D. and Kate Alford, unpublished).
Overall, the data in this study are consistent with those obtained in BMDM from TTP−/−/MK2−/− mice (25). TNF mRNA was similarly stable in TTP−/− and TTP−/−/MK2−/− BMDM (25). However in the present study, whilst TNF mRNA was relatively stable in TTP−/− cells either in the presence or absence of SB 202190, a small effect of the p38 inhibitor on TNF mRNA stability was observed. It is possible that additional ARE-binding proteins other than TTP might be responsible for this effect. The TTP homologue, BRF1 (45), is one such protein. So far, we have failed to detect BRF1 protein in TTP−/− BMDM, although BRF2, another TTP family member, does appear to be expressed in these cells. The ARE-binding protein, KSRP, which regulates certain ARE-containing transcripts in myocytes may also be involved (46). It should be stressed, however, that p38 MAPK-dependent regulation of TNF mRNA stability appeared to be mainly regulated by TTP, or in the case of the other p38 MAPK-regulated mRNAs examined in this study, exclusively regulated by TTP.
We found that whilst IL-10 mRNA was relatively stable in BMDM treated with LPS for 4h, rapid decay of the mRNA occurred following the addition of p38 MAPK inhibitor. Thus as for other mRNAs of the inflammatory response such as COX-2 (5) p38 MAPK controls IL-10 expression by activating its transcription and by stabilising its mRNA. The inhibition of steady-state IL-10 mRNA upon p38 MAPK blockade in TTP−/− BMDM is consistent with the regulation of IL-10 transcription by p38 MAPK. As for wild-type cells, chronic p38 MAPK blockade in TTP−/− BMDM, inhibited LPS-induced steady-state IL-6, IL-1α, and IL-1β mRNAs. The fact that in TTP−/− cells SB202190 regulates the steady-state levels of these mRNAs but not their stability is not a result of the higher concentration of inhibitor used for chronic inhibition because acute blockade of p38 MAPK activity (employed in actinomycin D chases) is achieved with a concentration of only 1 μM. It is likely that as for IL-10 and COX-2, the transcription of all of these mRNAs is p38 MAPK-dependent. Transcription of TNF by contrast may not regulated by p38 MAPK in murine macrophages since in these cells p38 MAPK inhibitors do not inhibit steady-state TNF mRNA. This contrasts the situation in human cells of myeloid lineage in which TNF mRNA is inhibited upon p38 MAPK blockade (5,47), likely due to regulation of TNF transcription by p38 MAPK.
COX-2 and IL-6 protein were inhibited by SB202190 in both wild-type and TTP−/− cells as expected given that their mRNAs are also inhbited. Despite the lack of an effect on TNF mRNA, TNF protein was inhibited by p38 MAPK blockade, but to a lesser extent in TTP−/− BMDM. This difference has been reported previously (25) and may be due to regulation of TNF translation by TTP.
The use of p38 MAPK inhibitor in actinomycin D chases allowed us to examine a possible correlation between TTP and p38 MAPK targets. Since many of the mRNAs examined were relatively stable in the absence of inhibitor in both wild-type and TTP−/− cells, p38 MAPK inhibition revealed targets of TTP which would otherwise not have been identified. This may be why of the mRNAs found to be regulated by TTP and p38 MAPK in this study, only TNF has previously been identified as a TTP target in experiments employing TTP−/− P cells. In fact, of the mRNAs tested, only TNF mRNA was found to be stabilised in TTP−/− BMDM in the absence of the inhibitor. It would be interesting to search for additional TTP targets in the future by performing microarray analyses of wild-type and TTP−/− cells treated with actinomycin D in the presence or absence of p38 MAPK inhibitor. The fact that TTP is more promiscuous than originally thought may compromise the utility of TTP-targeted therapies aimed at treating chronic inflammatory diseases.
Supplementary Material
Acknowledgments
We are grateful to Dr Matt Brook for the TNF riboprobe template and to Prof. Robert Schreiber for provision of anti-TNF antibody. We also thank Dr Keith Ray for useful discussions. We warmly acknowledge Olga Boruc and Allen Hazlehurst for technical assistance. Corina Tudor was supported by a CASE studentship from the Biotechnology and Biological Sciences Research Council and GlaxoSmithKline plc. We are also grateful to the Medical Research Council and the Arthritis Research Campaign for support.
References
- 1.Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Muller M, Gaestel M, Resch K, Holtmann H. Embo J. 1999;18:4969–4980. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lasa M, Mahtani KR, Finch A, Brewer G, Saklatvala J, Clark AR. Mol Cell Biol. 2000;20:4265–4274. doi: 10.1128/mcb.20.12.4265-4274.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ridley SH, Dean JL, Sarsfield SJ, Brook M, Clark AR, Saklatvala J. FEBS Lett. 1998;439:75–80. doi: 10.1016/s0014-5793(98)01342-8. [DOI] [PubMed] [Google Scholar]
- 4.Miyazawa K, Mori A, Miyata H, Akahane M, Ajisawa Y, Okudaira H. Journal Of Biological Chemistry. 1998;273:24832–24838. doi: 10.1074/jbc.273.38.24832. [DOI] [PubMed] [Google Scholar]
- 5.Dean JL, Brook M, Clark AR, Saklatvala J. J Biol Chem. 1999;274:264–269. doi: 10.1074/jbc.274.1.264. [DOI] [PubMed] [Google Scholar]
- 6.Wang SW, Pawlowski J, Wathen ST, Kinney SD, Lichenstein HS, Manthey CL. Inflamm Res. 1999;48:533–538. doi: 10.1007/s000110050499. [DOI] [PubMed] [Google Scholar]
- 7.Brook M, Sully G, Clark AR, Saklatvala J. FEBS Lett. 2000;483:57–61. doi: 10.1016/s0014-5793(00)02084-6. [DOI] [PubMed] [Google Scholar]
- 8.Tebo J, Der S, Frevel M, Khabar KS, Williams BR, Hamilton TA. J Biol Chem. 2003;278:12085–12093. doi: 10.1074/jbc.M212992200. [DOI] [PubMed] [Google Scholar]
- 9.Mavropoulos A, Sully G, Cope AP, Clark AR. Blood. 2005;105:282–288. doi: 10.1182/blood-2004-07-2782. [DOI] [PubMed] [Google Scholar]
- 10.Frevel MA, Bakheet T, Silva AM, Hissong JG, Khabar KS, Williams BR. Mol Cell Biol. 2003;23:425–436. doi: 10.1128/MCB.23.2.425-436.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carballo E, Lai WS, Blackshear PJ. Science. 1998;281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 12.Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ. Immunity. 1996;4:445–454. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 13.Carballo E, Lai WS, Blackshear PJ. Blood. 2000;95:1891–1899. [PubMed] [Google Scholar]
- 14.Ogilvie RL, Abelson M, Hau HH, Vlasova I, Blackshear PJ, Bohjanen PR. J Immunol. 2005;174:953–961. doi: 10.4049/jimmunol.174.2.953. [DOI] [PubMed] [Google Scholar]
- 15.Lai WS, Parker JS, Grissom SF, Stumpo DJ, Blackshear PJ. Mol Cell Biol. 2006;26:9196–9208. doi: 10.1128/MCB.00945-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Worthington MT, Pelo JW, Sachedina MA, Applegate JL, Arseneau KO, Pizarro TT. J Biol Chem. 2002;277:48558–48564. doi: 10.1074/jbc.M206505200. [DOI] [PubMed] [Google Scholar]
- 17.Blackshear PJ, Lai WS, Kennington EA, Brewer G, Wilson GM, Guan X, Zhou P. J Biol Chem. 2003;278:19947–19955. doi: 10.1074/jbc.M301290200. [DOI] [PubMed] [Google Scholar]
- 18.Brewer BY, Malicka J, Blackshear PJ, Wilson GM. J Biol Chem. 2004;279:27870–27877. doi: 10.1074/jbc.M402551200. [DOI] [PubMed] [Google Scholar]
- 19.Neininger A, Kontoyiannis D, Kotlyarov A, Winzen R, Eckert R, Volk HD, Holtmann H, Kollias G, Gaestel M. J Biol Chem. 2002;277:3065–3068. doi: 10.1074/jbc.C100685200. [DOI] [PubMed] [Google Scholar]
- 20.Mahtani KR, Brook M, Dean JL, Sully G, Saklatvala J, Clark AR. Mol Cell Biol. 2001;21:6461–6469. doi: 10.1128/MCB.21.9.6461-6469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, Sturgill TW. J Biol Chem. 2004;279:10176–10184. doi: 10.1074/jbc.M310486200. [DOI] [PubMed] [Google Scholar]
- 22.Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, Anderson P. Embo J. 2004;23:1313–1324. doi: 10.1038/sj.emboj.7600163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carballo E, Cao H, Lai WS, Kennington EA, Campbell D, Blackshear PJ. J Biol Chem. 2001;276:42580–42587. doi: 10.1074/jbc.M104953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kontoyiannis D, Kotlyarov A, Carballo E, Alexopoulou L, Blackshear PJ, Gaestel M, Davis R, Flavell R, Kollias G. Embo J. 2001;20:3760–3770. doi: 10.1093/emboj/20.14.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hitti E, Iakovleva T, Brook M, Deppenmeier S, Gruber AD, Radzioch D, Clark AR, Blackshear PJ, Kotlyarov A, Gaestel M. Mol Cell Biol. 2006;26:2399–2407. doi: 10.1128/MCB.26.6.2399-2407.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai WS, Kennington EA, Blackshear PJ. Mol Cell Biol. 2003;23:3798–3812. doi: 10.1128/MCB.23.11.3798-3812.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lykke-Andersen J, Wagner E. Genes Dev. 2005;19:351–361. doi: 10.1101/gad.1282305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dean JL, Sarsfield SJ, Tsounakou E, Saklatvala J. J Biol Chem. 2003;278:39470–39476. doi: 10.1074/jbc.M306345200. [DOI] [PubMed] [Google Scholar]
- 29.Foey AD, Parry SL, Williams LM, Feldmann M, Foxwell BM, Brennan FM. J Immunol. 1998;160:920–928. [PubMed] [Google Scholar]
- 30.Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, Flavell RA. Proc Natl Acad Sci U S A. 2006;103:2274–2279. doi: 10.1073/pnas.0510965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma W, Lim W, Gee K, Aucoin S, Nandan D, Kozlowski M, Diaz-Mitoma F, Kumar A. J Biol Chem. 2001;276:13664–13674. doi: 10.1074/jbc.M011157200. [DOI] [PubMed] [Google Scholar]
- 32.Sheehan KC, Ruddle NH, Schreiber RD. J Immunol. 1989;142:3884–3893. [PubMed] [Google Scholar]
- 33.Alford KA, Glennie S, Turrell BR, Rawlinson L, Saklatvala J, Dean JL. J Biol Chem. 2007;282:6232–6241. doi: 10.1074/jbc.M610987200. [DOI] [PubMed] [Google Scholar]
- 34.Clark AR, Dean JL, Saklatvala J. FEBS Lett. 2003;546:37–44. doi: 10.1016/s0014-5793(03)00439-3. [DOI] [PubMed] [Google Scholar]
- 35.Newton R, Seybold J, Liu SF, Barnes PJ. Biochem Biophys Res Commun. 1997;234:85–89. doi: 10.1006/bbrc.1997.6586. [DOI] [PubMed] [Google Scholar]
- 36.Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Immunity. 1999;10:387–398. doi: 10.1016/s1074-7613(00)80038-2. [DOI] [PubMed] [Google Scholar]
- 37.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 38.Phillips K, Kedersha N, Shen L, Blackshear PJ, Anderson P. Proc Natl Acad Sci U S A. 2004;101:2011–2016. doi: 10.1073/pnas.0400148101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 40.Brook M, Tchen CR, Santalucia T, McIlrath J, Arthur JS, Saklatvala J, Clark AR. Mol Cell Biol. 2006;26:2408–2418. doi: 10.1128/MCB.26.6.2408-2418.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sully G, Dean JL, Wait R, Rawlinson L, Santalucia T, Saklatvala J, Clark AR. Biochem J. 2004;377:629–639. doi: 10.1042/BJ20031484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dixon DA, Tolley ND, King PH, Nabors LB, McIntyre TM, Zimmerman GA, Prescott SM. J Clin Invest. 2001;108:1657–1665. doi: 10.1172/JCI12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sengupta S, Jang BC, Wu MT, Paik JH, Furneaux H, Hla T. J Biol Chem. 2003;278:25227–25233. doi: 10.1074/jbc.M301813200. [DOI] [PubMed] [Google Scholar]
- 44.Mukhopadhyay D, Houchen CW, Kennedy S, Dieckgraefe BK, Anant S. Mol Cell. 2003;11:113–126. doi: 10.1016/s1097-2765(03)00012-1. [DOI] [PubMed] [Google Scholar]
- 45.Stoecklin G, Colombi M, Raineri I, Leuenberger S, Mallaun M, Schmidlin M, Gross B, Lu M, Kitamura T, Moroni C. Embo J. 2002;21:4709–4718. doi: 10.1093/emboj/cdf444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Briata P, Forcales SV, Ponassi M, Corte G, Chen CY, Karin M, Puri PL, Gherzi R. Mol Cell. 2005;20:891–903. doi: 10.1016/j.molcel.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 47.Campbell J, Ciesielski CJ, Hunt AE, Horwood NJ, Beech JT, Hayes LA, Denys A, Feldmann M, Brennan FM, Foxwell BM. J Immunol. 2004;173:6928–6937. doi: 10.4049/jimmunol.173.11.6928. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.