

Abstract
Phenylalanine Ammonia Lyase (PAL) gene which plays a key role in bio-synthesis of medicinally important compounds, Rutin/quercetin was sequence characterized for its efficient genomics application. These compounds possessing anti-diabetic and anti-cancer properties and are predominantly produced by Fagopyrum spp. In the present study, PAL gene was sequenced from three Fagopyrum spp. (F. tataricum, F. esculentum and F. dibotrys) and showed the presence of three SNPs and four insertion/deletions at intra and inter specific level. Among them, the potential SNP (position 949th bp G>C) with Parsimony Informative Site was selected and successfully utilised to individuate the zygosity/allelic variation of 16 F. tataricum varieties. Insertion mutations were identified in coding region, which resulted the change of a stretch of 39 amino acids on the putative protein. Our Study revealed that autogamous species (F. tataricum) has lower frequency of observed SNPs as compared to allogamous species (F. dibotrys and F. esculentum). The identified SNPs in F. tataricum didn’t result to amino acid change, while in other two species it caused both conservative and non-conservative variations. Consistent pattern of SNPs across the species revealed their phylogenetic importance. We found two groups of F. tataricum and one of them was closely related with F. dibotrys. Sequence characterization information of PAL gene reported in present investigation can be utilized in genetic improvement of buckwheat in reference to its medicinal value.
Introduction
Rutin and Quercetin are plant metabolites having antioxidant property and play a significant role in combating diabetes [1]. Diabetes is a chronic metabolic disorder resulted in mortality of over one million people globally [2]. Besides diabetes, rutin helps in reducing severity of colon carcinogenesis [3] and hypertension [4]. Rutin is neither present in cereals nor in pseudocereals [5]. Buckwheat is the only field crop species which possess rutin in the form of its secondary metabolite product [6]. Buckwheat generally grows at high altitude mountainous area, which derives the evolutionary mechanism of protecting itself against UV rays [7]. Rutin and quercetin are synthesized by a cascade of enzymes, in which Phenylalanine Ammonia Lyase (PAL; E.C 4.3.1.5) gene (PAL gene) is the first enzyme, which catalyses the conversion of precursor amino acid ‘phenylalanine’ to ‘trans-cinnamic acid’. Subsequently, several cascade of enzymes catalyzes further on each substrates until dihydroquercetin and finally Flavonol Synthase (FLS) converts dihydroquercetin to quercetin and rutin [8] (Fig 1).
Fig 1. Description of PAL gene and rutin synthesis pathway.

PAL, Phenylalanine Ammonia Lyase; C4H, Cinnamate 4-Hydroxylase; 4CL1 & 2, 4-Coumaroyl CoA Ligase; CHS, Chalcone Synthase; CHI, Chalcone Isomerase; F3H, Flavone 3-Hydroxylase; F3′H, Flavonoid 3′-Hydroxylase; FLS1&2, Flavonol Synthase; ANS, Anthocyanidin Synthase; Source [8].
F. tataricum, a pseudo cereal commonly known as ‘tartary buckwheat’ is rich in rutin [9]. During the culinary preparations, rutin (flavonol 3-O-rutinoside) is hydrolysed to a bitter compound called, quercetin, which gives strong bitter taste [10]. It is therefore very crucial to analyse Fagopyrum spp. with reference to genes involving in rutin and quercetin production. PAL gene is an important candidate involves in rutin/quercetin production and is studied extensively in this report. Tatary buckwheat contains 40 mg/g of flavonoids compared to common buckwheat (F. esculentum with 10 mg/g), among which rutin occupies a major portion [11]. SNPs are present in plant genomes at a high frequency and can be utilized efficiently as molecular markers for complex trait [12]. Allelic characterization of PAL gene is an important strategy for the genetic improvement of Fagopyrum spp. concerning the enhancement of rutin and quercetin content. PAL gene sequence information of F. tataricum (2864bp) and F. dibotrys (2583 bp) is well known [13] but, variation at single nucleotide polymorphism (SNP) level have not been reported yet. SNP variation in PAL gene can be identified in different accessions of the Fagopyrum spp. for their deployment in genetic improvement program. Amplification of the gene to identify SNPs can be used through cost effective strategies like Tetra primer ARMS PCR [14] for allele mining.
The genus Fagopyrum belongs to the family, Polygonaceae and consists of about 16 species [15] including common buckwheat (F. esculentum), tartary buckwheat and wild perennial buckwheat (F. cymosum or F. dibotrys). Among these two species, F. tartaricum and F. esculentum are domesticated. F. esculentum is the most commonly used species for human consumption, so called, ‘common buckwheat’ or ‘sweet buckwheat’ followed by F. tataricum, which is comparatively bitter in taste (therefore, also known as ‘bitter buckwheat’).
F. esculentum is widely grown in the temperate regions of Eurasia and North America, while the cultivation of tartary buckwheat is confined to Himalayan hills and some parts of Southern China. The latter areas are recognised as the natural habitat of the Fagopyrum genus including its wild relatives [16]. PAL gene has not been studied in most wild relatives of Fagopyrum genus. However, since past few decades research efforts have been given on F. dibotrys, led to utilize this species extensively for the characterization of this gene [11]. Rutin is a phenolic compound present in high concentrations in ‘tartary buckwheat’ and to limited extent in ‘common buckwheat’ [17]. Very little information is available concerning the genetic analysis of different species of the genus Fagopyrum, which led this genus remain underutilized. F. esculentum and F. dibotrys are allogamous, whereas F. tartaricum is an autogamous species. Morphological similarities suggested a greater closeness between F. dibotrys and F. esculentum [18, 19]. Conversely, recent RFLP-cpDNA molecular analysis revealed that F. dibotrys is more closely related to F. tataricum compared to F. esculentum [20]. An in-depth characterization of different Fagopyrum species with important genes (such as PAL gene) will lead to an increased taxonomic understanding and ultimately helps in their genetic enhancement as a crop of economic value.
Results
Allele mining of PAL gene in F. tataricum and related species
The molecular profiling of PAL gene from different accessions of F. tataricum, F. esculentum and F. dibotrys, led to decipher the species specific allelic sequence variations in the form of SNPs and/or Indels (Fig 2 and S1 Fig).
Fig 2. Amplification of PAL gene fragment (approximately 2.6 kb size) from F. tataricum genotypes (Gol-Golden, Don-Donan, Jak-Jakar, Chy-Chumey, Cha- N7605Chumoa, PI73-PI481673, PI35-PI427235, FAG50-FAG50, PI72-PI481672).

In addition to the species specific sequence signatures intra-specific variations were also found. SNPs alleles were designated with letters ‘A’ and ‘B’ (Fig 3). Furthermore, two accessions exhibited the presence of three insertions at exon2 (Fig 3 and S2 Fig) causing the variation of a stretch of amino acids with respect to its putative protein and designated as allele ‘A1’ (Table 1).
Fig 3. A synonymous mutation and insertion induced frame shift mutation in PAL gene of F. tataricum.

Table 1. Fagopyrum spp. PAL gene allele designation with Genbank accession numbers.
| F. tataricum accessions | Allele | Genbank | Size | SNPs/Allele description |
|---|---|---|---|---|
| F. tataricum Golden | A | KF286895 | 860bp | g.[949G>C;1114_1115insG;1346C>T] |
| F. tataricum Donan | A | KF286896 | 811bp | g.[949G>C;1114_1115insG;1346C>T] |
| F.tataricum Jakar | B | KF286897 | 751bp | g.[949C>G;1114_1115insG;1346T>C] |
| F. tataricum Chumey | B | KF286898 | 880bp | g.[949C>G;1114_1115insG;1346T>C] |
| F. tataricum Chumoa | B | KF286899 | 780bp | g.[949C>G;1114_1115insG;1346T>C] |
| F.tataricum PI481673 | B | KF286900 | 795bp | g.[949C>G;1114_1115insG;1346T>C] |
| F. tataricum PI427235 | A | KF286901 | 720bp | g.[1114_1115insG;1346C>T] |
| F. tataricum FAG50 | A1 | KF680943 | 2188bp | g.[949G>C;1017A>G;1114_1115insG;1346C>T;1722_1723insA;1744_1745insC;1835_1836insT] |
| F.tataricum PI481672 | A1 | KF408290 | 2156bp | g.[949G>C;1017A>G;1114_1115insG;1346C>T;1722_1723insA;1744_1745insC; 1835_1836insT] |
| F. esculentum Botan | E1 | KC792588 | 620bp | g.[685G>A;697G>C; 707G>A;709C>T;841G>C] |
| F. esculentum Karmen | E2 | KC792587 | 983bp | g.[685A>G;697G>C;707G>A;709C>T;841G>C] |
| F. esculentum Koto | E1 | KC792586 | 949bp | g.[685G>A;697G>C; 707G>A;709C>T;841G>C] |
| F. esculentum Luba | E2 | KC792585 | 983bp | g.[685A>G;697G>C;707G>A;709C>T;841G>C] |
| F. esculentum Koban | E2 | KC792584 | 969bp | g.[685A>G;697G>C;707G>A;709C>T;841G>C] |
| F. esculentum Svityazyanka | E2 | KC792583 | 983bp | g.[685A>G;697G>C;707G>A;709C>T;841G>C] |
| F. dibotrys FCA1_FAG135 | F1 | KF408292 | 2141bp | g.[1122C>T;1254G>C;1458C>G;2220C>A; 2432G>C] |
| F. dibotrys FCA3_FAG135 | F1 | KF408293 | 2133bp | g.[1122C>T;1254G>C;1458C>G;2220C>A;2432G>C] |
| F. dibotrys FDD1_FAG142 | F2 | KF408291 | 2164bp | g.[1122C>T;1254C>G;1458C>G;2220C>A;2432G>C] |
| F. dibotrys FCB3_FAG135 | F3 | KF680944 | 2120bp | g.[1122C>T;1254C>G;1458C>G;2220C>A;2432C>G] |
Effect of SNPs/Indel on putative protein
Annotation of Allele ‘A1’ from F. tataricum sequences revealed three insertion mutations, found to cause frame shift of 39 amino acids in ORF of PAL gene. This frame shift resulted in altered amino acids stretch on putative protein corresponding to exon2 in two F. tataricum accessions (Figs 3 and 4). Amplified sequences of the other two Fagopyrum species were submitted to Genbank and allele designated with the SNPs causing the change of amino acid as shown in Table 1.
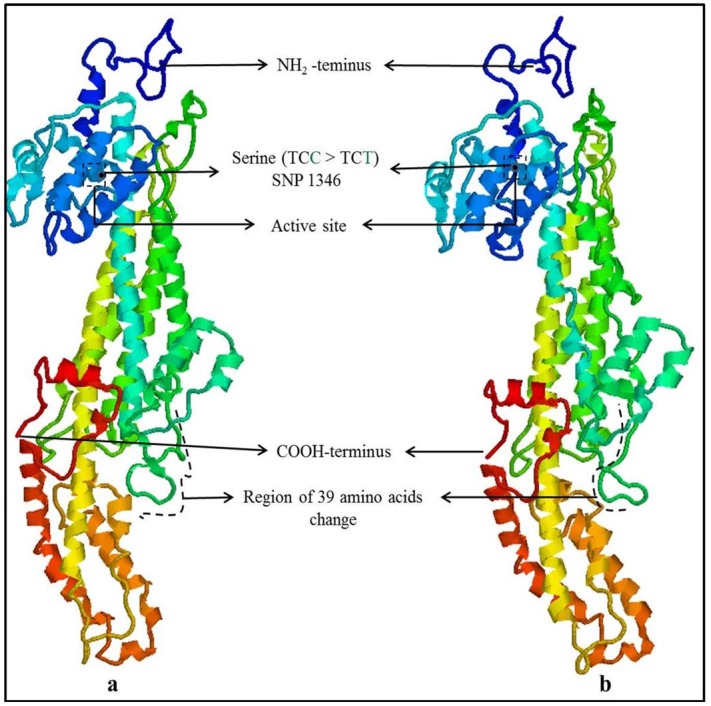
Fig 4. Protein modelling of PAL gene Allele ‘A1’ and PAL gene reference protein describing the structural similarity in active site and other region.
SNP analysis in natural populations of F. tataricum
A SNP at 949th base pair position in PAL gene (Fig 5) was found to be present in homozygous and heterozygous conditions in 10 and 6 accessions of F. tataricum respectively.
Fig 5. Agarose gel picture depicting the allelic variation among sixteen F. tataricum varieties differing G>C.

Heterozygous (GC alleles with both 297and 244 bp amplicons) and homozygous (G with only 297 bp amplicon). The size of the common outer amplicon was 486 bp. Varieties used: J-Jakar, Y-Chumey, M- C8816 Malong, PI-PI481672, D-Donan, G-Golden, H-Hei Qiao-4, I-Ishisoba, O-01Z100014, K-C9717 Kuer, P3-PI481673, X-Xinong 9909, Q—Q000120, H-Hei Feng, P5-PI427235.
Subsequent intra-varietal analysis revealed that among 77 genotypes, 50 and 27 samples showed homozygosity and heterozygosity for this SNP respectively (Fig 6). Further, analysis suggested the existence of strong linkage disequilibrium between SNP positions at 949th and 1346th bp. The SNP at 949th bp had more homozygotes in 16 F. tataricum varieties of diverse origin as indicated in Fig 6.
Fig 6. Gel picture differentiating between homozygous and heterozygous alleles at the 949th bp position in F. tataricum at intra- varietal level.
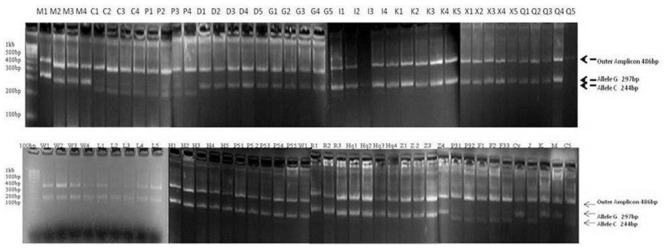
Accessions utilized: M1-M4: C8816Malong, C1-C4: N7605Chumoa, P1-P4: PI481672, D1-D5: Donan, G1-G5: Golden, I1-I4-: Ishisoba, K1-K5: C9717 Kuer, X1-X5: Xinong 9909, Q1-Q5: Q0001120, W1-W4: Wei 93–8, L1-L5: N8614 Lukla, H1-H5:Hei Feng, P51-P55: PI427235, R1-R3: RCAT061058, Hq1-Hq4: Hei Qiao-4, Z1-Z4: 01Z100014, P31-P32: PI481673, F1-F2: FAG 50, F33: FAG 33, Cy: Chumey, J: Jakar, K: Kuer, M: Malong, C5: Chumoa.
Interspecies sequence analysis
Interspecies polymorphic site analysis revealed the presence of more polymorphic sites in F. dibotrys followed by F. esculentum and F. tataricum. Phylogenetic analysis indicated towards taxonomic closeness of F. tataricum and F. esculentum, which was further proved with the presence of relevant SNPs and indel mutations (S3 Fig). However, parsimony informative sites (PIS) with Linkage Disequilibrium (LD) were not collinear to their exact nucleotide positions among these three species, although some PIS were sharing identity with other species (Table 2).
Table 2. Parsimony Informative Sites (PIS) and other sites among three species.
Legend: PS: Parsimony Informative Sites (PIS), MS: Monomorphic Sites (MS), SV: Singleton Variable, Sites, SV2V: Singleton Variable Sites with Two Variants (SV2V), PIS2V: Parsimony Informative Sites with Two Variants, SV3V: Singleton Variable with Two Variants, PIS3V: Parsimony Informative sites with Three Variants.
| Species | Sequences/Sites analysed | PS | MS | SV | PIS | SV2V | PIS2V | SV3V | PIS3V | LD sites | χ2 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| F. tataricum | 8/742bp | 3 | 690 | 3 | 2 | 1 | 2 | 0 | 0 | 3 | 1 |
| F. esculentum | 5/742bp | 11 | 723 | 9 | 2 | 9 | 2 | 0 | 0 | 55 | 17 |
| F. dibotrys | 5/742bp | 19 | 659 | 14 | 5 | 13 | 5 | 1 | 0 | 153 | 18 |
| All aligned | 18/742bp | 116 | 553 | 12 | 104 | 12 | 98 | 0 | 6 | - | - |
Gene flow and genetic differentiation resulted into three haplotypes among the accessions of F. tataricum. Observed homozygous and heterozygous individuals from Tetra primer ARMS PCR were subjected to Hardy Weinberg Equilibrium analysis, which revealed 60 and 33 genotypes with alleles GG and GC with allele frequencies of 62.5% and 37.5% respectively (Fig 5). Further screening of this SNP in intra-varietal genotypes revealed homozygous alleles (64.93%) compared to heterozygous (35.06%) as shown in Fig 6. Phylogenetic study indicated the presence of two F. tataricum groups sharing each SNP sites (949 and 1346th) in LD and PIS separately with F. dibotrys and F. esculentum (Fig 7). Divergence time tree clearly explained the relative and early divergence of the ancestor species belonging to the clade of Fagopyrum spp. and Medicago trancatula than the ancestor species of rest of the dicots clade (Fig 8).
Fig 7. Phylogenetic tree with SNP consistency of each species and accessions.

Figure shows that the SNPs in LD with PIS sharing identity among F. tataricum, F. esculentum and F. dibotrys.
Fig 8. Time tree describing the relative divergence time between Fagopyrum spp. and other dicot species.
Through sequencing, the heterozygosity at 949th bp position was not identified. However, using Tetra primer ARMS PCR, the presence of heterozygous genotypes (with GC allele) from the natural populations was observed. Interestingly, one of the homozygote allele CC was not found through this method. These results are in agreement with Hardy Weinberg Equilibrium. Practically it was not possible to assess the homozygous individuals with CC allele. This allele was predicted to be in frequency of 0.03 (q2 = 0.03) through Hardy Weinberg Equilibrium. The χ2 value was 4.33 with significant P-value of 0.0374 (P<0.05). Using the allele frequency of p allele and q allele (0.82 and 0.18), the genotype frequency was calculated according to the Hardy Weinberg Equilibrium (p2+2pq+q2). Thus, p2 = 0.6732, 2pq = 0.1476 and q2 = 0.03.
Discussion
Sequence characterization of the PAL gene was carried out from Fagopyrum spp. in this study, which plays an important role in rutin and quercetin bio-synthesis pathway. Species specific sequence signatures were observed showing evolutionary significance of Fagopyrum genus as well as putative protein structure. Three insertion mutations and three SNPs were identified in F. tataricum. Among three SNPs, one was singleton variant and other two are PIS, one at 949th and other at 1346th bp positions. SNPs at 949th and 1346th bp position were in intron1 and exon 2 respectively in the PAL gene.
The three insertion mutations in PAL gene caused a variation of stretch of 39 amino acids in exon2 of ORF in comparison with reference PAL protein, ACT68010 (Figs 3 and 4). These insertion mutations caused frame shift of 39 amino acids resulted into different protein isoform as implicated in this present study in accordance with the previous reports [21, 22]. Altered protein due to change of 39 amino acids likely resulting for the evolution of adaptive proteins [23] and may cause structural and functional changes. Theoretical predictions of physico-chemical properties revealed that the protein of allele ‘A1’ (altered protein of 39 amino acids) possessed 57 positively charged residues (Arginine + Lysine) with 6.19 theoretical isoelectric focusing point (pI), while the reference protein possessed 53 positively charged residues with 5.81 pI. The instability index of the variant region with 39 amino acids of allele ‘A1’ alone considered as unstable one, as instability index (II) calculated was 76.08, which exceeded the instability index limit of 40 [24]. The transition but synonymous mutation observed at 1346th position did not change the amino acid ‘serine’. Further, comparison of the putative protein of PAL allele A1 (AHC29062) in reference to PAL putative protein (Protein ID: ACT68010) and indicated for no change in active site (GTITASGDLVPLSYIAG). However, protein modelling suggested a significant alteration in the protein structure and thereby the possible alteration of physico-chemical properties.
The amino acid change in exon2 of F. esculentum is shown in Fig 9. There were five amino acids change and two of them were conservatively altered (Glutamine to Glutamic acid, Valine to Isoleucine) and changes in other three amino acids were non conservative (Proline to Asparagine, Histidine to Arginine, Cysteine to Arginine). Similarly, five amino acid changes were observed in F. dibotrys (Fig 10). In F. dibotrys exon2, the SNPs caused two conservative changes in amino acids (Glutamine to Glutamic acid, Glutamic acid to Aspartic acid), whereas other SNPs caused non conservative alteration (Cysteine to Arginine, Valine to Lysine, Methionine to Lysine). Although, the positions of amino acid change were not collinear between F. esculentum and F. dibotrys. In F. tataricum, no amino acid change was detected from the observed synonymous mutation/SNPs variation, while three insertion mutations caused the change of long stretch amino acids. Apart from these non-silent mutations, there were more than 30 SNPs silent mutations observed in both F. esculentum and F. dibotrys, while in F. tataricum only one silent mutation was observed. Overall, we found more SNP mutations in allogamous species F. esculentum and F. dibotrys, than autogamous F. tataricum. Conversely, indel mutations were observed only in F. tataricum (not in F. dibotrys and F.esculentum) which caused a major change in putative protein (Fig 3). The SNP and indel mutations observed in different F. tataricum, F. dibotrys and F. esculentum indicate towards the evolutionary role of PAL gene in Fagopyrum spp.
Fig 9. Description of conservative and non-conservative amino acid change due to SNP mutation in PAL gene of F. esculentum.
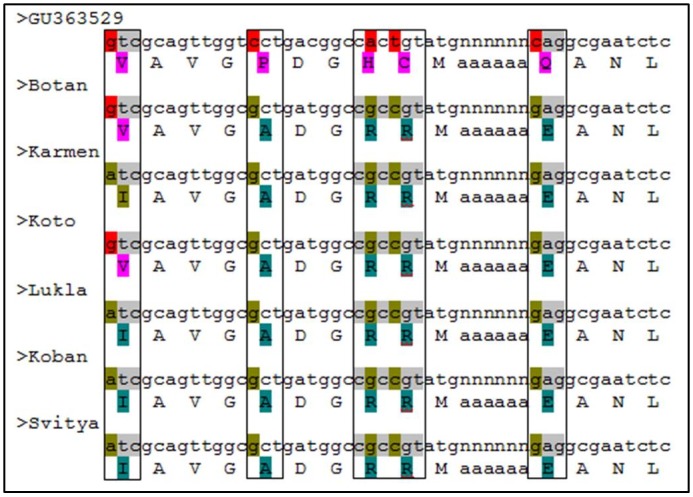
Fig 10. Description of conservative and non-conservative amino acid change due to SNP mutation in PAL gene of F. dibotrys.

The sequences of F. tataricum were represented as two sub-groups (group 1 and 2) according to the 949th and 1346th bp SNPs (Table 3). Genetic diversity within and between the two groups revealed that the group one is more diverged as compared to sub-group two. Genetic differentiation of both assigned sub-groups was statistically significant with pairwise comparison. Haplotype based statistics for the genetic differentiation of these two groups was significant with PM test (Table 3). This finding was further supported by Fst estimate and effective migrants (Nm) indicated towards an absolute migration with low gene flow (Table 3). Similar trend of haplotype diversity was also previously reported [25]. These results clearly indicated the phylogenetic importance of two tightly linked PIS at 949th and 1346th bp SNP positions.
Table 3. Genetic diversity, differentiation and gene flow analysis in F. tataricum.
| Genetic Diversity | ||||||
|---|---|---|---|---|---|---|
| Sub-groups | Sequences | Segregating sites | Haplotypes | Hd | K | PiJC |
| SNP1 G>C and C>T | 5 | 1 | 2 | 0.4 | 0.4 | 0.0006 |
| SNP2 C>G and T>C | 4 | - | 1 | - | - | - |
| Both SNPs | 9 | 3 | 3 | 0.66 | 1.33 | 0.0019 |
| Genetic differentiation of both SNP Sub-groups | ||||||
| χ2 | Hst | Kst | Kst* | Z* | Snn | |
| Estimated value | 9 | 0.64 | 0.83333 | 0.76873* | 2.0644 | 1* |
| Value of PM test | 0.0111* | 0.0190* | 0.0100 * | 0.0100 * | 0.0100 * | 0.0100 * |
| Gene flow of both SNP Sub-groups | ||||||
| Haplotype data | Sequence Data | |||||
| Gst | DeltaSt | GammaSt | Nst | Fst | ||
| Estimated value | 0.64 | 0.00145 | 0.85 | 0.9092 | 0.9091 | |
| Nm | 0.14 | 0.04 | 0.04 | 0.02 | 0.03 |
Hd-Haplotype Diversity, K- Average Nucleotide Differences, PiJC- Nucleotide diversity with Jukes Cantor corrections, χ2- Chi Square test, Hst- Haplotype based statistics, Kst- Sequence based statistics, Z*- Rank Statistics, Snn- Near Neighbour Statistics, Gst- Differentiation of population, GammaSt- Gamma Statistics, Nst- N statistics, Fst- Fixation index, Nm- Effective number of migrants, PM- Permutation test with 1000 replicates,
* 0.05<P.
Putative PAL gene protein of Fagopyrum spp. (generated in our study) was aligned with the protein of PAL gene from other dicot spp. PAL protein, which aligned from Fagopyrum spp. and other dicots led to identify the conserved signature motif ‘GTITASGDLVPLSYIAG’. Further, we calculated relative divergence time (0.8), which revealed an early divergence of the ancestor species of the clade of Fagopyrum spp. and Medicago trancatula from the ancestor species of the clade other dicot spp. (0.7) subjected to analysis. Besides, within a clade, the divergence time revealed an early divergence of Fagopyrum spp. (0.1) than Medicago trancatula (0.0). It is noteworthy fact that both M. trancatula and Fagopyrum spp. are well known for rutin production [26, 6], whereas in most other dicots, it has been predominantly associated with lignin and anthocyanin production [27, 28, 29]. In particular, 8 amino acids were identical between these two species corresponding to F. tataricum PAL protein 642th to 652th amino acid positions: ‘ARTLYNNGASG’ rather than other species. Therefore, protein sequence alignment clearly revealed the close proximity of amino acids of Fagopyrum spp. with Medicago trancatula, which is highly likely associated with rutin bio-synthesis pathway.
There were two SNPs in F. tataricum (SNP at 949th and 1346th bp position) showing LD and PIS and one of them (SNP at 949th bp position) showed association with agronomically important traits. SNPs at 949th and 1346th bp were located in intron (only intron of this gene) and exon2 respectively. The SNP at 949th position was always found to be in LD with 1346th, a mutation in the first site is always paired with the presence of SNP in the second site (i.e. 1346th bp). Interestingly, heterozygosity at these sites (SNP at 949th and 1346th bp position) showed correlation with increased seed number, reduced plant height and 100-kernel weight (Table 4). It is a well-established fact that SNPs at splicing sites or branch points of intron may affect the splicing of intron and exon. As the result mRNA transcript may be abnormal, because of these kinds of mutational consequences of important sites at intron. But in this study, we found a mutation apart from these splicing sites or branch points, so functionally it has no direct role, while the SNP/mutation in intron (949th position) always paired with 1346th exon2 SNP due to LD. If there is alteration in SNP at 949th bp (intron) then there will be alteration in exon at 1346th bp due to LD. Based on these facts we hypothesize that that SNP mutation in exon have ‘functional agronomic’ role. However a definitive test would further confirm this.
Table 4. F. tataricum intraspecies accessions zygosity and phenotypic traits assessment.
| Traits | AVS (numbers) | 100 KW (mg) | APH (cm) |
|---|---|---|---|
| HZ—GG | 256.44 | 2536.94 | 205 |
| HT—GC | 282 | 2056.6 | 151.6 |
| Std. Dev | 4.38 | 4.43 | 4.05 |
| Sum Sq | 3.938 | 3.937 | 3.437 |
| Mean Sq | 0.28 | 0.28 | 0.28 |
| LSD mean | 1.43 | 1.43 | 1.43 |
| LSD CV | 3.50e-15 | 7.36e-15 | 28.39 |
| MSerror | 2.54e-33 | 1.12e-32 | 0.16 |
| F test | 1.109e+32 *** | 2.516e+31*** | 1.719 Ns |
AVS- Average Seeds Per Plant, 100KW- 100 Kernel Weight, APH—Average Plant Height;
***0.001<P, Ns-Not Significant.
Numerous studies have been focused on SNP analysis of PAL gene in different plant species to improve the yield with reference to rutin, anthocyanin, lignin or relevant metabolites [30, 31, 32, 33]. Among Fagopyrum spp. total flavonoid content is commonly higher in F. tataricum than F. esculentum. Among released F. tataricum varieties, ‘Donan’ is very popular and known for high thousand kernel weight as revealed in our study (data not presented). This variety can be utilized as a potential germplasm source for medicinal application.
Polymorphic sites in Fagopyrum spp. at inter and intra species level
Through interspecies sequence analysis of the three Fagopyrum species, we identified PIS and other useful sites (Table 2). Disparity index revealed the existence of homogenous substitution pattern between F. tataricum and F. dibotrys with significant heterogeneity between F. esculentum and F. dibotrys (S1 Table). Distance matrix index values also revealed that the distance between F. tataricum and F. esculentum is more than F. dibotrys. The distance index between F. dibotrys and F. tataricum was between 4–5%, while the distance index with between F. dibotrys and F. esculentum was 15–16% (S2 Table, Fig 7). Similar results were presented in previous reports [34].
F. esculentum and F. tataricum had two PIS, while in F. dibotrys four PIS were observed. Intra-specific SNPs were maximum in F. dibotrys (18) followed by F. esculentum (11) and lest in F. tataricum (3). SVs were also least in F. tataricum than other two allogamous species. In these three species balancing selection maintained the monomorphic sites at 553 positions and thus the variations of only 116 positions allowed to discriminate these species. In contrast, the adaptive mutation reduced the variations of these 553 positions, which are remaining unchanged during evolution (Table 2). Besides, the gene exhibited significant variation with 42 bp deletion in F. tataricum and F. dibotrys as shown in S3 Fig (corresponding to the insertion in F. esculentum).
In F. tataricum three pair of sites with LD was observed and among them, the one between 949th and 1346th bp was statistically significant (S3 Table). Allelic pattern at this LD site in PAL gene have been depicted in Fig 7. LD event in F. tataricum classified this species in two different groups (groups I and II). The SNP allele of F. tataricum group II at 949th bp (Cross species comparison site 952) showed identity in F. esculentum and F. dibotrys at this locus, indicating that this allele was contributed to F. tataricum by F. dibotryis/F. esculentum, while group I allele from some other progenitor. Similar observation for F. tataricum group I allele at 1346th bp (cross species comparison site 1395) supported to above mentioned conclusion. Noticeably, these PIS and/or recombinations were found within 400 bp region of PAL gene. There were other LD events present in this gene among different Fagopyrum spp. as indicated in the S4 Fig. LD sites were more in allogamous species (F. dibotrys and F. esculentum) than F. tataricum. F. tataricum group II was closer to F. dibotrys than group I as shown in Fig 7. It clearly revealed the importance of SNPs with LD and PIS of PAL gene in evolution. These SNP and indel variations clearly indicated that F. tataricum is more closely related to F. dibotrys than F. esculentum (Fig 7 and S3 Fig). The species specific sequence signature in PAL gene of three Fagopyrum spp. has emphasized the phylogenetic importance of this gene.
There were three types of inter-specific SNPs: (i) which represented LD and PIS (ii) other which showed only LD and not PIS and (iii) those which only represented PIS. With reference to the SNPs, which represented both LD and PIS in F. tataricum were species specific i.e. across the species they were not comparable (Table 2). SNPs in two positions, which showed LD of F. tataricum, are not sharing identity in other species, while one SNP among these two were sharing identity in either species. SNPs with PIS alone shared more identity between F. tataricum and F. dibotrys than F. esculentum. These results indicate that species specific SNPs are under selection pressure, when they are in LD. The breakage of LD due to mutation, genetic drift and absence of selection pressure might disturb these SNPs. SNP at the 949th bp position had two alleles ‘GG’ and ‘CC’. Interestingly, in the natural population of F. tataricum we could detect only one homozygote ‘GG’ and heterozygote ‘GC’. The ‘CC’ homozygote was neither identified through sequencing the gene nor through following Tetra primer ARMS PCR strategy. Following the Hardy Weinberg Equilibrium, we predicted the frequency of ‘CC’ homozygote (0.03%) to be rare. This was the most probable reason for not identifying the rare allele ‘CC’ in present study.
Present study provides an in-depth sequence characterization of PAL gene in Fagopyrum spp. which is known for its medicinal value. The sequence information concerning the SNPs/alleles can be used for the identification of elite cultivars from germplasm collections of F. tataricum and related species within the genus Fagopyrum as well as the species from other genus of plant kingdom. Certain insertion/deletions caused major variations of amino acids in F. tataricum possibly due to genomic plasticity events in this species, which harbored beyond normal mutations and thus caused enormous variations. Comparative genomics of these kinds of alleles with other species will excavate the rare mutations in other species. Overall analysis clearly suggested towards an evolutionary significance of PAL gene in the genus Fagopyrum. Informations presented in this report can be efficiently utilized in genetic improvement of Fagopyrum spp. with respect to its medicinal relevance.
Materials and Method
Genotypes and DNA extraction
Sixteen accessions of F. tataricum were utilized for the screening of inter and intra-specific diversity. To facilitate the understanding of the evolutionary relationship, five accessions of F. esculentum and five of F. dibotrys were also included. The genetic material was either obtained from different sources as shown in Table 5.
Table 5. Buckwheat varieties and accessions utilized for the screening of inter- and intra-specific diversity: origin and seed source.
| Species/variety | Origin | Source |
|---|---|---|
| Fagopyrum tataricum | ||
| Golden | Bosnia-Hercegovina | Parco Scientifico e Tecnologico del Molise, Campobasso, Italy |
| Q0001120 | China | Department of Biology, Honghe University, Yunnan, China |
| PI481672, PI481673 | Bhutan | Northeast Regional PI Station, USDA, Agricultural Research Service, Plant Genetic Resources Unit, Geneva, New York, USA |
| PI427235 | Nepal | Northeast Regional PI Station, USDA, Agricultural Research Service, Plant Genetic Resources Unit, Geneva, New York, USA |
| Hei Feng, Hei Qiao-4, Wei 93–8, Xinong 9909 | China | Hodowli Róslin Palikije, Wojciechów, Poland |
| 01Z5100014 | USA | Department of Gene Bank, Division of Genetics and Plant Breeding, Research Institute of Crop Production, Prague-Ruzyne, Czech Republic |
| C8816Malong, C9717 Kuer | China | Plant Germ-Plasm Institute, Graduate School of Agriculture, Kyoto University, Japan |
| N7605Chumoa, N8614 Lukla | Nepal | Plant Germ-Plasm Institute, Graduate School of Agriculture, Kyoto University, Japan |
| Donan, Ishisoba | Japan | Plant Genetic Resources Laboratory, Dept. of Upland Agriculture, National Agricultural Research Center for Hokkaido Region, Shinsei, Memuro-cho, Kasai-gun, Hokkaido, Japan |
| FAG 50 | China | The Leibniz Institute of Plant Genetics and Crop Plant Research (IPK) in Gatersleben, Germany |
| RCAT 061058 | Unknown | The Institute for Agrobotany at Tápiószele, Hungary |
| Fagopyrum esculentum | ||
| Karmen, Svityazyanka | Belarus | RUP The Institute of Arable Farming and Plant Breeding of the National Academy of Sciences of Belarus, Zhodino, Minsk, Belarus |
| Koban, Koto | Canada | Kade Research Ltd., Morden, Manitoba, Canada |
| Luba | Poland | Hodowli Róslin Palikije, Wojciechów, Poland |
| Fagopyrum dibotrys | ||
| FAG 135 | Unknown | The Leibniz Institute of Plant Genetics and Crop Plant Research (IPK) in Gatersleben, Germany |
| FCA3_FAG135 | Unknown | IPK, Gatersleben, Germany |
| FCB3_FAG135 | Unknown | IPK, Gatersleben, Germany |
| FCA1_FAG135 | Unknown | IPK, Gatersleben, Germany |
| FDD1_FAG142 | Unknown | IPK, Gatersleben, Germany |
In order to analyse the intra-varietal zygosity, about four genotypes of each F. tataricum accession were germinated in petri plates, transferred to pots and grown in a greenhouse.
For each genotype, approximately 100 mg of fresh leaves were collected from 4 weeks old plantlets and ground with liquid nitrogen. Total DNA was extracted by CTAB method [35], quantified using MaestroNano Micro-Volume Spectrophotometer (Cat. No. MN-913, Maestrogen) and further diluted with sterile distilled water to obtain a DNA template with a concentration of 50 ng/μl. Similar methodology was followed for the extraction of DNA from individual genotypes of F. esculentum and F. dibotrys accessions as mentioned in the Table 5.
Polymerase Chain Reaction and Sequencing
Specific forward and reverse primers for F. tataricum PAL gene were designed (S4 Table) using reference sequence available at GenBank [13]. Primers, synthesised by Sigma Aldrich S.r.l. (Milano, Italy), allowed amplifying the whole gene, from start to stop codon, within a single Polymerase Chain Reaction (PCR). Alternatively, additional couples of primers were also designed to anneal with different regions, so that the fragments obtained, when overlapped, would cover the whole length of the gene. The PCR reaction volume was fixed at 25 μl and included the following reagents: 2 μl of dNTP 200 uM, 1.5 μl of 3 mM MgCl2, 2.5 μl of 1X Reaction buffer, 0.2 μl of 1 Unit Bioline Taq, 1 μl of 1 pM Forward primer, 1 μl of 1 pM Reverse primer, 15.8 μl of sterile distilled water and 1 μl of DNA template.
The PCR amplification was performed on a Mastercycler® pro (Eppendorf) thermocycler using the following cycling program Initial denaturation at 94°C for 5 minutes, 35 cycles consisting of 1 minute denaturation at 94°C, 1 minute annealing at 57°C and, 1.5 minutes extension at 72°C, and final extension at 72°C for 10 minutes. Samples were stored at 4°C overnight and subsequently added with 2 μl of MaestroSafe Nucleic Acid loading dye (Cat. No. MR-031201, Maestrogen). Amplified fragments were resolved using 2% agarose gel electrophoresis at 90 V for 90 minutes. Each time the expected size band was visualized through an UltraSlim LED Illuminator (Cat. No. SLB-01W, Maestrogen) identified thanks to the comparison with a 1 kb molecular-weight size marker (DNA ladder) (AccuRuler) and excised from the gel with the aid of a clean scalpel. Excised fragments were purified using a Sigma Aldrich GenElute agarose gel purification kit following the manufacturer’s directions.
The concentration of purified fragments was measured with a MaestroNano Micro-Volume Spectrophotometer (Cat. No. MN-913, Maestrogen) and diluted to 56 ng/ μl. 1 μl of the solution was added with 13 μl of sterile distilled water and 1 μl of 10 μM appropriate primer. The reaction mixture obtained was sent for sequencing with AB1 sequencer by Ylichron/Genechron, Rome. Previously synthesised internal primers were used for sequencing (S4 Table).
Utilization of the sequences for SNPs identification and phylogenetic analysis
Chromatograms were screened using Finch TV (Geospiza Inc., USA) chromatogram viewer software. Sequences of the expected fragment were aligned using Clustal W [36] and the presence of SNPs and insertion deletion mutations was manually detected. Among these the potential SNP (949, G>C) with Parsimony Informative Site (PIS) was selected and utilised as a basic platform for designing Tetra primer ARMS PCR. Phylogenetic analysis and Relative Divergence Time were done using MEGA (Molecular Evolutionary Genetic analysis software) [37]. Using PAL gene/alleles generated in this study and with reference gene sequences from NCBI, a Phylogenetic tree was constructed through Maximum Likelihood method with Jukes Cantor (JC) model and 1000 bootstrap resampling. Besides, F. tataricum putative PAL protein (AHC29062) was subjected to BLASTp against non-redundant (nr) protein database at NCBI and 98–100% query coverage with 85%-99% similarity range based dicot orthologous sequences were retrieved and aligned using Clustal X [36]. Subsequently excluding gaps and missing parameter, Time tree was generated through RelTime using Maximum Likelihood method with Jones-Tailor-Thorns (JTT) model and 1000 bootstrap resampling [38]. Nucleotide substitutions were assessed through disparity index [39] using Monte Carlo test with 500 replicates. Genetic analysis was done using a computational algorithm Gamma statistics for gene flow estimates of haplotypes [40], DeltaST [41], Nst [42], Fst [43] of sequence gene flow estimates and other analysis were done using DNAsP V5 [44]. The Hardy Weinberg Equilibrium was assessed with OEGE, Hardy-Weinberg Equilibrium calculator [45] using number of homozygous and heterozygous genotypes resulted from Tetra primer ARMS PCR. Tetra primers were designed using the tools/program available at the web server http://primer1.soton.ac.uk/primer1.html [14]. Tetra primer ARMS PCR reaction master mix and primers are shown in S5 and S6 Tables respectively. Inter and intraspecific SNPs with PIS were subjected for evolutionary analysis. Tetra primers of the SNP locus 949 are Forward Outer Primer 949 (FOP 949), Reverse Outer Primer 949 (ROP 949), Forward Inner Primer 949G (FIP 949G) and Reverse Inner Primer 949C (RIP 949C). To maximize the allele amplification, a single base pair mismatch was introduced at 3’ of inner primers according to the Tetra primer ARMS PCR designing strategy The amplification of the position 949 was achieved with simple profile PCR program as follows: Step 1: 94°C Initial denaturation for 5 Minutes, Step 2: 35 cycles of 25 seconds of denaturation at 94°C, 35 seconds of annealing at 60°C, 30 seconds of extension at 72°C, Step 3: Final extension of 10 minutes at 72°C. The outer band amplicon size was size 484 bp, the G allele and C allele amplicon size was 297bp and 244 bp respectively. In order to improve the amplification the concentration of outer and inner primers were maintained at 1:2 ratio (10 μM of Outer primer and 20 μM of Inner Primer). The amplified products were resolved and visualized using 5% agarose gel. Further primers were designed and the same methodology was applied to amplify either whole PAL gene or fragments of F. esculentum and F. dibotrys and clear chromatogram derived FASTA file fragments were assembled using CAP3 [46].
Protein modelling was done using Geno3D [47] and visualized and annotated with Rasmol [48]. Active site finding was done with Scanprosite tools and the documentation of protein physico- Chemical parameters including instability index was calculated using Protparam tool at Expasy server http://web.expasy.org/tools/protparam/protparam-doc.html [24].
Phenotypic analysis study
All phenotypic and genotypic data was imported to MS-Excel and the results were compared with homozygous and heterozygous alleles for a SNP position with parsimony informative site and linkage disequilibrium. The statistical analysis of phenotypic traits with respect to zygosity was done using R program [49].
Conclusion
F. tataricum and F. esculentum are medicinally important species besides the nature of being pseudocereal food resource crops. Genetics and genomics studies are being focused widely for these two species to enhance their medicinally important flavonoid compounds rutin and quercetin. We here report that the medicinally important PAL gene has an evolutionary significance in Fagopyrum spp. Further, we also provided a detailed sequence characterization of this gene which led to identify novel SNP and indel variations. Informations generated in this report can be efficiently utilized in genetic improvement of the under-utilized domesticated Fagopyrum spp. for nutraceutical food resource.
Supporting Information
(TIF)
Legend: a) SNP 949th position G>C b) SNP 1346th position C>T c) SNP 1017th position G>A d) Insertion of G between 1114th and 1115th positions e) Insertion A between 1722nd and 1723th f) Insertion C between 1744th and 1745th positions g) Insertion of T between 1835th and 1836th positions.
(TIF)
(TIF)
(TIF)
Bold letters are significant, P<0.05.
(DOCX)
(DOCX)
Nucleotides represented in capital letters, are the sites in LD and corresponding positions of other species in represented in small letters. D: Linkage Disequilibrium, D’ = Correlation Coefficient of the pairing SNPs, R2 = Coefficient of determination, χ2 = Chi square test, B = Bonferroni corrections, F = Fisher test, **P<0.01, *P<0.05.
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Acknowledgments
Authors duly acknowledge the financial support of ENEA International Fellowship, Italy. We also extend our sincere thanks to all researchers of Biotechnology and Agroindustry Division (formerly called UTAGRI), ENEA, Italy, for their time to time support and encouragement. Authors also wishes to express their sincere gratitude to Prof. Enrico Porceddu and Prof. Mario Enrico Pè for their constant encouragement and support.
Data Availability
All data are presented as supporting information
Funding Statement
Funding for this work came from an ENEA International Fellowship (2012-14), http://www.enea.it/en/job-study/international-fellowship.
References
- 1.Lee CC, Hsu WH, Shen SR, Cheng YH, Wu SC, Fagopyrum tataricum (Buckwheat) Improved High-Glucose-Induced Insulin Resistance in Mouse Hepatocytes and Diabetes in Fructose-Rich Diet-Induced Mice. Exp. Diabetes Res. 2012;2012:375673 10.1155/2012/375673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization, 2013. Available: http://www.who.int/en/. Accessed 27 November 2015.
- 3.Liu Z, Ishikawa W, Huang X, Tomotake H, Kayashita J, Watanabe H, et al. (2001) A buckwheat protein product suppresses 1,2-dimethylhydrazine-induced colon carcinogenesis in rats by reducing cell proliferation. J Nutr 131(6):1850–3. [DOI] [PubMed] [Google Scholar]
- 4.Ðurendić-Brenesel M, Popović T, Pilija V, Arsić A, Milić M, Kojić D, et al. (2013) Hypolipidemic and antioxidant effects of buckwheat leaf and flower mixture in hyperlipidemic rats. Bosnian J Basic Med 13(2):100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kreft S, Knapp M, Kreft I (1999) Extraction of Rutin from Buckwheat (Fagopyrum esculentum Moench) Seeds and Determination by Capillary Electrophoresis. J. Agric. Food Chem. 47, 4649–4652. [DOI] [PubMed] [Google Scholar]
- 6.Ohsawa R, Tsutsumi T (1995) Inter-varietal variations of rutin content in common buckwheat flour (Fagopyrum esculentum Moench). Euphytica 86: 183–189. [Google Scholar]
- 7.Kreft I, Fabjani N, Germ M (2003) Rutin in buckwheat—Protection of plants and its importance for the production of functional food, Fagopyrum 20: 7–11. [Google Scholar]
- 8.Li X, Park NI, Xu H, Woo SH, Park CH, Park SU (2010) Differential expression of flavonoid biosynthesis genes and accumulation of phenolic compounds in common buckwheat (Fagopyrum esculentum). Journal of Agric Food Chem. 58 (23): 12176–12181. [DOI] [PubMed] [Google Scholar]
- 9.Cho KS, Yun BK, Yoon YH, Hong SY, Mekapogu M, Kim KH, et al. (2015) Complete Chloroplast Genome Sequence of Tartary Buckwheat (Fagopyrum tataricum) and Comparative Analysis with Common Buckwheat (F. esculentum). PLoS ONE. May 12; 10 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yasuda T (2001) Development of tartary buckwheat noodles through research on rutin degrading enzymes and its effect on blood fluidity Advances in Buckwheat Research (II), Chinchon, Korea, IBRA, 499–502. [Google Scholar]
- 11.Li SQ, Zhang QH (2001) Advances in the development of functional foods from buckwheat. Crit Rev Food Sci Nutr. 41:451–464. [DOI] [PubMed] [Google Scholar]
- 12.Deschamps S, Campbell MA (2010) Utilization of next-generation sequencing platforms in plant genomics and genetic variant discovery. Mol Breeding. 25: 553–570. [Google Scholar]
- 13.Li CL, Bai YC, Chen H, Zhao HX, Shao JR, et al. (2012) Cloning, Characterization and Functional Analysis of a Phenylalanine Ammonia-lyase Gene (FtPAL) from Fagopyrum tataricum Gaertn. Plant Mol Biol Rep 30 (5): 1172–1182. [Google Scholar]
- 14.Ye S, Dhillon S, Ke X, Collins AR, Day INM (2001) An efficient procedure for genotyping single nucleotide polymorphisms. Nucleic Acids Res 29 (17): e88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohnishi O (1998) Search for the wild ancestor of buckwheat I. Description of new Fagopyrum (Polygonaceae) species and their distribution in China and the Himalayan hills. Fagopyrum 15: 18–28. [Google Scholar]
- 16.Ohnishi O (1995) Discovery of new Fagopyrum species and its implication for the studies of evolution of Fagopyrum and of the origin of cultivated buckwheat. 175–190 in Current Advances in Buckwheat Research. Vol. I-III. Proc. 6th Int. Symp. On Buckwheat in Shinshu, 24–29 August, (T. Matano and A. Ujihara, eds.). Shinshu University Press.
- 17.Kreft S, Knapp M, Kreft I (1999) Extraction of rutin from buckwheat (Fagopyrum esculentum Moench) seeds and determination by capillary electrophoresis. Journal of Agric Food Chem 47 (11): 4649–52. [DOI] [PubMed] [Google Scholar]
- 18.Ohnishi O, Matsuoka Y (1996) Search for the wild ancestor of buckwheat II. Taxonomy of Fagopyrum (Polygonaceae) species based on morphology, isozymes and cpDNA variability. Genes Genet Syst 71: 383–390. [Google Scholar]
- 19.Steward AN (1930) The Polygonaceae of eastern Asia. Contributions from Gray Herbarium of Harvard University; 88: 1–129. [Google Scholar]
- 20.Yamane K, Yasui Y, Ohnishi O (2003) Intraspecific cpDNA variations of diploid and tetraploid perennial buckwheat, Fagopyrum cymosum (Polygonaceae), Am J Bot 90,3,339–346. 10.3732/ajb.90.3.339 [DOI] [PubMed] [Google Scholar]
- 21.Tian D, Wang Q, Zhang P, Araki H, Yang S, Kreitman M, et al. (2008) Single-nucleotide mutation rate increases close to insertions/deletions in eukaryotes. Nature 455:105–108. 10.1038/nature07175 [DOI] [PubMed] [Google Scholar]
- 22.Vakhrusheva AA, Kazanov MD, Mironov AA, Bazykin GA (2011) Evolution of prokaryotic genes by shift of stop codons. J Mol Evol. 72:138–146. 10.1007/s00239-010-9408-1 [DOI] [PubMed] [Google Scholar]
- 23.Williams LE, Wernegreen JJ (2013) Sequence context of indel mutations and their effect on protein evolution in a bacterial endosymbiont. Genome Biol Evol 5 (3): 599–605. 10.1093/gbe/evt033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, et al. (2005) Protein Identification and Analysis Tools on the ExPASy Server; (In) Walker John M. (ed): The Proteomics Protocols Handbook, Humana Press; 571–607. [Google Scholar]
- 25.Beaty TH, Fallin MD, Hetmanski JB, McIntosh I, Chong SS, Ingersoll R, et al. (2005) Haplotype Diversity in 11 Candidate Genes Across Four Populations. Genetics 1711: 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kowalska I, Stochmal A, Kapusta I, Janda B, Pizza C, Piacente S, et al. (2007) Flavonoids from barrel medic (Medicago truncatula) aerial parts. J Agric Food Chem. 10;55(7):2645–52. [DOI] [PubMed] [Google Scholar]
- 27.Hamberger B, Ellis M, Friedmann M, Souza C, Barbazuk B, Douglas CJ (2007) Genome-wide analyses of phenylpropanoid-related genes in Populus trichocarpa, Arabidopsis thaliana and Oryza sativa: the Populus lignin toolbox and conservation and diversification of angiosperm gene families. Can J Bot. 85:1182–1201. [Google Scholar]
- 28.Bagal UR, Leebens-Mack JH, Lorenz WW, Dean JF (2012) The phenylalanine ammonia lyase (PAL) gene family shows a gymnosperm-specific lineage. BMC Genomics. 13(Suppl 3):S1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boss PK, Davies C, Robinson SP (1996) Analysis of the Expression of Anthocyanin Pathway Genes in Developing Vitis vinifera L. cv Shiraz Grape Berries and the Implications for Pathway Regulation. Plant Physiol. 111(4):1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verdu CF, Guyot S, Childebrand N, Bahut M, Celton JM, Gaillard S, et al. (2014) QTL Analysis and Candidate Gene Mapping for the Polyphenol Content in Cider Apple, PLoS One. 9 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peukert M, Weise S, Röder MS, Matthies IE (2013) Development of SNP markers for genes of the phenylpropanoid pathway and their association to kernel and malting traits in barley. BMC Genetics.14:97 10.1186/1471-2156-14-97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Jong F, Hanley SJ, Beale MH, Karp A (2015) Characterisation of the willow phenylalanine ammonia-lyase (PAL) gene family reveals expression differences compared with poplar. Phytochemistry. 117:90–97. 10.1016/j.phytochem.2015.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dillon SK, Nolan M, Li W, Bell C, Wu HX, Southerton SG (2010) Allelic Variation in Cell Wall Candidate Genes Affecting Solid Wood Properties in Natural Populations and Land Races of Pinus radiata. Genetics. 185:4, 1477–1487. 10.1534/genetics.110.116582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yasui Y, Ohnishi O (1998b) Phylogenetic relationships among Fagopyrum species revealed by nucleotide sequences of the ITS region of the nuclear rRNA gene. Genes Geneti Syst 73: 201–210. [DOI] [PubMed] [Google Scholar]
- 35.Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res. 8(19):4321–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics, 23, 2947–2948. [DOI] [PubMed] [Google Scholar]
- 37.Kumar S, Dudley J (2007) Bioinformatics software for biologists in the genomics era. Bioinformatics 23:1713–1717. [DOI] [PubMed] [Google Scholar]
- 38.Tamura K, Battistuzzi FU, Billing-Ross P, Murillo O, Filipski A, Kumar S (2012) Estimating divergence times in large molecular phylogenies. Proc Natl Acad Sci USA. 109(47):19333–19338. 10.1073/pnas.1213199109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar S, Gadagkar SR (2001) Disparity Index: A simple statistic to measure and test the homogeneity of substitution patterns between molecular sequences. Genetics 158: 1321–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nei M (1973). Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70: 3321–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nei M. (1982). Evolution of human races at the gene level, pp. 167–181. In Bonne-Tamir B., Cohen T., and Goodman R. M. (eds.), Human genetics, part A: The unfolding genome. Alan R. Liss, New York. [PubMed] [Google Scholar]
- 42.Lynch M, Crease TJ (1990). The analysis of population survey data on DNA sequence variation. Mol Biol Evol. 7: 377–394. [DOI] [PubMed] [Google Scholar]
- 43.Hudson RR. Slatkin M, Maddison WP (1992). Estimation of levels of gene flow from DNA sequence data. Genetics 132: 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25 (11): 1451–1452. 10.1093/bioinformatics/btp187 [DOI] [PubMed] [Google Scholar]
- 45.Rodriguez S, Gaunt TR, Day INM (2009) Hardy-Weinberg Equilibrium Testing of Biological Ascertainment for Mendelian Randomization Studies. Am J Epidemiol. 169, 4, 505–514. 10.1093/aje/kwn359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang X, Madan A (1999) CAP3: A DNA sequence assembly program. Genome Res. 9, 868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Combet C, Jambon M, Deléage G, Geourjon C (2002) Geno3D: Automatic comparative molecular modelling of protein. Bioinformatics 18, 213–214. [DOI] [PubMed] [Google Scholar]
- 48.Sayle RA, Milner-White EJ (1995) RasMol: Biomolecular graphics for all Trends Biochem Sci. 20, 9, 374. [DOI] [PubMed] [Google Scholar]
- 49.R Development Core Team (2008). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: ISBN 3-900051-07-0, URL http://www.R-project.org [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(TIF)
Legend: a) SNP 949th position G>C b) SNP 1346th position C>T c) SNP 1017th position G>A d) Insertion of G between 1114th and 1115th positions e) Insertion A between 1722nd and 1723th f) Insertion C between 1744th and 1745th positions g) Insertion of T between 1835th and 1836th positions.
(TIF)
(TIF)
(TIF)
Bold letters are significant, P<0.05.
(DOCX)
(DOCX)
Nucleotides represented in capital letters, are the sites in LD and corresponding positions of other species in represented in small letters. D: Linkage Disequilibrium, D’ = Correlation Coefficient of the pairing SNPs, R2 = Coefficient of determination, χ2 = Chi square test, B = Bonferroni corrections, F = Fisher test, **P<0.01, *P<0.05.
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
All data are presented as supporting information