

Abstract
Objective: To outline the epidemiological profile and prognosis for Ewing's sarcoma in the Brazilian population. Material and Methods: The medical records of 64 patients with intraosseous Ewing's sarcoma who were treated at the Pediatric Oncology Institute, IOP-GRAACC-Unifesp, between 1995 and 2010, were retrospectively evaluated. Results: The statistical analysis on the data obtained did not correlate factors such as sex, trauma, pathological fracture and time taken for case diagnosis with the treatment outcome. Factors such as initial metastasis, lung metastasis, tumor site, age, recurrence and type of surgery showed results corroborating what has been established in the literature. Conclusion: The prognosis in cases of Ewing's sarcoma was mainly influenced by the presence of metastases at the time of diagnosis.
Keywords: Sarcoma, Ewing; Bone Neoplasms; Prognosis
INTRODUCTION
Ewing's tumor was described by James Ewing in 1921, and is a high-grade osteolytic malignant neoplasm(1). It is the third commonest primary malignant bone tumor, after multiple myeloma and osteosarcoma. Among children and young adults, it is the second in frequency after osteosarcoma, and in the population under the age of 15 years, it is the most frequent type2, 3, 4, 5. Cases are mainly diagnosed in the second decade of life, while 20–30% are in the first decade and occurrences are rare in individuals over the age of 30 years and under the age of five years(6). It is extremely rare in Afrodescendent and Asian populations and is slightly more frequent among males(4). The estimated five-year survival in cases of localized disease, i.e. non-metastatic cases, is between 50 and 70%, according to the worldwide literature7, 8, 9. The emergence of chemotherapy in the 1970s and its evolution have contributed towards improvements in prognoses; however, in cases of metastatic disease, the prognosis remains unfavorable and survival continues to be between 18 and 30%1, 8, 10, 11, 12. The lungs are the commonest site for metastases (50%) and, in isolated occurrences, the results presented are slightly better than in cases of extrapulmonary metastasis. Symptoms such as pain and edema have been attributed mainly to the growth of bone lesions, and these tend to be the first sign of the disease. The time that elapses until the diagnosis is made ranges from three to nine months, according to the literature, thus delaying the start of oncological treatment13, 14.
Ewing's tumor most frequently affects the pelvis, femur, tibia, humerus and thoracic wall, but it may affect any bone. It is commonest in the long bones, such as the femur and tibia, and is typically a diaphyseal lesion12, 15.
METHOD
This was a retrospective study based on reviewing the database of all the patients registered in the Oncological Orthopedics Sector of the Federal University of São Paulo, and in this university's Institute of Pediatric Oncology (IOP-GRAACC-Unifesp). Between January 1, 1995, and December 31, 2010, 64 patients with Ewing's tumor underwent oncological treatment at our clinic. All the medical files were reviewed to seek data on sex, age, staging, clinical signs and symptoms, surgical procedures, location of the lesions, anatomopathological results, local recurrence and oncological clinical outcome. All the patients received neoadjuvant and adjuvant chemotherapy. All the patients underwent local lesion control, which was by means of surgery, radiotherapy, or both.
For the statistical analysis, the relationships between age, sex, site, presence of pathological fractures, radiotherapy with late metastatic outcomes, late pulmonary metastasis, recurrence and death were evaluated. The chi-square, Fisher and likelihood ratio tests were used. To analyze estimates of mean length of survival, the Kaplan-Meier function and the results from log-rank test comparisons were used. All the data were analyzed using the SPSS software, version 17.0 (SPSS, Inc., Chicago, IL, USA), and P-values < 0.05 were considered to be statistically significant.
RESULTS
We found 26 female patients (40.6%) and 38 male patients (59.4%). Their mean age was 14.7 years (ranging from four to 30 years).
Among the 64 patients who underwent oncological treatment, eight (12.5%) progressed to death and no orthopedic treatment was performed; three (4.6%) underwent amputation; nine (14.0%) only underwent radiotherapy; and 44 (68.7%) underwent resection of the lesion. Among the latter, 18 cases (40.9%) were managed with surgery alone and 26 (59.1%) with surgery followed by radiotherapy. Over the course of the treatment, seven patients (38.8%) who had been treated with surgery alone, 11 (42.3%) with surgery and radiotherapy and four (44.4%) with radiotherapy alone evolved with metastasis. None of the patients who underwent amputation presented metastasis during the follow-up. There was local recurrence in two patients (11.1%) who had been treated with surgery alone and in seven (26.9%) who had undergone surgery and radiotherapy. Among the patients who underwent limb-preserving surgery and reconstruction, an endoprosthesis was used in 10 (22.7%), followed by biological techniques (Figure 1 A-C) in nine (20.4%) and tumor resection without reconstruction or other surgical techniques in 25 (56.8%). Among the patients who presented local recurrence, six (50%) evolved with late metastasis; of these, three patients (50%) presented pulmonary and non-pulmonary metastasis and three (50%) presented non-pulmonary metastasis. In relation to age, seven (23.3%) of the patients aged < 12 years presented metastases at the time of diagnosis, and 12 (40%) presented late metastasis over the course of the treatment. Among the patients over the age of 12 years, 12 (35.2%) presented metastases at the time of diagnosis, and 11 (35.2%) presented late metastasis during the follow-up. The tumor affected the appendicular skeleton in 39 patients (61.0%) and the axial skeleton in 25 (39.0%). Of these cases, respectively 18 (46.1%) and five (20.0%) evolved with metastases. At the end of the period analyzed, 31 patients (48.4%) did not present any evidence of disease, 11 (17.1%) were alive with metastases in the lungs or other locations and 22 (34.3%) had died due to their disease.
Figure 1.
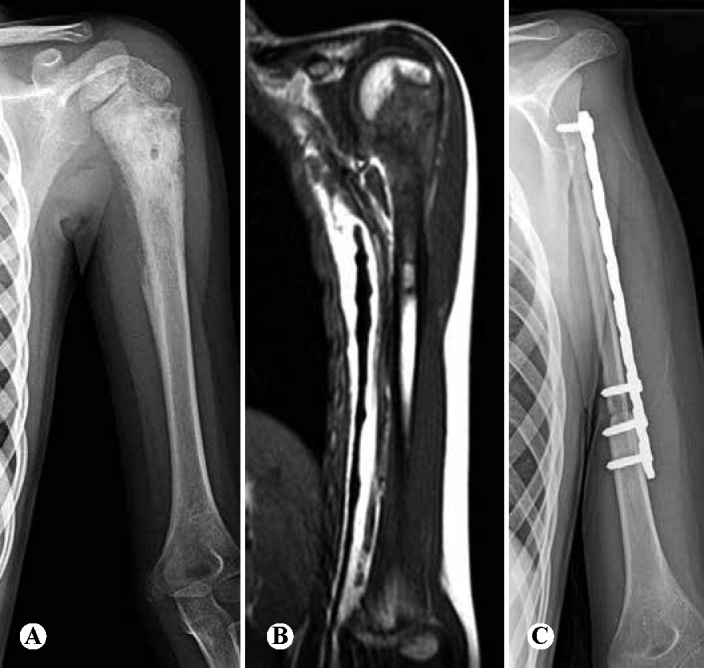
Ten-year-old patient with pain in the proximal region of the left humerus for three months: (A) radiograph showing mixed lesion (osteolytic and osteoblastic), with periosteal reactions in “sunray” pattern and presence of Codman triangle; (B) magnetic resonance imaging showing lesion in the proximal third of the humerus, invading the epiphysis, and also an extraosseous mass; (C) radiograph after resection of the lesion and reconstruction using vascularized fibular tissue, while preserving the fibular proximal epiphysis to enable growth.
The mean length of time that elapsed until the patients in this study were diagnosed was approximately six months. Thirty-four patients were diagnosed with Ewing's tumor within six months and among these, 15 (44.1%) presented late metastasis and 14 (41.1%) died. For 30 patients, the diagnosis was made after more than six months and among these, six (20%) presented late metastasis and six (20%) progressed to death.
DISCUSSION
Ewing's tumor is the third commonest malignancy of bone origin, in order of incidence, only behind multiple myeloma and osteossarcoma. In the population under the age of 15 years, it is the leader in incidence. Our patients’ ages were concentrated between four and 30 years. Age is a prognostic factor in the literature16, 17, and this was observed in our sample, with lower five-year survival among patients over the age of 12 years (50.2%), compared with patients under the age of 12 years (71.7%) (P = 0.075). The mean age of the patients with pulmonary metastasis was slightly lower than the mean age among patients with extrapulmonary metastasis (respectively, P = 0.501 and P = 0.261), thus contrasting with local recurrence, which occurred in slightly older patients (P = 0.592).
We did not observe any differences in sex, trauma or pathological fractures, in relation to pulmonary metastases, extrapulmonary metastases, local recurrence and total number of deaths (Table 1). We did not study the size of the lesion or the degree of tumor necrosis, since the data were not available in the files. These parameters are difficult to measure in cases of Ewing's tumor because soft tissues are frequently a component and because of differences between the imaging examinations.
Table 1.
Comparative analysis on epidemiological factors and outcomes.
| Extrapulmonary metastasis* | Pulmonary metastasis* | Recurrence* | Death* | |
|---|---|---|---|---|
| Sex | 0.728 | >0.999 | 0.53 | 0.615 |
| Trauma | 0.529 | >0.999 | 0.42 | 0.745 |
| Fracture | 0.619 | 0.546 | >0.999 | 0.118 |
P values.
The general estimated five-year survival was 59.5% (Figure 2), and in correlations with sex, location and type of non-chemotherapeutic treatment, no statistical differences were presented (respectively, P = 0.361; P = 0.464 and P = 0.238).
Figure 2.
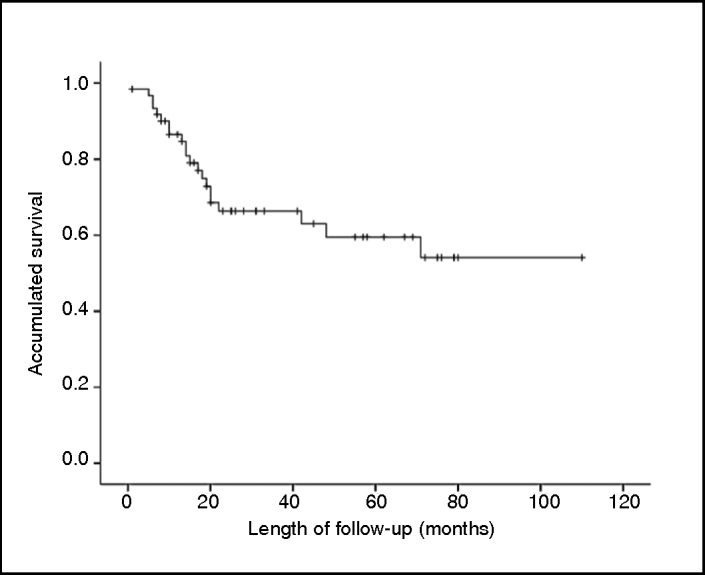
Survival of the patients treated due to Ewing's tumor.
The most frequent location in the appendicular skeleton was in the femur (46%), followed by the tibia (35%) and humerus (0%). In relation to the axial skeleton, the innominate bone was affected in 44% of the cases, followed by the sacrum and spine in 20%. In the literature, an axial location for Ewing's tumor (in the pelvis or spine) is a classical criterion for a poor prognosis(16). In our sample, late metastasis occurred more frequently in appendicular locations (45%; P = 0.051), in relation to axial locations (20.0%), which contradicts the hypothesis of worse prognosis for lesions occurring in the axial skeleton, although this was probably due to the small number of patients. However, death occurred more commonly in cases with axial tumor locations (37.5%; P = 0.603), in comparison with appendicular locations (32.5%).
In cases of localized disease, the current treatment may achieve a five-year disease-free survival rate of 50–70%. However, among patients with initial metastasis, this number only reaches 30%(0,12). The lungs are the commonest site for metastases (50%), and pulmonary metastases present better prognosis than observed for extrapulmonary metastases2, 11, 14. In our sample, 57.1% of the patients with initial metastases died, whereas only 23.3% died among the patients who did not present initial metastasis (P = 0.007).
Ewing's tumor, like other small and round-cell tumors, responds well to radiotherapy, and local control can be achieved through this therapeutic method alone(18). However, complications relating to radiotherapy in pediatric patients, such as lesions in growth cartilage or the risk of developing other secondary forms of neoplasia, restrict this method to inoperable tumors, which are mostly located in the pelvis and spine. Another indication related to local adjuvant treatment, when the surgical margins are seen to be compromised following tumor resection.
Among the patients in our sample who received radiotherapy, 41.7% evolved with late metastasis 41.7% (P = 0.195), 25.7% with local recurrence (P = 0.094) and 30.9% with death (P = 0.446). The prognosis for these patients was worse than among patients who did not receive this type of therapy: respectively 25.9%, 7.4% and 29.6%. Comparisons between the groups should be analyzed carefully, since patients who receive indications for radiotherapy tend to present larger tumors in locations that are less favorable for resection(10).
Despite aggressive treatment, it is reported in the literature that around 20 to 40% of the patients with localized disease and 00% of the patients with metastases evolve to death due to disease recurrence or progression(19). Most of the cases of recurrence occur after the termination of the treatment, such that 00% occur more than two years after the initial diagnosis. The time that elapses until recurrence is a major prognostic factor, such that the survival rate from recurrences occurring more than two years after the initial diagnosis is 25%, but it is less than 10% among cases of early recurrence11, 20. In our sample, the mean survival among the patients who presented recurrence was 46 months, in contrast with the 73 months of survival among the patients who did not present recurrence (P = 0.495).
Despite the established importance of early diagnosis, the time that elapsed until diagnosis in our sample did not influence any of the variables: late metastasis (P = 0.219), late pulmonary metastasis (P = 0.903), local recurrence (P = 0.521), death (P = 0.660) or five-year survival (P = 0.796). This leads us to believe that correct treatment is a more important factor than the time that elapses before it starts. This also corroborates the classical recommendation that oncological patients should be referred to treatment centers, instead of starting with procedures such as biopsies in non-specialist hospitals, in order to accelerate reaching a diagnosis.
Classically, Ewing's tumor presents genetic abnormalities that can be identified in 80% of the cases, represented by the reciprocal translocation between chromosomes 11 and 22, i.e. t(11;22) (q24;q12), which results in expression of the protein EWS/FLI-1(21). Presence of this translocation in the tumor is a diagnostic factor for Ewing, but it is not prognostic. Presence of the same translocation in the patient's peripheral blood, or in bone marrow aspirate away from the area of tumor, is a sign of disseminated disease and therefore of poor prognosis(22). In our clinic, genetic analysis on this translocation began in 2008, but it has not yet been possible to assess these data.
CONCLUSION
From this study, we concluded that factors like sex, trauma, pathological fractures and time elapsed until the diagnosis did not modify the patients’ outcomes. Age at the time of the diagnosis was important, but it was also not decisive with regard to the result from the treatment. The majority of the patients underwent orthopedic surgical treatment with preservation of the affected limb. The results were worse for patients with tumors in axial locations. Presence of an initial metastasis was the main factor for worse evolution among the patients, and this was statistically significant. The results should be assessed carefully, because the limited number of patients makes it difficult to trace out a more accurate panorama of Ewing's tumor in our population.
ACKNOWLEDGEMENTS
We are grateful for the collaboration and assistance of Rogério Ruscitto do Prado, a statistician in the Department of Orthopedics and Traumatology, Federal University of São Paulo (EPM/Unifesp).
Footnotes
Work performed in the Department of Orthopedics and Traumatology, Federal University of São Paulo (DOT-EPM/Unifesp), São Paulo, SP, Brazil.
REFERENCES
- 1.Ewing J. Diffuse endothelioma of Bone. Proc New York Path Soc. 1921;21:17–24. [Google Scholar]
- 2.Esiashvili N, Goodman M, Marcus RB., Jr Changes in incidence and survival of Ewing sarcoma patients over the past 3 decades: Surveillance Epidemiology and End Results data. J Pediatr Hematol Oncol. 2008;30(6):425–430. doi: 10.1097/MPH.0b013e31816e22f3. [DOI] [PubMed] [Google Scholar]
- 3.Leavey PJ, Mascarenhas L, Marina N, Chen Z, Krailo M, Miser J. Prognostic factors for patients with Ewing sarcoma (EWS) at first recurrence following multi-modality therapy: A report from the Children's Oncology Group. Pediatr Blood Cancer. 2008;51(3):334–338. doi: 10.1002/pbc.21618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li S, Siegal GP. Small cell tumors of bone. Adv Anat Pathol. 2010;17(1):1–11. doi: 10.1097/PAP.0b013e3181bb6b9c. [DOI] [PubMed] [Google Scholar]
- 5.Scotlandi K, Remondini D, Castellani G, Manara MC, Nardi F, Cantiani L. Overcoming resistance to conventional drugs in Ewing sarcoma and identification of molecular predictors of outcome. J Clin Oncol. 2009;27(13):2209–2216. doi: 10.1200/JCO.2008.19.2542. [DOI] [PubMed] [Google Scholar]
- 6.Bernstein M, Kovar H, Paulussen M, Randall RL, Schuck A, Teot LA. Ewing's sarcoma family of tumors: current management. Oncologist. 2006;11(5):503–519. doi: 10.1634/theoncologist.11-5-503. [DOI] [PubMed] [Google Scholar]
- 7.Burdach S, Meyer-Bahlburg A, Laws HJ, Haase R, van Kaik B, Metzner B. High-dose therapy for patients with primary multifocal and early relapsed Ewing's tumors:results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol. 2003;21(16):3072–3078. doi: 10.1200/JCO.2003.12.039. [DOI] [PubMed] [Google Scholar]
- 8.Hunold A, Weddeling N, Paulussen M, Ranft A, Liebscher C, Jürgens H. cyclophosphamide in patients with refractory or relapsed Ewing tumors. Pediatr Blood Cancer. 2006;47(6):795–800. doi: 10.1002/pbc.20719. [DOI] [PubMed] [Google Scholar]
- 9.Scotlandi K, Perdichizzi S, Bernard G, Nicoletti G, Nanni P, Lollini PL. Targeting CD99 in association with doxorubicin: an effective combined treatment for Ewing's sarcoma. Eur J Cancer. 2006;42(1):91–96. doi: 10.1016/j.ejca.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 10.Grier HE, Krailo MD, Tarbell NJ, Link MP, Fryer CJ, Pritchard DJ. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing's sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med. 2003;348(8):694–701. doi: 10.1056/NEJMoa020890. [DOI] [PubMed] [Google Scholar]
- 11.Huang M, Lucas K. Current therapeutic approaches in metastatic and recurrent ewing sarcoma. Sarcoma. 2011;2011:863210. doi: 10.1155/2011/863210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miser JS, Goldsby RE, Chen Z, Krailo MD, Tarbell NJ, Link MP. Treatment of metastatic Ewing sarcoma/primitive neuroectodermal tumor of bone: evaluation of increasing the dose intensity of chemotherapy-a report from the Children's Oncology Group. Pediatr Blood Cancer. 2007;49(7):894–900. doi: 10.1002/pbc.21233. [DOI] [PubMed] [Google Scholar]
- 13.Barker LM, Pendergrass TW, Sanders JE, Hawkins DS. Survival after recurrence of Ewing's sarcoma family of tumors. J Clin Oncol. 2005;23(19):4354–4362. doi: 10.1200/JCO.2005.05.105. [DOI] [PubMed] [Google Scholar]
- 14.Schleiermacher G, Peter M, Oberlin O, Philip T, Rubie H, Mechinaud F. Increased risk of systemic relapses associated with bone marrow micro-metastasis and circulating tumor cells in localized ewing tumor. J Clin Oncol. 2003;21(1):85–91. doi: 10.1200/JCO.2003.03.006. [DOI] [PubMed] [Google Scholar]
- 15.Jedlicka P. Ewing Sarcoma, an enigmatic malignancy of likely progenitor cell origin, driven by transcription factor oncogenic fusions. Int J Clin Exp Pathol. 2010;3(4):338–347. [PMC free article] [PubMed] [Google Scholar]
- 16.Pradhan A, Grimer RJ, Spooner D, Peake D, Carter SR, Tillman RM. Oncological outcomes of patients with Ewing's sarcoma: is there a difference between skeletal and extra-skeletal Ewing's sarcoma? J Bone Joint Surg Br. 2011;93(4):531–536. doi: 10.1302/0301-620X.93B4.25510. [DOI] [PubMed] [Google Scholar]
- 17.Rosito P, Mancini AF, Rondelli R, Abate ME, Pession A, Bedei L. Cooperative Study for the treatment of children and young adults with localized Ewing sarcoma of bone: a preliminary report of 6 years of experience. Cancer. 1999;86(3):421–428. doi: 10.1002/(sici)1097-0142(19990801)86:3<421::aid-cncr10>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 18.Donati D, Yin J, Di Bella C, Colangeli M, Bacci G, Ferrari S. Local and distant control in non-metastatic pelvic Ewing's sarcoma patients. J Surg Oncol. 2007;96(1):19–25. doi: 10.1002/jso.20752. [DOI] [PubMed] [Google Scholar]
- 19.Huang HY, Illei PB, Zhao Z, Mazumdar M, Huvos AG, Healey JH. Ewing sarcomas with p53 mutation or p16/p14ARF homozygous deletion: a highly lethal subset associated with poor chemoresponse. J Clin Oncol. 2005;23(3):548–558. doi: 10.1200/JCO.2005.02.081. [DOI] [PubMed] [Google Scholar]
- 20.Subbiah V, Anderson P. Targeted therapy of Ewing's Sarcoma. Sarcoma. 2011;2011:686985. doi: 10.1155/2011/686985. Epub 2010 Oct 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dei Tos AP, Dal Cin P. The role of cytogenetics in the classification of soft tissue tumours. Virchows Arch. 1997;431(2):83–94. doi: 10.1007/s004280050073. [DOI] [PubMed] [Google Scholar]
- 22.Balamuth NJ, Womer RB. Ewing's sarcoma. Lancet Oncol. 2010;11(2):184–192. doi: 10.1016/S1470-2045(09)70286-4. [DOI] [PubMed] [Google Scholar]