

Abstract
Whereas the roles of the canonical wingless-type MMTV (mouse mammary tumor virus) integration site family (WNT) signaling pathway in the regulation of ovarian follicle growth and steroidogenesis are now established, noncanonical WNT signaling in the ovary has been largely overlooked. Noncanonical WNTs, including WNT5a and WNT11, are expressed in granulosa cells (GCs) and are differentially regulated throughout follicle development, but their physiologic roles remain unknown. Using conditional gene targeting, we found that GC-specific inactivation of Wnt5a (but not Wnt11) results in the female subfertility associated with increased follicular atresia and decreased rates of ovulation. Microarray analyses have revealed that WNT5a acts to down-regulate the expression of FSH-responsive genes in vitro, and corresponding increases in the expression of these genes have been found in the GCs of conditional knockout mice. Unexpectedly, we found that WNT5a regulates its target genes not by signaling via the WNT/Ca2+ or planar cell polarity pathways, but rather by inhibiting the canonical pathway, causing both β-catenin (CTNNB1) and cAMP responsive element binding (CREB) protein levels to decrease via a glycogen synthase kinase-3β-dependent mechanism. We further found that WNT5a prevents follicle-stimulating hormone and luteinizing protein from up-regulating the CTNNB1 and CREB proteins and their target genes, indicating that WNT5a functions as a physiologic inhibitor of gonadotropin signaling. Together, these findings identify WNT5a as a key regulator of follicle development and gonadotropin responsiveness.—Abedini, A., Zamberlam, G., Lapointe, E., Tourigny, C., Boyer, A., Paquet, M., Hayashi, K., Honda, H., Kikuchi, A., Price, C., Boerboom, D. WNT5a is required for normal ovarian follicle development and antagonizes gonadotropin responsiveness in granulosa cells by suppressing canonical WNT signaling.
Keywords: female fertility, conditional gene targeting, microarray
The pituitary gonadotropins follicle-stimulating hormone (FSH) and luteinizing hormone (LH) represent the major endocrine regulators of ovarian function and are essential for follicle development beyond the secondary stage (1, 2). In addition to the endocrine level of control, follicle development is regulated by several paracrine and autocrine factors produced within the ovary itself. These factors notably include IGF-1, steroid hormones, prostaglandins, epidermal growth factor–like molecules and several TGF-β superfamily members, all of which are indispensable for normal ovarian function and female fertility. The gonadotropins can affect the expression and function of these intraovarian factors; conversely, these factors can modulate follicular responses to the gonadotropins (1–3).
Recent studies have established the wingless-type MMTV (mouse mammary tumor virus) integration site (WNT) family of secreted glycoproteins as yet another class of signaling molecules that act to modulate and coordinate follicular responses to the gonadotropins and whose activities are indispensable for ovarian function and female fertility. During FSH-stimulated follicle growth, the WNT/β-catenin (CTNNB1)–signaling pathway (also known as the canonical pathway) appears to play a central role. The binding of WNT to a cognate frizzled (FZD) receptor results in the stabilization of the signaling effector CTNNB1, which then translocates to the nucleus and associates with several transcription factors to alter the transcriptional activity of target genes (4, 5). In granulosa cells (GCs), CTNNB1 has been shown to bind the transcription factor NR5A1 (steroidogenic factor-1), resulting in the up-regulation of cytochrome P450 family 19, subfamily a, polypeptide1 (Cyp19a1; aromatase) expression in vitro (6). Further in vitro experiments using the Cre/LoxP system to knock down CTNNB1 expression in primary cultured GCs have confirmed its requirement for FSH-induced estrogen synthesis (7). The identity of the WNT signaling molecule that acts upstream of CTNNB1 in this context remains unclear. WNT2 is expressed in GCs, has been shown to stimulate proliferation in vitro, and signals via the CTNNB1 pathway (8, 9), but its in vivo role has not yet been established. Likewise, WNT4 is essential for antral follicle development, and its overexpression results in increased Cyp19a1 expression in vitro; however, its conditional inactivation does not affect estrogen biosynthesis in vivo (10). Another possibility is that, in addition to (or perhaps instead of) WNTs, the activation of CTNNB1 signaling in GCs is caused by FSH itself. Indeed, FSH has been found to induce the (Ser552/Ser665) phosphorylation of CTNNB1 in a PKA-dependent manner, resulting in an increase in Lhcgr transcriptional activity (11). Another recent study expanded on the role of CTNNB1 during follicle growth in a transgenic mouse model in which CTNNB1 was constitutively activated in the GCs of antral follicles (12). The results showed a marked enhancement of FSH action, inducing an overexpression of FSH-responsive genes including Cyp19a1, Nr5a1, Fshr, and Ccnd2 and promoting follicle growth (12).
Considerable progress has also been made in the elucidation of the roles of WNT signaling in LH-regulated processes. LH induces the expression of several WNT signaling effectors in both granulosa and cumulus cells, including WNT4, the receptors FZD1 and -4, and secreted frizzled-related protein (SFRP)-2 and -4 (1, 2). The conditional inactivation of Wnt4 in GCs in vivo blunts the induction of genes involved in ovulation and steroidogenesis (Adamts1, Ptgs2, Star, and Cyp11a1) in response to human chorionic gonadotropin (hCG)/LH, and results in lower circulating progesterone (P4) levels (10). Female Fzd4-null mice are sterile, at least in part because of the abnormal development and function of the corpus luteum (CL), including decreased P4 synthesis and altered morphology, gene expression, and vasculogenesis (13). In addition, Fzd1-null mice are subfertile, and gene expression analyses of cumulus-oocyte complexes (COCs) reveal a blunted response of both oocyte and cumulus genes to LH/hCG (14). As for FSH, LH may also activate CTNNB1 signaling independent of WNT/FZD. Using a bovine luteal cell model, Roy et al. (15) observed that LH induces the phosphorylation (and inactivation) of glycogen synthase kinase (GSK)-3β in a PKA-dependent manner. As phosphorylation of CTNNB1 by GSK3β normally causes its degradation, LH thereby stabilizes CTNNB1, resulting in the transcriptional up-regulation of STAR and increased P4 synthesis.
WNTs are frequently categorized according to pathways by which they signal. Certain WNTs, such as WNT1 and -3a, preferentially (or exclusively) activate the canonical pathway, whereas WNT5a and -11 are associated with noncanonical pathways in mammalian cells. The 2 best characterized noncanonical pathways are the planar cell polarity (PCP) and WNT/Ca2+ pathways (16, 17). In the latter, the WNT-FZD complex interacts with heterotrimeric G proteins to activate PLC, resulting in increased intracellular Ca2+ concentrations and the activation of Ca2+-dependent effectors including PKC, calmodulin-dependent kinase (CAMK)-II, and calcineurin (18–21). In the PCP pathway, the WNT-FZD complex activates 2 parallel pathways involving the small GTPases, RHO and RAC (22), which in turn activate disheveled-associated activator of morphogenesis (DAAM)-1, RHO-associated kinase (ROCK) and cJUN kinase (JNK) (23–26). DAAM1, ROCK, and JNK then act via different effectors, mainly to modify the actin cytoskeleton, leading to cytoskeletal rearrangement (27). However, JNK-mediated phosphorylation of the transcription factor c-JUN also occurs in certain contexts, resulting in the transcriptional activation of target genes (17, 28).
Little is known of noncanonical WNT signaling in the ovary. Wnt5a and Wnt11 are expressed in mouse GCs (29), and Wnt5a transcripts have been detected in human luteinized GCs and cumulus cells (30, 31). We have recently identified Wnt5a as a gene whose expression is down-regulated by FSH in a bovine primary GC culture model (32). We found that exogenous WNT5a suppressed FSH-stimulated estradiol (E2) and P4 secretion and CYP19A1 expression, but stimulated the expression of FOS and JUN by signaling via the PCP pathway. Although the physiologic relevance of these findings is not clear, it has been proposed that WNT5a plays a role in suppressing steroidogenesis during follicular atresia. The objectives of the present study were to determine the physiologic roles of noncanonical WNTs in the mouse ovary and their mechanisms of action. To this end, we used conditional targeting of WNT5a and -11 as a primary experimental approach.
MATERIALS AND METHODS
Animal models, fertility trials, ovulation rate, and follicle counting
C57BL/6J mice [referred to herein as wild-type (WT)] were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and Wnt5atm1.1Homy (RBRC04609, hereafter Wnt5aflox) mice were obtained from Riken BRC (Tsukuba, Ibaraki, Japan). Genotyping analyses for the Wnt5a alleles were performed by PCR, with DNA from tail biopsies and the primers 5′- GTGAGGGACTGGAAGTTGCAGGA-3′, 5′-CAACGGAAACATCCGAGGTCTCT-3′, and 5′-CCAGCCACTGGCTTTAGAGAAAGT-3′, using the following cycling conditions: 2 min at 95°C for 1 cycle, 30 s at 95°C, 30 s at 68°C, and 30 s at 72°C for 35 cycles; and 7 min at 72°C, for 1 cycle. Primers were designed to generate PCR products of 398 bp for the floxed allele, 335 bp for the Cre-recombined allele, and 243 bp for the WT allele. Genotype analyses of Wnt11tm1.1Khay (33) (hereafter, Wnt11flox), Amhr2tm3(cre)Bhr (34) (hereafter, Amhr2cre) and Tg(CYP19A1-cre)1Jri (35) (hereafter, CYP19-cre) mice were performed as has been described (33–35). To assess female fertility, 8-wk-old females of experimental and control genotypes were housed with 8-wk-old WT males and monitored daily for litters. Litter sizes were recorded at birth. The males were removed after 6 mo, and the experiment was terminated 22 d later to allow for the final litter. To determine ovulation rates, 8–10-wk-old females of experimental and control genotypes were housed with WT males and monitored daily for the presence of a vaginal plug. Females were then euthanized, and the oviducts were removed and placed in sterile saline under a dissection microscope. COCs were released by tearing open the ampullae of the oviducts with forceps and were counted. Follicle counting was performed on formalin-fixed, paraffin-embedded ovaries. In all cases, only the left ovary from each animal was analyzed. Serial sections were prepared at a thickness of 5 μm, and every fifth section was stained with hematoxylin and eosin. Follicles in which the oocyte nucleus was visible were counted, classified according to Pedersen’s system, and scored as healthy or atretic, as described (10). All animal procedures were approved by the institutional animal care and use committee and conformed to the International Guiding Principles for Biomedical Research Involving Animals.
GC isolation and culture
All materials were obtained from Thermo Scientific-Life Technologies (Burlington, ON, Canada), unless otherwise stated. Ovaries were obtained from immature (21- to 26 d-old) mice 48 h after administration of equine (e)CG (Folligon, 5 IU i.p.; Intervet, Kirkland, QC, Canada) followed or not by hCG (Chorulon, 5 IU i.p.; Intervet) for 4–12 h. GCs were isolated by placing the ovaries in HBSS and puncturing them with 26 gauge needles to release the GCs (36). GCs for primary culture were obtained from eCG-treated immature mice, as described above; pooled; and suspended, in modified Eagle’s medium containing 1% fetal bovine serum, 0.25 nM sodium pyruvate, and 3 mM l-glutamate. GCs were seeded in 48-well culture plates (divided such that each well received GCs equivalent to 0.7 ovary) and incubated for 3 h at 37°C with 5% CO2. Culture medium was then replaced with serum-free medium for 2 h before addition of recombinant WNT11 or -5a (R&D Systems, Minneapolis, MN, USA). For some experiments, small-molecule inhibitors (Tocris Bioscience, Bristol, United Kingdom) were added to the culture medium 1 h before addition of WNT5a. These included SP600125 (cat. no. 1496; 100 μm), NSC23766 (cat. no. 2161; 100 μm), KN-93 (cat. no. 1278; 2.5 μm), Xestrospongin C (cat. no. 1280; 5 μm), GF109203X (cat. no. 0741; 30 μm), and SB216763 (cat. no. 1616; 1 μm). To study the effects of WNT5a on gonadotropin signaling, GCs were pretreated with WNT5a (3.5 μg/ml) for 1 h before addition (or not) of recombinant human FSH or LH (50 ng/ml; National Hormone and Peptide Program, Torrance, CA, USA).
Either immediately after isolation or after culture, GCs were lysed with RLT buffer (Qiagen, Montreal, QC, Canada) or Laemmli loading buffer and stored at −80°C before RNA extraction or SDS-PAGE.
Real-time RT-PCR and microarray analyses
Total RNA from GCs was extracted with the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. Reverse transcription was performed with 200 ng RNA and the SuperScript Vilo cDNA synthesis kit (Thermo Scientific-Invitrogen, Carlsbad, CA, USA). Real-time PCR was performed with SsoAdvanced Universal SYBR Green Supermix (172-5274; Bio-Rad, Mississauga, ON, Canada) and a CFX96 Touch instrument (Bio-Rad). Each PCR reaction consisted of 7.5 μl of SsoAdvanced SYBR Green PCR Master Mix, 2.3 μl of water, 4 μl of cDNA sample, and 0.6 μl (10 pmol) of gene-specific primers (Supplemental Table S1). Cycling conditions were 3 min at 95°C followed by 40 cycles of 15 s at 95°C, 30 s at 60°C, and 30 s at 72°C. To quantify relative gene expression, the cycle threshold (Ct) of target gene amplification was normalized to the expression level of a housekeeping gene (Rpl19), according to the ratio, R = ECt Rpl19/ECt target, where E is the amplification efficiency for each primer pair.
Microarray analyses were performed in triplicate on RNA samples from cultured GCs treated (or not) with 3.5 μg/ml recombinant WNT5a or -11 for 3 h, as described above, and using Agilent 8×60k Mouse Gene Expression Arrays (Agilent Technologies, Inc., Mississauga, ON, Canada). All steps of RNA quality control, probe synthesis, hybridization, washing, and array scanning were performed by the McGill University (Genome Quebec) Innovation Center (Montreal, QC, Canada). Microarray data were analyzed with FlexArray 1.6.1 software (Genome Quebec). P > 0.05 and 2.0-fold change cutoffs were used for identification of differentially expressed genes (WNT5a- or WNT11-treated vs. untreated control).
Immunohistochemistry
Paraformaldehyde-fixed, paraffin-embedded ovaries were sectioned at a thickness of 4 μm. Immunohistochemical analysis was performed with an anti-cleaved caspase 3 antibody (9661S; Cell Signaling Technology, Danvers, MA, USA) incubated at a 1:100 dilution overnight at 4°C. Subsequent detection steps were performed with the Vectastain Elite ABC kit and the 3,3′ diaminobenzidine peroxidase substrate kit (Vector Laboratories, Burlingame, CA, USA), as has been described (37). The slides were lightly counterstained with hematoxylin before mounting.
Immunoblot analysis
Immunoblot analysis was performed as described elsewhere (38), using primary antibodies against nonphosphorylated (Ser33/Ser37/Thr41) (i.e., active) CTNNB1 (cat. no. 8814), phospho-CTNNB1 (Ser552) (cat. no. 9566s), total CTNNB1 (cat. no. 8480), phospho-cAMP responsive element binding (CREB) (cat. no. 9191), total CREB (cat. no. 9197), phospho-JNK (cat. no. 9251), total JNK (cat. no. 9252), phospho-JUN (cat. no. 2361, 1:500), total JUN (cat. no. 9165, 1:500), phospho-CAMKΙΙ (cat. no. 3361), total CAMKΙΙ (cat. no. 4436), phospho-GSK3β (cat. no. 9323), total GSK3β (cat. no. 12456), phospho-CAPZIP [capping (actin filament) muscle Z-line interacting protein] (cat. no. 07-2190; EMD-Millipore, Billerica, MA, USA) CAPZIP (cat. no. 07-2189; 1:250; EMD-Millipore) or ACTB (cat. no. sc-47778; 1:10000; Santa Cruz Biotechnology, Dallas, TX, USA) diluted in 5% bovine serum albumin. All antibodies mentioned were obtained from Cell Signaling Technology (Danvers, MA, USA) and used at a 1:1000 dilution, unless otherwise specified. After the membranes were washed 3 times with Tween 20–Tris-buffered saline (TTBS), they were incubated for 1 h at room temperature with anti-rabbit horseradish peroxidase–conjugated IgG (GE Healthcare Life Sciences, Baie d’Urfé, QC, Canada) diluted 1:10,000 in 5% nonfat dry milk in TTBS. In some instances, membranes were sequentially stripped and reprobed with several antibodies. Protein bands were visualized by ECL (EMD Millipore) and quantified with the ChemiDoc MP detection system and Image Lab software (Bio-Rad).
Steroid hormone measurement
Blood samples were collected by cardiac puncture before euthanasia. E2 and P4 levels in the serum were determined by ELISA and radioimmunoassay, respectively, and FSH and LH were measured with the Pituitary Panel Multiplex kit (EMD Millipore). All assays were performed by the Ligand Assay and Analysis Core Laboratory of the University of Virginia (Charlottesville, VA, USA).
Statistical Analyses
All statistical analyses were performed with Prism 4.0a (GraphPad Software Inc., La Jolla, CA, USA) software. All the data sets (ovary weights, follicle numbers, mRNA expression, protein expression, and steroid hormone levels) were subjected to the F test to determine the equality of variances. Data were transformed to logarithms if they were not normally distributed. Two-tailed t tests were used when 2 experimental groups were compared or ANOVA (with Tukey’s multiple comparisons posttest) to compare 3 or more groups. All data are presented as means ± sem.
RESULTS
Wnt5a expression in GCs is essential for normal female fertility
To study Wnt5a and Wnt11 expression in GCs and their regulation by gonadotropins in vivo, immature mice were treated with (FSH mimetic) eCG for 48 h, to stimulate follicle growth, followed by (LH mimetic) hCG, to provoke ovulation and luteinization (ovulation occurs 12–14 h after hCG in this model). Results showed a gradual increase in Wnt5a mRNA levels throughout follicle development that reached statistical significance near the time of ovulation (Fig. 1). Conversely, eCG did not affect Wnt11 expression, but hCG induced a transient increase in Wnt11 mRNA that returned to basal levels by the time of ovulation. These results indicate that Wnt5a and Wnt11 are differentially regulated by gonadotropins during follicle development.
Figure 1.

In vivo regulation of Wnt5a and Wnt11 mRNA levels in GCs by gonadotropins. Immature (21- to 26-d-old) female mice were injected with eCG (5 IU, i.p.) 44-48 h before the administration of hCG. GCs were isolated by needle puncture. mRNA levels were determined by RT-qPCR and normalized to the housekeeping gene Rpl19 (n = 4 animals/time point). Data are means ± sem. Different letters show statistically significant differences between strains. P < 0.05.
To study the physiologic functions of Wnt5a and Wnt11 in GCs, we generated conditional knockout (cKO) mice by mating strains bearing floxed alleles to both the CYP19-cre and Amhr2cre strains. This strategy was designed to target distinct stages of follicle development, as the Amhr2cre knockin allele drives Cre activity in GCs mainly from the secondary stage onward (34), whereas the CYP19-Cre transgene drives Cre expression from the antral stage (35). RT-quantitative (q)PCR analyses of GCs from eCG-treated immature mice showed a significant (P < 0.05) reduction (∼4–6-fold) in Wnt5a and Wnt11 mRNA levels in corresponding cKO mice relative to controls, using either the Amhr2cre or the CYP19-Cre strains (Fig. 2; Supplemental Fig. S1). No compensatory overexpression of Wnt11 was observed in the Wnt5a cKO model, or vice versa. Six-month mating trials were then conducted, to study the effects of Wnt5a and Wnt11 loss on female fertility. Conditional inactivation of Wnt5a in the Amhr2cre strain resulted in a ∼52% decrease in average litter sizes and a ∼20% decrease in the CYP19-Cre strain (P < 0.05; Table 1; Supplemental Table S2). Conversely, loss of Wnt11 did not result in a loss of fertility with either Cre driver, and the concomitant inactivation of Wnt5a and Wnt11 did not result in a greater loss of fertility than the inactivation of Wnt5a alone (Supplemental Tables S3, S4, S5). These results indicate that Wnt5a expression in GCs is necessary for normal female fertility and that its loss at the preantral stage (i.e., driven by the Amhr2cre) results in a more severe phenotype than when it is lost at later stages. Wnt11, on the other hand, appears dispensable for female fertility, and its expression in GCs cannot compensate for the loss of Wnt5a.
Figure 2.
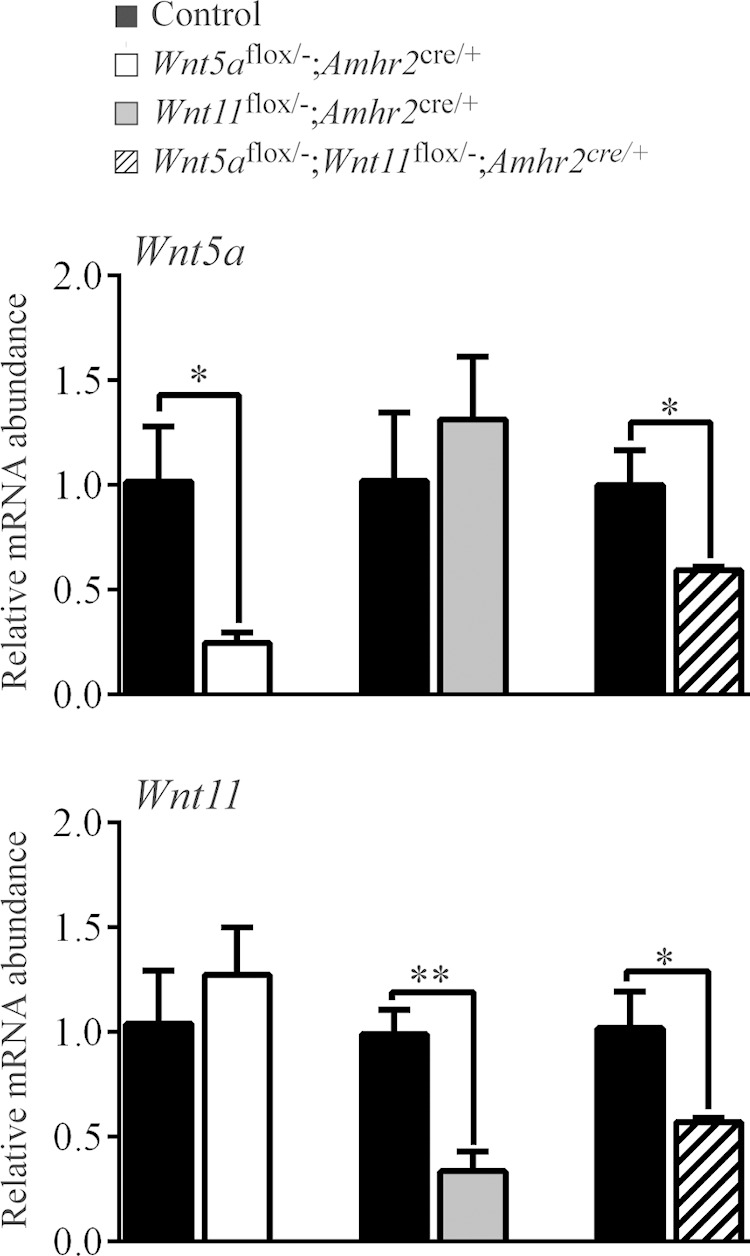
Wnt5a and Wnt11 knockdown efficiency in the cKO models. Expression of Wnt5a and Wnt11 was determined by RT-qPCR in GCs isolated from immature eCG-treated mice of the indicated genotypes. Expression of each transcript was normalized to the housekeeping gene Rpl19. Control, corresponding genotype lacking the Amhr2cre allele. Values for each experimental genotype are expressed relative to their corresponding control (n = 4 animals/genotype). Data are means ± sem. *P < 0.05 and **P < 0.01, statistically significant differences between strains.
TABLE 1.
Mating trials
| Litter (n) | Amhr2cre/+ | Wnt5aflox/− | Wnt5aflox/−;Amhr2cre/+ |
|---|---|---|---|
| Total litters | 38 | 34 | 24 |
| Total pups | 278 | 282 | 96 |
| Litter size | 7.32 ± 0.30a | 8.29 ± 0.32a | 4.00 ± 0.38b |
Data are means ± sem. n = 6 per group. a,bDifferent superscript letters indicate significant differences. P < 0.05.
Wnt5aflox/−;Amhr2cre/+ mice have defects in follicle development and ovulation
Histopathological examinations of the ovaries and reproductive tracts were conducted in adult mice for all cKO models. No morphologic defects were detected in the ovaries, uteri, or oviducts of any genotype, except in the Wnt5aflox/−;Amhr2cre/+ and Wnt5aflox/−;Wnt11flox/−;Amhr2cre/+ females, in which the ovaries appeared smaller, with a reduced number of antral follicles and CLs (Fig. 3 and data not shown). Wnt5aflox/−;Amhr2cre/+ mice (but none of the others) also had reduced ovary weights compared to the controls (P < 0.05; Table 2 and data not shown).
Figure 3.

Wnt5aflox/−;Amhr2cre/+ mice have smaller ovaries and increased follicular atresia. A) Representative images of Wnt5aflox/−;Amhr2cre/+ (control) and Wnt5aflox/−;Amhr2cre/+ (mutant) ovaries from 42-d-old animals. B) Activated caspase-3 immunohistochemistry of sections of ovaries of 6-wk-old mice of the indicated genotypes. Negative control, the control reaction in which the primary antibody was omitted.
TABLE 2.
Ovary weights
| Age group | Wnt5aflox/− | Wnt5aflox/−;Amhr2cre/+ |
|---|---|---|
| Immature+eCG | ||
| n | 12 | 12 |
| Weight (mg) | 5.53 ± 0.22a | 3.83 ± 0.24b |
| Adult | ||
| n | 12 | 14 |
| Weight (mg) | 5.38 ± 0.22a | 4.18 ± 0.22b |
Data are means ± sem. a,bDifferent superscript letters indicate significant differences. P < 0.05.
To provide a quantitative analysis of folliculogenesis, follicles were counted in serial histologic sections of ovaries from Wnt5aflox/−;Amhr2cre/+ mice at various ages. Five-day-old Wnt5aflox/−;Amhr2cre/+ mice had a normal number of primordial and primary follicles, indicative of a normal ovarian reserve and postnatal follicle formation (Fig. 4A). However, at 42 d of age, the number of primordial, healthy secondary, and healthy antral follicles decreased to 46, 44, and 28% of controls, respectively (P < 0.05; Fig. 4B). A similar reduction in the number of healthy secondary and antral follicles was observed at 8 mo of age, but primordial follicles further decreased to 20% of the number in the control, and primary follicles were also significantly reduced (P < 0.05; Fig. 4C). At 42 d of age, fully 78% of antral follicles were atretic in the Wnt5aflox/−;Amhr2cre/+ group vs. 36% in controls. Increased atresia was also evidenced by activated caspase-3 immunohistochemistry, which showed a much greater incidence of GC apoptosis in secondary and antral follicles in ovaries from Wnt5aflox/−;Amhr2cre/+ mice (Fig. 3B). Circulating FSH levels in Wnt5aflox/−;Amhr2cre/+ mice were comparable to those in controls at 42 d and 8 mo of age (data not shown), indicating that follicle loss was not caused by the lack of gonadotropin support.
Figure 4.

Progressive follicle loss and increased follicular atresia in the ovaries of Wnt5aflox/−;Amhr2cre/+ mice at 5 d (A), 42 d (B), and 8 mo (C). Ovaries (n = 6 per genotype at each age) were serially sectioned, and all follicles from every fifth section were counted; categorized as primordial, primary, secondary, or antral; and scored as healthy or atretic. Data represent raw follicle counts and were not adjusted to estimate the total ovarian follicle population. Data are means ± sem. Different letters above histograms indicate statistically significant differences between strains. P < 0.05.
Based on these results, we suspected that the subfertility of Wnt5aflox/−;Amhr2cre/+ mice was caused by the decrease in healthy antral follicles, leading to a reduction in the number of ovulations occurring per cycle. To verify this finding, Wnt5aflox/−;Amhr2cre/+ females were euthanized after mating, and COCs were retrieved from the oviducts and counted. The results showed a 59% decrease in recovered COCs in Wnt5aflox/−;Amhr2cre/+ females compared to the number in the controls (P < 0.05; Table 3). Subfertility in these mice was therefore attributed mainly to a reduction in the number of follicles that attained the ovulatory stage.
TABLE 3.
Ovulatory rates
| COCs (n) | Wnt5aflox/− | Wnt5aflox/−;Amhr2cre/+ |
|---|---|---|
| Total COCs | 37 | 15 |
| COCs/mouse | 9.25 ± 0.62a | 3.75 ± 1.93b |
Data are means ± sem. n = 4 per group. a,bDifferent superscript letters indicate significant differences. P < 0.05.
WNT5a down-regulates the expression of FSH-responsive genes
To determine the mechanism of action of WNT5a, we first sought to identify its transcriptional targets. GCs from eCG-primed immature mice were placed in culture and treated (or not) with recombinant WNT5a or -11, and global changes in gene expression analyzed by microarray. WNT5a treatment increased the expression of 542 genes and decreased the expression of 1656 genes by at least 2-fold over the control (Supplemental data, WNT5a microarray results Excel file 1). We observed that many of the genes most highly down-regulated by WNT5a were key FSH-responsive genes associated with GC steroidogenesis, differentiation, and proliferation. The regulation of a subset of these by WNT5a was confirmed by RT-qPCR (Fig. 5). The expression of many of these FSH-responsive genes was also found to be increased in GCs from eCG-treated Wnt5aflox/−;Amhr2cre/+ mice (Fig. 6), confirming them to be bona fide targets of WNT5a signaling in vivo. The increase in Cyp19a1 expression was also accompanied by increased serum E2 levels in these mice, as well as decreased P4 levels (Table 4).
Figure 5.

RT-qPCR confirmation of microarray data. Primary cultured GCs were treated or not with 3.5 μg/ml WNT5a for 3 h (n = 4 samples/group), and mRNA levels of the indicated genes were determined by RT-qPCR and normalized to the housekeeping gene Rpl19. Data are means ± sem. **P < 0.01 and ***P < 0.001, statistically significant differences in the effect of WNT5a treatment.
Figure 6.

Expression of WNT5a target genes is altered in Wnt5aflox/−;Amhr2cre/+ mice. RT-qPCR analysis of the indicated genes was performed on GC samples collected from immature mice of the indicated genotypes, either immediately after eCG treatment (48 h) or after an additional 12 h treatment with hCG (n = 4 animal/time point). All data were normalized to the housekeeping gene Rpl19. Data are means ± sem. *P < 0.05, **P < 0.01, and ***P < 0.001, significant difference vs. control.
TABLE 4.
Hormone assays
| Hormone | Wnt5aflox/− | Wnt5aflox/−;Amhr2cre/+ |
|---|---|---|
| E2 (pg/ml) | 76.08 ± 18.32a | 197.20 ± 40.79b |
| P4 (ng/ml) | 9.55 ± 2.14a | 1.83 ± 0.25b |
Data are means ± sem. n = 4 per group. a,bDifferent superscript letters indicate significant differences. P < 0.05.
Unlike WNT5a, microarray analyses revealed a very small number of genes that were regulated by WNT11 in vitro; 51 and 22 genes were up- or down-regulated, respectively, by at least 2-fold (Supplemental data, WNT11 microarray results Excel file 2). Only 1 gene, Il1b, was up-regulated by both WNT5a and -11 (Fig. 7). To further study potential functional redundancy between WNT5a and -11, GCs were treated with graded doses of WNT11, and its effects on the mRNA levels of WNT5a target genes were determined by RT-qPCR. Although WNT11 also down-regulated WNT5a target genes to some extent, it was generally much less potent than WNT5a itself (Fig. 7). Accordingly, the expression of WNT5a target genes was not changed in GCs from eCG-treated Wnt11flox/−;Amhr2cre/+ mice, nor were they further up-regulated in the GCs of Wnt5aflox/−;Wnt11flox/−;Amhr2cre/+ relative to Wnt5aflox/−;Amhr2cre/+ mice (Supplemental Fig. S2). WNT11 may therefore signal via mechanisms similar to those of WNT5a, but it does not seem to function in a manner redundant with WNT5a in the physiologic context.
Figure 7.

Regulation of WNT5a target genes by WNT11. Primary cultured GCs were treated or not with WNT11 or -5a at the indicated concentrations for 3 h (n = 4 samples/group), and mRNA levels of the indicated genes were determined by RT-qPCR and normalized to the housekeeping gene Rpl19. Data are means ± sem. Different letters above histograms indicate significant differences between treatments. P < 0.05.
WNT5a suppresses the canonical pathway in GCs
To determine the intracellular signaling mechanisms whereby WNT5a regulates its target genes, mouse GC primary cultures were treated with WNT5a on a time course, and the expression of mediators of the PCP signaling pathway was assessed. WNT5a did not affect JNK or cJUN expression or phosphorylation (Supplemental Fig. S3). Phosphorylation of CAPZIP, another substrate of JNK, was similarly unaffected. mRNA levels of cJUN transcriptional targets genes such as Fos and Jun did not change in response to WNT5a (Fig. 5), nor were they altered in the GCs of Wnt5aflox/−;Amhr2cre/+ mice (Fig. 6). Furthermore, pretreatment of GCs with the JNK inhibitor SP600125 or the RAC1 inhibitor NSC23766 did not prevent WNT5a from down-regulating the expression of its target genes (Supplemental Fig. S4). Similar experiments were conducted to determine the effects of WNT5a on WNT/Ca2+ pathway activity. WNT5a did not affect the expression or phosphorylation of CAMKII (Supplemental Fig. S3). Likewise, pretreatment of GCs with the PKC inhibitor GF109203X or the CAMKII inhibitor KN-93 or with Xestrospongin C (which prevents Ca2+ release by blocking the IP3 receptor) had no effect on the ability of WNT5a to down-regulate its target genes, although basal levels of Cyp19a1 expression were affected in some cases (Supplemental Fig. S4). Together, these results suggest that neither the PCP nor the WNT/Ca2+ pathway is a major mediator of WNT5a signaling in this model.
Beyond the activation of noncanonical signaling pathways, noncanonical WNTs are also known to suppress the canonical WNT pathway in certain contexts (21, 39–43). We therefore conducted a time course analysis of the effects of WNT5a on the expression of nonphosphorylated (Ser33/Ser37/Thr41) (i.e., active) CTNNB1 and total CTNNB1. The expression of both forms showed a rapid and dramatic decrease in response to WNT5a (Fig. 8). Destabilization of CTNNB1 can be induced by phosphorylation at its N-terminus by GSK3β (1, 2), by loss of phosphorylation at the C-terminus (Ser552/Ser665) (11, 44–46), or by both. Although levels of phospho-CTNNB1 (Ser552) decreased in response to WNT5a, the ratio of phospho-CTNNB1(Ser552) to total CTNNB1 remained unchanged, suggesting that C-terminal phosphorylation was not affected. Conversely, pretreatment of GCs with the GSK3 inhibitor SB216763 prevented WNT5a from decreasing CTNNB1 expression (Fig. 9), suggesting that WNT5a destabilizes CTNNB1 via a GSK3-dependent mechanism. However, this mechanism did not involve a rapid WNT5a-induced alteration in the expression of either total or phospho (Ser9) GSK3β (Fig. 8).
Figure 8.

WNT5a down-regulates canonical WNT signaling and CREB expression in GCs in vitro. Primary cultured GCs were treated or not with WNT5a (3.5 μg/ml) for the indicated times, and the expression of the indicated proteins and phosphoproteins was studied by immunoblot analysis. Representative immunoblots show 2 samples/treatment and time, with quantitative analyses of 4 samples/treatment and time. Values for the WNT5a-treated groups are expressed relative to their corresponding controls at each time point. ▪, control; □, WNT5a treatment. Data are means ± sem. *P < 0.05, **P < 0.01, and ***P < 0.001, effect of WNT5a treatment vs. control.
Figure 9.

WNT5a down-regulation of CTNNB1 and CREB is GSK3β dependent. Primary cultured GCs were treated with 3.5 μg/ml WNT5a for 15 min, with or without pretreatment with 1 μM SB216763 for 1 h, and the expression of CTNNB1 and CREB was studied by immunoblot analysis (n = 3 samples/treatment). Data are means ± sem. Different letters above histograms indicate significant differences between treatments. P < 0.05.
WNT5a down-regulates CREB expression and inhibits gonadotropin responsiveness
As is true of CTNNB1, the transcription factor CREB is both a critical mediator of gonadotropin signaling and a substrate for GSK3β (47). We therefore studied the effects of WNT5a on CREB expression in GCs and found that WNT5a down-regulated CREB protein levels, just as it did CTNNB1 levels (Fig. 8). Pretreatment with SB216763 prevented this effect, indicating that it was GSK3-dependent (Fig. 9).
As WNT5a was found to down-regulate FSH-responsive genes (Fig. 5) and destabilized CTNNB1 and CREB, both of which are key mediators of FSH and LH signaling (Figs. 8, 9) (6, 12), we hypothesized that WNT5a functions to antagonize gonadotropin responsiveness in GCs. To test this theory, GCs were pretreated (or not) with WNT5a for 1 h before addition of FSH or LH, and the expression of CTNNB1 and CREB and the mRNA levels of key FSH and LH target genes were determined. Both FSH and LH induced an increase in total, active and phospho-CTNNB1 (Ser552) protein levels in cultured GCs, and WNT5a greatly decreased both basal and gonadotropin-stimulated expression of all 3 forms (Fig. 10A). As reported (48), both FSH and LH also increased the levels of total and phosphorylated CREB (Ser133). As for CTNNB1, WNT5a pretreatment drastically down-regulated CREB expression in both untreated and gonadotropin-treated GCs. WNT5a pretreatment also inhibited (or abrogated) the ability of both FSH and LH to increase the mRNA levels of their target genes (Fig. 10B). The mRNA levels of the same LH target genes were increased in the GCs of Wnt5aflox/−;Amhr2cre/+ mice specifically 12 h after hCG treatment (Fig. 6), indicating that WNT5a also antagonizes LH action in vivo. Together, these results indicate that WNT5a functions as a physiologic antagonist of gonadotropin signaling.
Figure 10.

WNT5a suppresses gonadotropin signaling in GCs. A) Primary cultured GCs were treated or not with 50 ng/ml LH or FSH for 1 h, with or without pretreatment with 3.5 μM WNT5a for 1 h, and the expression of the indicated proteins and phosphoproteins was studied by immunoblot analysis. Representative immunoblots show 2 samples/treatment and time, and quantitative analyses show 4 samples/treatment. B) Primary cultured GCs were treated or not with 50 ng/ml LH or FSH for 2 h, with or without pretreatment with 3.5 μM WNT5a for 1 h, and the mRNA levels of the indicated FSH and LH target genes were studied by RT-qPCR. Expression of each transcript was normalized to the housekeeping gene Rpl19 (n = 4 samples/treatment). Data are means ± sem. Different letters above histograms indicate significant differences between treatments. P < 0.05.
DISCUSSION
The gonadotropins are essential endocrine regulators of ovarian follicle development, but their actions are dependent on several locally produced ovarian factors that serve to modulate their effects. In this study, we identified WNT5a as a novel secreted regulatory molecule that acts to antagonize the follicular response to gonadotropins, based on our key findings that WNT5a prevented FSH and LH from inducing their transcriptional target genes in vitro, whereas an enhanced transcriptional response of these same targets to eCG/hCG was observed in Wnt5a cKO mice in vivo. Wnt5a may therefore function to prevent premature or excessive growth, limit steroid hormone synthesis, prevent premature luteinization, and otherwise temper the effects of FSH and LH, as required, to ensure the proper development of the follicle.
Our inability to detect activation of noncanonical signaling by WNT5a in murine GCs was unexpected, particularly considering that we have recently reported that WNT5a signals via a RAC1/JNK/JUN pathway in cultured bovine GCs (32). Although the basis for this discrepancy remains unknown, the culture systems used differed between the 2 studies. Notably, the bovine cells were cultured from early antral follicles, whereas the murine cells were cultured from the preovulatory follicles of eCG-primed immature animals. As the WNT receptor complexes and signaling machinery present in GCs may vary considerably from one phase of follicle development to the next, the differences between the studies may indicate that WNT5a signals via different mechanisms (and hence may have different physiologic functions) at specific stages of folliculogenesis. Consistent with this notion, we found that the follicle development phenotype of the Wnt5aflox/−;Amhr2cre/+ model (in which secondary follicles are targeted) was much more severe than that of the Wnt5aflox/−;CYP19-Cre model (in which only antral stages are targeted), suggesting specific functions for WNT5a at the secondary stage of development. Further studies are needed to properly dissect the stage-specific roles and signaling mechanisms of WNT5a in GCs.
Several mechanisms have been proposed to explain how noncanonical WNTs act to decrease CTNNB1 levels. For instance, noncanonical WNTs have been shown to compete directly with canonical WNTs for binding to FZD receptor complexes, thereby attenuating the canonical CTNNB1 stabilization signal (39). WNT5a has been shown to increase the expression of SIAH ubiquitin ligase, resulting in the accelerated ubiquitination and degradation of CTNNB1 (40). WNT11 can also activate caspases that degrade CTNNB1 via an unknown mechanism (42). Although the mechanism whereby WNT5a causes CTNNB1 protein levels to decrease in GCs was not determined in this study, we believe that the very rapid (i.e., minutes long) nature of this effect suggests that competition with canonical WNTs for their receptors is likely. Indeed, GCs express several canonical WNTs (including WNT2 and -3a) (49, 50) and high levels of active CTNNB1, suggesting that a constitutive level of canonical WNT signaling normally occurs in the GCs of growing follicles (see the model in Fig. 11). Our data and results of other studies suggest that FSH (and LH) signaling via PKA can enhance this CTNNB1 signaling in at least 3 ways. First, PKA can phosphorylate (and thereby inhibit) GSK3β, preventing it from promoting the degradation of CTNNB1 (45, 51). PKA can also directly phosphorylate CTNNB1 at Ser552/Ser665, enhancing its stability and transactivation activity (11). Finally, PKA also phosphorylates CREB, which then binds cAMP response elements present in the promoters of many of the CTNNB1 target genes identified in the current study (4, 5). CREB binding protein has been shown to enhance the transcriptional properties of CTNNB1 in several cell types (12, 52, 53), suggesting an additional level of synergy between gonadotropin/PKA and canonical WNT signaling (Fig. 11). We propose that WNT5a disrupts these signaling processes by competing with canonical WNTs for their receptors. This disruption results in the dissociation of the CTNNB1 destruction complex from the receptor complex and the activation of GSK3β, possibly because of the loss of an inhibitory interaction between GSK3β and the WNT coreceptor low-density lipoprotein receptor-related protein 5/6 (LRP5/6) (54). GSK3β then phosphorylates CTNNB1 and possibly CREB, resulting in the proteasomal degradation of both, and effectively disrupting both the gonadotropin/PKA and canonical WNT pathways (Fig. 11). Gonadotropin signaling may therefore be regulated by the precise balance between WNT5a and canonical WNTs present within the follicular microenvironment.
Figure 11.

A working model of the mechanism of action of WNT5a in GCs. Left: canonical WNTs act in tandem with gonadotropin/PKA signaling in the GCs of growing follicles to stabilize CTNNB1 and CREB, resulting in the transcriptional activation of CTNNB1 and FSH/LH target genes. Right: WNT5a may antagonize canonical WNTs by competing for their receptor complex. The resultant disinhibition of GSK3β leads to the degradation of CTNNB1 and CREB, thereby antagonizing gonadotropin action. Whether GSK3β promotes CREB degradation through direct phosphorylation remains to be determined.
The conditional knockout of Wnt5a in GCs enhanced FSH signaling and resulted in an increase in the expression of FSH-responsive genes, many of which serve to promote follicle growth and survival. Despite this result, a marked increase in follicular atresia occurred in the Wnt5aflox/−;Amhr2cre/+ model, causing a decreased ovulation rate and subfertility, and leading to an accelerated depletion of the follicular reserve. Although we were unable to reconcile these apparently paradoxical findings, several explanations can be proposed. For instance, increased FSH responsiveness, E2 synthesis, and proliferation in GCs may result in a loss of coordination of follicle development between the GCs, theca cells, and oocytes, ultimately compromising the viability of the follicle. Another possibility is that WNT5a produced in GCs is essential in oocyte development, an idea that is supported by a recent study indicating that WNT5a plays a role in oocyte entry into meiosis during embryonic development (55). As suggested above, another possibility is that WNT5a plays roles during the secondary (or earlier) stages of follicle development that were not elucidated in our mechanistic studies. Indeed, our in vitro studies were conducted in gonadotropin-treated mice and were focused on late stages of follicle development; a mechanism of WNT5a action occurring specifically at the secondary stage would therefore have been overlooked in our study design. If this were true, increased atresia in the Wnt5aflox/−;Amhr2cre/+ model would be the result of a loss of WNT5a function other than its ability to suppress CTNNB1 and gonadotropin signaling in growing follicles. Further studies are needed to test these ideas.
In summary, we report for the first time the crucial role of WNT5a in follicle development and normal female fertility. We found that WNT5a acts to suppress gonadotropin signaling by antagonizing the canonical WNT signaling pathway, thereby causing the degradation of CTNNB1 and CREB, 2 key transcriptional regulators of gonadotropin-responsive genes. This work provides new insight into the role of the noncanonical WNTs in GCs and elucidates new signaling mechanisms that modulate gonadotropin action.
Supplementary Material
Acknowledgments
The authors thank Ms. M. Girard (Université de Montréal) for assistance with mouse colony management. This work was supported by Operating Grant MOP-102508 from the Canadian Institutes of Health Research and the Canada Research Chair in Ovarian Molecular Biology and Functional Genomics (to D.B.) and U. S. National Institutes of Health (NIH) The Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) Grant R03-HD058222 (to K.H.). The University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core is supported by NIH NICHD (SCCPRR) Grant U54-HD28934. A.A. was supported by a graduate studentship from the Réseau Québécois en Reproduction.
Glossary
- CAMK
calmodulin-dependent kinase
- CAPZIP
capping (actin filament) muscle Z-line interacting protein
- cKO
conditional knockout
- CL
corpus luteum
- COC
cumulus–oocyte complex
- CREB
cAMP responsive element binding
- CTNNB1
β-catenin
- Cyp19a1
cytochrome P450 family 19, subfamily a, polypeptide 1
- DAAM
disheveled associated activator of morphogenesis
- E2
estradiol
- eCG
equine chorionic gonadotropin
- FSH
follicle-stimulating hormone
- FZD
frizzled
- GC
granulosa cell
- GSK
glycogen synthase kinase
- hCG
human chorionic gonadotropin
- LH
luteinizing hormone
- P4
progesterone
- PCP
planar cell polarity
- qPCR
quantitative PCR
- ROCK
RHO-associated kinase
- SFRP
secreted frizzled-related protein
- TTBS
Tween 20–Tris-buffered saline
- WNT
wingless-type MMTV (mouse mammary tumor virus) integration site family
- WT
wild-type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
REFERENCES
- 1.Edson M. A., Nagaraja A. K., Matzuk M. M. (2009) The mammalian ovary from genesis to revelation. Endocr. Rev. 30, 624–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richards J. S., Pangas S. A. (2010) The ovary: basic biology and clinical implications. J. Clin. Invest. 120, 963–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Conti M., Hsieh M., Park J. Y., Su Y. Q. (2006) Role of the epidermal growth factor network in ovarian follicles. Mol. Endocrinol. 20, 715–723 [DOI] [PubMed] [Google Scholar]
- 4.Lapointe E., Boerboom D. (2011) WNT signaling and the regulation of ovarian steroidogenesis. Front. Biosci. (Schol. Ed.) 3, 276–285 [DOI] [PubMed] [Google Scholar]
- 5.Boyer A., Goff A. K., Boerboom D. (2010) WNT signaling in ovarian follicle biology and tumorigenesis. Trends Endocrinol. Metab. 21, 25–32 [DOI] [PubMed] [Google Scholar]
- 6.Parakh T. N., Hernandez J. A., Grammer J. C., Weck J., Hunzicker-Dunn M., Zeleznik A. J., Nilson J. H. (2006) Follicle-stimulating hormone/cAMP regulation of aromatase gene expression requires beta-catenin. Proc. Natl. Acad. Sci. USA 103, 12435–12440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hernandez Gifford J. A., Hunzicker-Dunn M. E., Nilson J. H. (2009) Conditional deletion of beta-catenin mediated by Amhr2cre in mice causes female infertility. Biol. Reprod. 80, 1282–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang H. X., Li T. Y., Kidder G. M. (2010) WNT2 regulates DNA synthesis in mouse granulosa cells through beta-catenin. Biol. Reprod. 82, 865–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finnson K. W., Kontogiannea M., Li X., Farookhi R. (2012) Characterization of Wnt2 overexpression in a rat granulosa cell line (DC3): effects on CTNNB1 activation. Biol. Reprod. 87, 12–, 1–8. [DOI] [PubMed] [Google Scholar]
- 10.Boyer A., Lapointe E., Zheng X., Cowan R. G., Li H., Quirk S. M., DeMayo F. J., Richards J. S., Boerboom D. (2010) WNT4 is required for normal ovarian follicle development and female fertility. FASEB J. 24, 3010–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Law N. C., Weck J., Kyriss B., Nilson J. H., Hunzicker-Dunn M. (2013) Lhcgr expression in granulosa cells: roles for PKA-phosphorylated β-catenin, TCF3, and FOXO1. Mol. Endocrinol. 27, 1295–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fan H. Y., O’Connor A., Shitanaka M., Shimada M., Liu Z., Richards J. S. (2010) Beta-catenin (CTNNB1) promotes preovulatory follicular development but represses LH-mediated ovulation and luteinization. Mol. Endocrinol. 24, 1529–1542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsieh M., Boerboom D., Shimada M., Lo Y., Parlow A. F., Luhmann U. F., Berger W., Richards J. S. (2005) Mice null for Frizzled4 (Fzd4−/−) are infertile and exhibit impaired corpora lutea formation and function. Biol. Reprod. 73, 1135–1146 [DOI] [PubMed] [Google Scholar]
- 14.Lapointe E., Boyer A., Rico C., Paquet M., Franco H. L., Gossen J., DeMayo F. J., Richards J. S., Boerboom D. (2012) FZD1 regulates cumulus expansion genes and is required for normal female fertility in mice. Biol. Reprod. 87, 104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roy L., McDonald C. A., Jiang C., Maroni D., Zeleznik A. J., Wyatt T. A., Hou X., Davis J. S. (2009) Convergence of 3′,5′-cyclic adenosine 5′-monophosphate/protein kinase A and glycogen synthase kinase-3beta/beta-catenin signaling in corpus luteum progesterone synthesis. Endocrinology 150, 5036–5045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Komiya Y., Habas R. (2008) Wnt signal transduction pathways. Organogenesis 4, 68–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Semenov M. V., Habas R., Macdonald B. T., He X. (2007) SnapShot: noncanonical Wnt signaling pathways. Cell 131, 1378.e1–e2 [DOI] [PubMed] [Google Scholar]
- 18.Kühl M., Sheldahl L. C., Malbon C. C., Moon R. T. (2000) Ca(2+)/calmodulin-dependent protein kinase II is stimulated by Wnt and Frizzled homologs and promotes ventral cell fates in Xenopus. J. Biol. Chem. 275, 12701–12711 [DOI] [PubMed] [Google Scholar]
- 19.Sheldahl L. C., Park M., Malbon C. C., Moon R. T. (1999) Protein kinase C is differentially stimulated by Wnt and Frizzled homologs in a G-protein-dependent manner. Curr. Biol. 9, 695–698 [DOI] [PubMed] [Google Scholar]
- 20.Saneyoshi T., Kume S., Amasaki Y., Mikoshiba K. (2002) The Wnt/calcium pathway activates NF-AT and promotes ventral cell fate in Xenopus embryos. Nature 417, 295–299 [DOI] [PubMed] [Google Scholar]
- 21.Ishitani T., Kishida S., Hyodo-Miura J., Ueno N., Yasuda J., Waterman M., Shibuya H., Moon R. T., Ninomiya-Tsuji J., Matsumoto K. (2003) The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin signaling. Mol. Cell. Biol. 23, 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wallingford J. B., Habas R. (2005) The developmental biology of Dishevelled: an enigmatic protein governing cell fate and cell polarity. Development 132, 4421–4436 [DOI] [PubMed] [Google Scholar]
- 23.Marlow F., Topczewski J., Sepich D., Solnica-Krezel L. (2002) Zebrafish Rho kinase 2 acts downstream of Wnt11 to mediate cell polarity and effective convergence and extension movements. Curr. Biol. 12, 876–884 [DOI] [PubMed] [Google Scholar]
- 24.Weiser D. C., Pyati U. J., Kimelman D. (2007) Gravin regulates mesodermal cell behavior changes required for axis elongation during zebrafish gastrulation. Genes Dev. 21, 1559–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Habas R., Dawid I. B., He X. (2003) Coactivation of Rac and Rho by Wnt/Frizzled signaling is required for vertebrate gastrulation. Genes Dev. 17, 295–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L., Yuan H., Xie W., Mao J., Caruso A. M., McMahon A., Sussman D. J., Wu D. (1999) Dishevelled proteins lead to two signaling pathways: regulation of LEF-1 and c-Jun N-terminal kinase in mammalian cells. J. Biol. Chem. 274, 129–134 [DOI] [PubMed] [Google Scholar]
- 27.Keller R., Davidson L. A., Shook D. R. (2003) How we are shaped: the biomechanics of gastrulation. Differentiation 71, 171–205 [DOI] [PubMed] [Google Scholar]
- 28.Saadeddin A., Babaei-Jadidi R., Spencer-Dene B., Nateri A. S. (2009) The links between transcription, beta-catenin/JNK signaling, and carcinogenesis. Mol. Cancer Res. 7, 1189–1196 [DOI] [PubMed] [Google Scholar]
- 29.Harwood B. N., Cross S. K., Radford E. E., Haac B. E., De Vries W. N. (2008) Members of the WNT signaling pathways are widely expressed in mouse ovaries, oocytes, and cleavage stage embryos. Dev. Dyn. 237, 1099–1111 [DOI] [PubMed] [Google Scholar]
- 30.Sanchez A. M., Viganò P., Quattrone F., Pagliardini L., Papaleo E., Candiani M., Panina-Bordignon P. (2014) The WNT/β-catenin signaling pathway and expression of survival promoting genes in luteinized granulosa cells: endometriosis as a paradigm for a dysregulated apoptosis pathway. Fertil. Steril. 101, 1688–1696 [DOI] [PubMed] [Google Scholar]
- 31.Kenigsberg S., Bentov Y., Chalifa-Caspi V., Potashnik G., Ofir R., Birk O. S. (2009) Gene expression microarray profiles of cumulus cells in lean and overweight-obese polycystic ovary syndrome patients. Mol. Hum. Reprod. 15, 89–103 [DOI] [PubMed] [Google Scholar]
- 32.Abedini A., Zamberlam G., Boerboom D., Price C. A. (2015) Non-canonical WNT5A is a potential regulator of granulosa cell function in cattle. Mol. Cell. Endocrinol. 403, 39–45 [DOI] [PubMed] [Google Scholar]
- 33.Hayashi K., Yoshioka S., Reardon S. N., Rucker E. B. III, Spencer T. E., DeMayo F. J., Lydon J. P., MacLean J. A. II (2011) WNTs in the neonatal mouse uterus: potential regulation of endometrial gland development. Biol. Reprod. 84, 308–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jorgez C. J., Klysik M., Jamin S. P., Behringer R. R., Matzuk M. M. (2004) Granulosa cell-specific inactivation of follistatin causes female fertility defects. Mol. Endocrinol. 18, 953–967 [DOI] [PubMed] [Google Scholar]
- 35.Fan H. Y., Shimada M., Liu Z., Cahill N., Noma N., Wu Y., Gossen J., Richards J. S. (2008) Selective expression of KrasG12D in granulosa cells of the mouse ovary causes defects in follicle development and ovulation. Development 135, 2127–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zeleznik A. J., Midgley A. R. Jr., Reichert L. E. Jr (1974) Granulosa cell maturation in the rat: increased binding of human chorionic gonadotropin following treatment with follicle-stimulating hormone in vivo. Endocrinology 95, 818–825 [DOI] [PubMed] [Google Scholar]
- 37.Rico C., Dodelet-Devillers A., Paquet M., Tsoi M., Lapointe E., Carmeliet P., Boerboom D. (2014) HIF1 activity in granulosa cells is required for FSH-regulated Vegfa expression and follicle survival in mice. Biol. Reprod. 90, 135 [DOI] [PubMed] [Google Scholar]
- 38.Boyer A., Paquet M., Laguë M. N., Hermo L., Boerboom D. (2009) Dysregulation of WNT/CTNNB1 and PI3K/AKT signaling in testicular stromal cells causes granulosa cell tumor of the testis. Carcinogenesis 30, 869–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bryja V., Andersson E. R., Schambony A., Esner M., Bryjová L., Biris K. K., Hall A. C., Kraft B., Cajanek L., Yamaguchi T. P., Buckingham M., Arenas E. (2009) The extracellular domain of Lrp5/6 inhibits noncanonical Wnt signaling in vivo. Mol. Biol. Cell 20, 924–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Topol L., Jiang X., Choi H., Garrett-Beal L., Carolan P. J., Yang Y. (2003) Wnt-5a inhibits the canonical Wnt pathway by promoting GSK-3-independent beta-catenin degradation. J. Cell Biol. 162, 899–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishitani T., Ninomiya-Tsuji J., Nagai S., Nishita M., Meneghini M., Barker N., Waterman M., Bowerman B., Clevers H., Shibuya H., Matsumoto K. (1999) The TAK1-NLK-MAPK-related pathway antagonizes signalling between beta-catenin and transcription factor TCF. Nature 399, 798–802 [DOI] [PubMed] [Google Scholar]
- 42.Abdul-Ghani M., Dufort D., Stiles R., De Repentigny Y., Kothary R., Megeney L. A. (2011) Wnt11 promotes cardiomyocyte development by caspase-mediated suppression of canonical Wnt signals. Mol. Cell. Biol. 31, 163–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee J. M., Kim I. S., Kim H., Lee J. S., Kim K., Yim H. Y., Jeong J., Kim J. H., Kim J. Y., Lee H., Seo S. B., Kim H., Rosenfeld M. G., Kim K. I., Baek S. H. (2010) RORalpha attenuates Wnt/beta-catenin signaling by PKCalpha-dependent phosphorylation in colon cancer. Mol. Cell 37, 183–195 [DOI] [PubMed] [Google Scholar]
- 44.Taurin S., Sandbo N., Qin Y., Browning D., Dulin N. O. (2006) Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. J. Biol. Chem. 281, 9971–9976 [DOI] [PubMed] [Google Scholar]
- 45.Hino S., Tanji C., Nakayama K. I., Kikuchi A. (2005) Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase stabilizes beta-catenin through inhibition of its ubiquitination. Mol. Cell. Biol. 25, 9063–9072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fang D., Hawke D., Zheng Y., Xia Y., Meisenhelder J., Nika H., Mills G. B., Kobayashi R., Hunter T., Lu Z. (2007) Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 282, 11221–11229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sutherland C. (2011) What Are the bona fide GSK3 Substrates? Int. J. Alzheimers Dis. 2011, 505607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mukherjee A., Park-Sarge O. K., Mayo K. E. (1996) Gonadotropins induce rapid phosphorylation of the 3′,5′-cyclic adenosine monophosphate response element binding protein in ovarian granulosa cells. Endocrinology 137, 3234–3245 [DOI] [PubMed] [Google Scholar]
- 49.Ricken A., Lochhead P., Kontogiannea M., Farookhi R. (2002) Wnt signaling in the ovary: identification and compartmentalized expression of wnt-2, wnt-2b, and frizzled-4 mRNAs. Endocrinology 143, 2741–2749 [DOI] [PubMed] [Google Scholar]
- 50.Hsieh M., Johnson M. A., Greenberg N. M., Richards J. S. (2002) Regulated expression of Wnts and Frizzleds at specific stages of follicular development in the rodent ovary. Endocrinology 143, 898–908 [DOI] [PubMed] [Google Scholar]
- 51.Fang X., Yu S. X., Lu Y., Bast R. C. Jr., Woodgett J. R., Mills G. B. (2000) Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. USA 97, 11960–11965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolf D., Rodova M., Miska E. A., Calvet J. P., Kouzarides T. (2002) Acetylation of beta-catenin by CREB-binding protein (CBP). J. Biol. Chem. 277, 25562–25567 [DOI] [PubMed] [Google Scholar]
- 53.Polakis P. (2000) Wnt signaling and cancer. Genes Dev. 14, 1837–1851 [PubMed] [Google Scholar]
- 54.Metcalfe C., Bienz M. (2011) Inhibition of GSK3 by Wnt signalling: two contrasting models. J. Cell Sci. 124, 3537–3544 [DOI] [PubMed] [Google Scholar]
- 55.Naillat F., Prunskaite-Hyyryläinen R., Pietilä I., Sormunen R., Jokela T., Shan J., Vainio S. J. (2010) Wnt4/5a signalling coordinates cell adhesion and entry into meiosis during presumptive ovarian follicle development. Hum. Mol. Genet. 19, 1539–1550 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.