

Abstract
Background:
Cardiovascular diseases, including dilated cardiomyopathy (DCM) and hypertension, are the leading cause of death worldwide. The role of mitochondrial DNA (mtDNA) in the pathogenesis of these diseases has not been completely clarified. In this study, we evaluate whether A8701G mutation is associated with maternally inherited hypertension and DCM in a Chinese pedigree of a consanguineous marriage.
Methods:
Fourteen subjects in a three-generation Han Chinese family with hypertension and DCM, in which consanguineous marriage was present in the parental generation, were interviewed. We divided all the family members into case (7 maternal members) and control group (7 nonmaternal members) for comparison. Clinical evaluations and sequence analysis of mtDNA were obtained from all participants. Frequency differences between maternal and nonmaternal members were tested to locate the disease-associated mutations.
Results:
The majority of the family members presented with a maternal inheritance of hypertension and DCM. Sequence analysis of mtDNA in this pedigree identified eight mtDNA mutations. Among the mutations identified, there was only one significant mutation: A8701G (P = 0.005), which is a homoplasmic mitochondrial missense mutation in all the matrilineal relatives. There was no clear evidence for any synergistic effects between A8701G and other mutations.
Conclusions:
A8701G mutation may act as an inherited risk factor for the matrilineal transmission of hypertension and DCM in conjunction with genetic disorders caused by consanguineous marriage.
Keywords: Dilated Cardiomyopathy, Hypertension, Mitochondria, Mitochondrial DNA Mutation
INTRODUCTION
Cardiovascular disease, which includes hypertension, coronary heart disease, dilated cardiomyopathy (DCM) and others, is the leading cause of death worldwide. Clinical trials have shown that hypertension is one of the potential risk factors for DCM.[1,2] Considering the central role of mitochondria in the production of energy and reactive oxygen species (ROS), mutations in mitochondrial DNA (mtDNA) are believed to affect the pathogenesis of hypertension.[3] Evidence supports the notion that somatic mtDNA damage is a good predictor of hypertension.[4] A8701G mutation in the mitochondrial ATP6 gene is the most common mutation underlying hypertension. Other previously identified mutations include A4401G mutation in the junction between tRNAGln and tRNAMet genes, A4435G mutation in the tRNAMet gene,[5,6] and A4295G and A4263G mutations in the tRNAIle gene.[7,8]
DCM is a heart muscle disorder which can be inherited as autosomal dominant, X-linked, or mitochondrial inheritance.[9] However, DCM is probably underdiagnosed and is believed to account for a much larger number of cases, as subjects may remain asymptomatic until a remarkable ventricular dysfunction has occurred. Although mtDNA defects have been described in an increasing number of cases of DCM, it has heretofore rarely been in association with the demonstrable A8701G mutation. DCM can be accompanied by hypertension. In these cases, DCM is different from hypertensive cardiomyopathy, which is characterized by hypertrophy and takes years to develop. So far, knowledge of the role of the mitochondrial genome in the pathogenesis of these diseases has not been completely clarified in Asian population.
Consanguineous marriage, or marriage between relatives, has received a lot of attention as a potential risk factor for some inherited disorders including hypertension.[10,11] On the other hand, very limited studies have been conducted to delve into the possible impact of consanguineous marriages and mtDNA on common multifactorial and polygenic adult disorders.
Family based design is widely used in genetic association studies to detect disease susceptibility variants. Here, we report a Han Chinese pedigree in which several members were diagnosed with maternally transmitted hypertension and DCM. The family based study identified a significantly associated mitochondrial variant such as A8701G mutation, which was enriched in this family, may be due to the consanguineous marriage in the first generation. The mitochondrial genome in this Chinese family belonged to the eastern Asian haplogroup M. A8701G mutation in mtDNA, which is considered pathogenic, is absent in healthy control subjects and usually accompanied by affected levels of respiratory chain enzyme activity.[12]
METHODS
Subjects
In total, we recruited 14 subjects from this family at the Department of Cardiology in the Northern People's Hospital (Jiangsu, China). Figure 1 shows the pedigree of the reported family and sums up the clinical features of the individuals affected by hypertension and DCM. DCM was defined according to World Health Organization (WHO)/International Society and Federation of Cardiology criteria.[13] We divided the family members into case (7 maternal members) and control group (7 nonmaternal members) for comparison. Members of the family were interviewed and evaluated to identify both personal and medical histories of clinical abnormalities. Informed consent, medical history, clinical evaluations, echocardiographic scanning, biochemical tests, and genetic analysis were obtained from all participants. The protocol was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the Northern People's Hospital.
Figure 1.
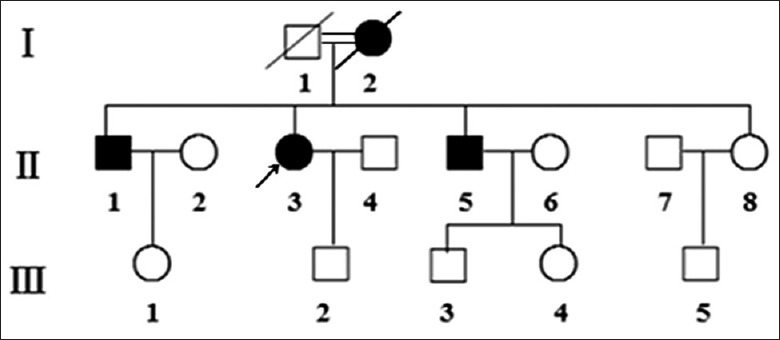
Pedigree of the Chinese family with hypertension and dilated cardiomyopathy. The degree of relationship between the proband's parents was the second cousin.
Measurements of blood pressure
Members of this Chinese family underwent a physical examination, laboratory assessment of cardiovascular disease risk factors, and routine electrocardiography. Blood pressure was measured by an experienced physician using a mercury column sphygmomanometer according to the WHO standardized criteria.[14] The first and fifth Korotkoff sounds were taken as indicative of systolic and diastolic blood pressure, respectively. The mean value of three systolic and diastolic blood pressure readings was used. According to the recommendation of the Joint National Committee on Detection, Evaluation, and Treatment of High Blood Pressure,[15] hypertension is defined as systolic blood pressure ≥140 mmHg or diastolic blood pressure ≥90 mmHg on at least three different occasions when measured in accordance with WHO criteria.
Mitochondrial DNA analysis
Genomic DNA was extracted from the peripheral blood of both case and control subjects. DNA was isolated using Promega Wizard Genomic DNA Purification Kit (Madison, WI, USA). The most likely sites for cardiovascular diseases as described previously[16] were screened using oligodeoxynucleotides 3777–4679 and 7908–8816 bp. The samples were incubated with extraction buffer (10 mmol/L Tris-HCl, 100 mmol/L ethylenediaminetetraacetic acid [EDTA], 0.5% sodium dodecyl sulfate, and 20 µg/ml RNAse, pH 8.0) and digested with proteinase K. DNA was extracted with phenol once, precipitated with ethanol, and recovered in TE0.1 (10 mmol/L Tris-HCl, 0.1 mmol/L EDTA, pH 8.0). The polymerase chain reaction (PCR) amplification of the mitochondrial tRNALys and tRNAIle gene was carried out, using the following primers: forward: 5′-ACGAGTACACCGACTACGGC-3′ and reverse: 5′- TGGGTGGTTGGTGTAAATGA-3′, forward: 5′-TGGCTCCTTTAACCTCTCCA-3′ and reverse: 5′-AAGGATTATGGATGCGGTTG-3′. The mitochondrial genomes of the proband carrying A8701G mutation were PCR amplified by sets of the light strand and heavy strand oligonucleotide primers as described elsewhere.[8] PCR was performed in 30 µl of the reaction mixture, containing 5.2 µl of PCR Master Mix (Qiagen; Hilden, Germany), 2.5 µl of each primer, 1 µl DNA sample, and 18.8 µl of water. PCR was carried out in a 9700 Thermocycler (Perkin-Elmer Applied Biosystems, Norwalk, USA) with one cycle of 95°C for 5 min, and then 35 cycles of 95°C for 30 s, 54°C for 30 s, and 72°C for 60 s with a full extension cycle of 72°C for 10 min. Each fragment was purified and subsequently analyzed by direct sequencing with ABI 3730 Sequence Analysis software (Applied Biosystems, Inc., Foster City, CA, USA) using the BigDye Terminator v1.1 kit (ABI Company, Carlsbad, CA, USA) and analyzed with SeqWeb program GAP (GCG) according to the updated consensus Cambridge sequence.[17]
Statistical analysis
Statistical analyses were performed using SPSS software (version 16.0; SPSS Inc., Chicago, IL, USA). Differences in categorical variables were assessed with Fisher's exact test. The level of P < 0.05 was considered statistically significant.
RESULTS
Clinical presentation
The proband (II-3) began suffering from hypertension at the age of 48 years old. She was referred to the Cardiology Clinic of Northern Hospital because of intermittent dizziness and exertional dyspnea at the age of 52 years old. Her blood pressure was 190/100 mmHg. Chest X-rays showed cardiomegaly with a cardiothoracic index of 0.6. The echocardiography examination revealed left chamber enlargement with impaired systolic left ventricular (LV) function (LV end-diastolic diameter, 60 mm; LV ejection fraction, 35%). Physical examination and laboratory assessment of cardiovascular disease risk factors were negative for other clinical abnormalities including diabetes, visual and hearing disorders, and renal and neurological impairments. Therefore, she exhibited a typical phenotype of essential hypertension and DCM. Six months later, she developed progressive cardiac failure due to LV dysfunction and mitral regurgitation despite optimal medical treatment. She was treated with a cardiac resynchronization therapy (CRT) device, after which there was a slight improvement in LV function (LV end-diastolic diameter, 57 mm, LV ejection fraction, 37%) [Figure 2]. Recent electrocardiogram (ECG) showed atrioventricular synchrony and QRS interval of 120 ms [Figure 3].
Figure 2.
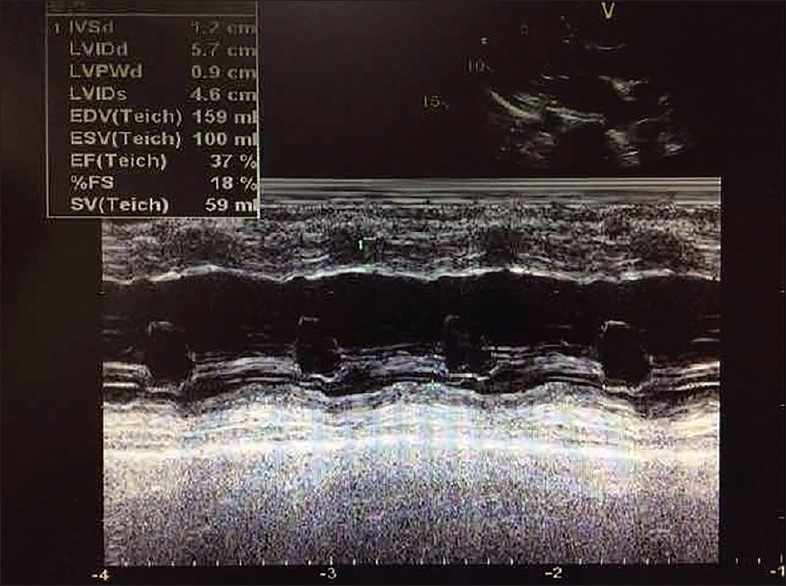
M-mode echocardiography of the proband (II-3) after cardiac resynchronization therapy.
Figure 3.

Electrocardiogram showing pacemaker pacing and QRS of 120 ms of II-3.
The proband's family originates from Jiangsu Province in Eastern China, and the majority of the family members presented with a maternal inheritance of hypertension and DCM. The age at onset ranged from 45 to 60 years [Figure 1]. The degree of relationship between the proband's parents was the second cousin. There was no evidence that any member of the family had any nonfamily related factors to account for the diseases. The proband, her brothers, and mother had a combination of DCM and hypertension. Her mother was known to have been treated with CRT, but died suddenly due to severe heart failure. Clinical features of some family members are summarized in Table 1. Two (II-1, II-3) matrilineal relatives had LV hypertrophy on the recorded ECG. In addition, the proband showed an increased interventricular septal thickness. As indicated in Table 2, the level of uric acid in three subjects (II-1, II-3, and II-8), the level of fasting blood glucose in 1 subject (II-5), and the level of triglyceride in three subjects (II-1, II-5, and II-8) exceeded the standard levels. The level of LV end-diastolic diameter, LV ejection fraction, and brain natriuretic peptide in three subjects (II-1, II-3, II-5) were above the standard levels. None of other matrilineal relatives, except the proband, exhibited an increase in total cholesterol. In addition, no other organ system involvement was clinically significant.
Table 1.
Clinical characteristics of some members in the reported Chinese family
| Subjects | Sex | Age of test (years) | Age of onset (years) | Systolic pressure (mmHg) | Diastolic pressure (mmHg) | ECG | IVST (mm) |
|---|---|---|---|---|---|---|---|
| I-2 | Female | 80 | 60 | 180 | 100 | SB | 9 |
| II-1 | Male | 54 | 50 | 170 | 103 | LVH | 11 |
| II-2 | Female | 53 | NA | 130 | 80 | N | NA |
| II-3 | Female | 52 | 48 | 190 | 100 | SB | 11 |
| II-4 | Male | 53 | NA | 120 | 65 | N | NA |
| II-5 | Male | 49 | 44 | 150 | 96 | MI | 10 |
| II-6 | Female | 48 | 47 | 159 | 105 | N | 11 |
| II-7 | Male | 47 | NA | 130 | 75 | N | NA |
| II-8 | Female | 47 | 45 | 147 | 98 | N | 9 |
| III-1 | Female | 26 | NA | 120 | 80 | N | 9 |
| III-2 | Male | 25 | NA | 110 | 70 | N | 8 |
| III-3 | Male | 24 | NA | 130 | 64 | N | NA |
| III-4 | Female | 22 | NA | 115 | 81 | N | NA |
| III-5 | Male | 19 | NA | 106 | 69 | N | NA |
Pretreatment blood pressures. NA: Not applicable; IVST: Interventricular septal thickness; N: Electrocardiography was normal; LVH: Electrocardiography showed left ventricular hypertrophy; MI: Myocardial ischemia; SB: Sinus bradycardia; ECG: Electrocardiogram.
Table 2.
Past histories and Laboratory examinations for some members of the reported Chinese family
| Items | II-1 | II-3 | II-5 | II-8 | Chinese reference |
|---|---|---|---|---|---|
| Past histories | |||||
| Therapy | Yes | Yes | Yes | Yes | NA |
| Tobacco | Yes | No | Yes | No | NA |
| Alcohol | No | No | Yes | No | NA |
| Laboratory examinations | |||||
| FPG (mmoL/L) | 5.6 | 4.6 | 7.9 | 4.8 | 3.9–6.1 |
| TC (mmoL/L) | 5.12 | 6.18 | 3.80 | 4.30 | 2.44–5.17 |
| TG (mmoL/L) | 2.42 | 1.69 | 1.92 | 2.30 | 0.40–1.70 |
| HDL (mmoL/L) | 0.78 | 0.81 | 1.34 | 1.13 | 1.16–1.42 |
| LDL (mmoL/L) | 3.04 | 1.60 | 2.97 | 3.08 | 2.10–3.10 |
| UA (µmoL/L) | 487 | 540 | 446 | 465 | 143–463 |
| CR (µmoL/L) | 78.9 | 99.9 | 93.9 | 84.4 | 40.0–110.0 |
| BUN (mmoL/L) | 6.89 | 5.50 | 5.05 | 3.85 | 1.70–8.30 |
| LVEDD (mm) | 68 | 65 | 63 | 56 | 35–56 |
| LVEF (%) | 40 | 45 | 48 | 55 | 50–80 |
| BNP (pg/ml) | 1090 | 1790 | 830 | 388 | 0–400 |
FPG: Fasting blood glucose; TC: Total cholesterol; TG: Triglyceride; HDL: High-density lipoprotein cholesterol; LDL: Low-density lipoprotein cholesterol; UA: Uric acid; CR: Creatinine; BUN: Blood urea nitrogen; LVEDD: Left ventricular end-diastolic diameter; LVEF: Left ventricular ejection fraction; BNP: Brain natriuretic peptide; NA: Not applicable.
Analysis of mitochondrial sequence variants
Because the pedigree analysis suggested a maternal inheritance pattern, we investigated the function of the mitochondrial genome of family members. For this purpose, DNA fragments spanning the mtDNA at positions 3777–4679 and 7908–8816 were PCR amplified, and each fragment was purified and subsequently analyzed by direct sequencing. We looked into the variants in two lists of pathogenic variants from MitoMap (http://www.mitomap.org/) and mtDB (http://www.genpat.uu.se/mtDB/).[18,19] The mtDNA variants identified by the sequencing in all the family members are listed in Table 3. Fisher's exact frequency difference test showed that A8701G mutation [Figure 4], a known missense single nucleotide polymorphisms (SNP) substituting threonine for alanine in the ATP6 gene and belongs to haplogroup M, was the only significant variant, with a P value of 0.005. Among seven other private variants in this matriline, three (m.3970 C>T, m.3915 G>A, and m.4248 T>C) were located in the MT-ND1 gene, m.8363 G>T in the MT-RNAlys gene, m.4435 A to G in the MT-RNAmet gene, m.8414 C>T in the ATP8 gene, and m.8793 T>C in the ATP6 gene. All of these private variants were previously reported and had no potential pathogenicity except for m.8701A>G, which was one of the primary high blood pressure mutations. There was no clear evidence for a synergistic effect between m.8701A>G and other mutations in this matriline. In control group, 1 base change in the tRNA gene and 2 amino-acid substitutions were found.
Table 3.
mtDNA variants in the reported Chinese Han family
| Gene | Position | Replacement | Previously reported | Frequency | P | |
|---|---|---|---|---|---|---|
| Case group | Control group | |||||
| ND1 | 3970 | C to T | Yes | 1 | 1 | NS |
| 3915 | G to A | Yes | 2 | 0 | NS | |
| 4248 | T to C | Yes | 1 | 0 | NS | |
| tRNAMet | 4435 | A to G | Yes | 3 | 1 | NS |
| tRNALys | 8363 | G to T | Yes | 1 | 0 | NS |
| ATP8 | 8414 | C to T | Yes | 4 | 3 | NS |
| ATP6 | 8793 | T to C | Yes | 1 | 0 | NS |
| ATP6 | 8701 | A to G | Yes | 7 | 1 | 0.005 |
NS: Nonsignificant using Fisher’s exact test. A8701G in ATP6 gene (threonine to alanine) was statistically different after adjusting age, gender, and body surface area (Fisher’s exact test P = 0.005). mtDNA: Mitochondrial DNA.
Figure 4.
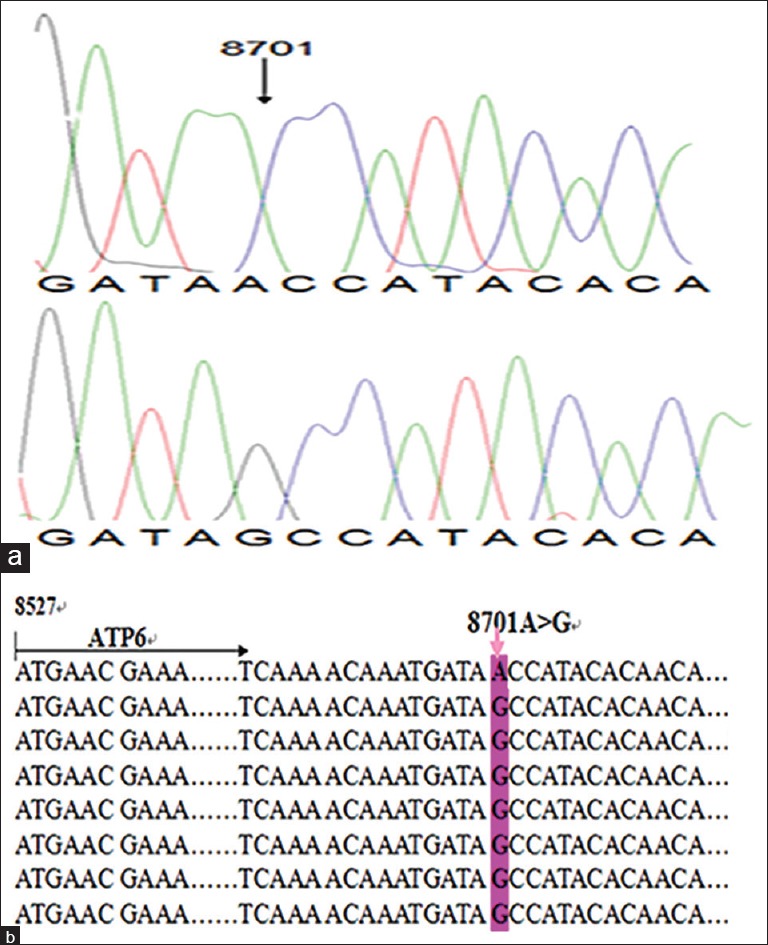
Identification of the 8701A>G mutation in the mitochondrial ATP6 gene. (a) Partial sequence chromatograms of the ATP6 gene from affected individual II-3 and a married-in control II-4. An arrow indicates the location of the base changes at position 8701. (b) The location of the 8701A>G mutation in the mitochondrial ATP6 gene. Pink arrow indicates the position of the ATP6 gene mutation.
DISCUSSION
The association between hypertension and DCM of mitochondrial A8701G is apparently unique in this pedigree. Mother to son transmission was observed, but the high degree of consanguinity suggests recessive inheritance is more likely. It has been shown that consanguineous marriages have a higher prevalence of certain adult diseases and genetic disorders than nonconsanguineous marriages.[20] In accordance with previous reports, our findings showed that consanguineous marriage was an important factor causing illnesses in the offspring of this family. Nevertheless, hypertension and DCM as the clinical phenotype presented only in the maternal lineage of this pedigree. The pathogenesis of the role of genetic disorders caused by consanguineous marriage could not be clarified. The inherited pattern also provides a clear indication that the mtDNA variant contributes to the phenotype.
Although deleterious mtDNA variants are generally heteroplasmic and induce severe clinical symptoms early at onset, mild mutations are often homoplasmic, can be observed in a general population, and may predispose to specific symptoms of diseases such as type 2 diabetes mellitus, Alzheimer's disease, and obesity in various ethnic groups.[21] Clinical and genetic evaluation revealed the varying severity and age at onset in both hypertension and DCM. In this study, the age at onset for the affected matrilineal relatives in the family was between 45 and 60 years old, with an average of 50 years. It was previously reported that 20% of the cases of DCM were likely familial.[22] This mutation in structural gene has also been reported in patients with DCM and hypertension.[23] Multiple types of mutations in mtDNA that cause dysfunction of oxidative phosphorylation (OXPHOS) enzymes have been investigated in patients with cardiomyopathy and hypertension or a combination of the two.[6,7] Mitochondrial OXPHOS defects resulting in the reduction of adenosine triphosphate (ATP) generation and longstanding increase of ROS production may be responsible for the pathogenesis of hypertension, cardiac contractile dysfunction and, subsequently, heart failure and death. In this family, the proband's mother died of cardiac failure several days after surgery and could not be investigated further. Three offspring carrying the mutant were diagnosed with hypertension and DCM, means the transmission was compatible with a maternally inherited trait. Apart from the involvement of variants of the maternally inherited mitochondrial genome, knowledge of the pathogenesis of mitochondrial disorder as a cause of cardiomyopathy remains incomplete.
Our results indicated that hypertension and DCM were prominent features in maternally transmitted disorders in conjunctions with mtDNA A8701G mutation. To date, there is very limited information on the importance of this mutation in cardiovascular disease in general.[24] The underlying contributing factors in the family remain poorly understood, but mtDNA A8701G mutation is potentially an explanatory variable. Due to the high mutation rate of mtDNA, it has accumulated sequential mutations stratified into different mtDNA lineages or haplogroups that have been used as genetic markers of human evolution.[19] mtDNA A8701G mutation in this Chinese pedigree belongs to the Asian haplogroup M. It was characteristically heteroplasmic and was not observed in healthy control subjects during the study period. In the mitochondrial SNP (mtSNP) 8701A/G, there is a 59th amino-acid substitution from threonine to alanine at ATPase6, the F0 subunit of complex V. Using trans-mitochondrial hybrid cells, mtSNP A8701G was shown to cause abnormal results in impaired mitochondrial pH and intracellular calcium dynamics and is suspected to be associated with the pathogenesis of some diseases.[25] Complex V or ATP synthase, the final enzyme of the OXPHOS system, enables protons to flow back to the matrix and uses the released energy to synthesize ATP. It is composed of at least 16 subunits, of which 2 (ATP6 and ATP8) are encoded by the mtDNA. So far, the most common noted result of ATP6 mutation has been confirmed to be neuropathy, ataxia and retinitis pigmentosa, or maternally inherited Leigh's syndrome.[26] A previous study illustrated that mutations of the ATP6 gene were associated with a deficiency in the stability of complex V.[27] More recently, the nucleotide change A to G at position 8701 in ATP6 gene, part of the ATP synthase protein, has been demonstrated to reduce ATP synthesis and significantly impair the assembly or stability of the ATP synthase.[28] To our understanding, the present report of hypertension and DCM in a Chinese pedigree is directly caused by a maternally inherited point mutation in mtDNA and expands the knowledge of genetic heterogeneity.
It is now clear that the mtDNA mutation may play an important role in the pathophysiology of hypertension and DCM. In this family, the prevalence of diseases showed a marked age dependence. The proband's son and younger sister were carrying the same mutant but did not show any clinical presentations of disease. When heteroplasmy reaches the cardiac tissue threshold level, mitochondrial ATP generation becomes insufficient for physiological cardiac function and cardiomyopathy develops. Thus, the impaired mitochondrial function with aging might arise later in life in blood pressure and symptoms of DCM, particularly during the later decades of human life. Those asymptomatic carriers that are at increased risk of disease may need regular screening such as echocardiograms to monitor the early progression of the disorder. Based on studies of heart diseases, it is believed that early pharmacological therapy including angiotensin converting enzyme inhibitors could be very effective in limiting the progression of a given disorder. Further studies will also be needed to elucidate the molecular mechanisms by which A8701G polymorphism of the mtDNA affects age-related mitochondrial functional changes. The homoplasmic level, moderate nature of mitochondrial dysfunction, late onset, and incomplete penetrance of hypertension observed in the affected subjects on maternal linkage harboring A8701G mutation, showed that the mutation is an inherited contributing factor necessary for the development of disease but may be insufficient to produce a clinical picture on its own. Although most of the known mutations appear to be specific to individual probands, the incomplete penetrance of other clinical abnormalities arises from homoplasmic mtDNA mutations including hypertension-associated mtDNA A4435G mutation, sensorineural hearing loss-associated 12SrRNA A1555G mutation, and Leber's hereditary optic neuropathy-associated G11778A mutation.[29,30] The novel known A4435G mutation in tRNAMet gene, as shown in Table 3, may influence the transcription of tRNA, thereby affecting the steady level of protein synthesis. In addition, the base pair change C to T at position 8414 in ATP8 gene was also found in matrilineal relatives in this family. The mutations most likely affect the function of protein synthesis.
As a lifestyle-related disease, both genetic and environmental factors may contribute to the clinical phenotype of heart disease. For instance, patient II-5, who became a heavy smoker only after the first symptoms of diabetes, developed hypertension, cardiomyopathy, and other symptoms possibly related to chronic alcohol abuse in conjunction with the mitochondrial dysfunction. Many factors could contribute to the observed phenotypes in the studied family with the proband having both a more severe phenotype and more severe clinical syndrome than her two brothers. Sequencing of the 8701 mutation failed to identify other sequence variations that could account for the varying phenotypes in these subjects. Variability in clinical expression can also be explained by nuclear genetic background, environmental and epigenetic factors, and personal lifestyle interactions.[31,32,33,34] Integrated research could disclose some of the ways that genetic and environmental factors lead to the expression of proteins controlling the causal processes of hypertension.[35] A8701G mutation should be added to the list of predicted risk factors for future molecular investigation. Further studies should be performed to confirm its detrimental effect with more data closely related to mitochondrial function. So far, genetic counseling approaches for familial disease are still underdeveloped, and consensus guidelines remain meager. As incomplete penetrance is a feature of this disease, mutation carriers who remain asymptomatic should be counseled that they are at risk for the development of cardiac symptoms and advised to consider salt and fluid restriction, treatment of hypertension, limitation of alcohol intake, control of body weight, and physical exercise. In addition, a detailed examination and investigation will be required to define the role of A8701G mutation, and genetic factors caused by consanguineous marriage might affect this family with maternally inherited hypertension and DCM.
There are some study limitations in this study. A family trio sequencing study identified a mtDNA A8701G mutation associated with maternally inherited hypertension and DCM in a Chinese pedigree of a consanguineous marriage. A mitochondrial chromosome sequencing-based association study looked into the sequence variations that could account for the varying phenotypes in these subjects. The findings are helpful for counseling families with cardiovascular disorders and studies on the cardiovascular disorders associated mtDNA mutations. While the variability in clinical expression can also be explained by many factors, including nuclear genetic background, environmental and epigenetic factors, and personal lifestyle choices, the mechanism underlying the relationship between A8701G and maternally inherited hypertension and DCM is still unclear. Further integrated research could reveal how genetic and environmental factors, together with protein expression, control the causal processes of hypertension.
In conclusion, our data reinforced the previous observation that mitochondrial dysfunction caused by A8701G mutation has the potential to contribute, either singly or synergistically, to the pathophysiology of cardiovascular diseases. This mutation was heteroplasmic in blood and was accompanied by slightly reduced deficiency of mitochondrial respiratory chain enzymes. There has been little evidence supporting a direct association of cardiovascular diseases with the 8701 variant of mtDNA before our study. Consequently, our findings will be helpful for counseling patients with familial DCM and hypertension. From a lifestyle modification viewpoint, avoiding consanguineous marriages in addition to refraining from environmental risk factors should be emphasized.
Financial support and sponsorship
This work was supported by grants from the Science Committee (No. BS2004532), Health Department of Jiangsu Province (No. Z200514), and Program for Jiangsu Province Outstanding Medical Talented Leader (No. JS2006038).
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
We are grateful to the parents of the patients and their family, nurses and technical staff from the Department of Cardiology in Northern People's Hospital.
Footnotes
Edited by: Yuan-Yuan Ji
REFERENCES
- 1.Murtuza B, Fenton M, Burch M, Gupta A, Muthialu N, Elliott MJ, et al. Pediatric heart transplantation for congenital and restrictive cardiomyopathy. Ann Thorac Surg. 2013;95:1675–84. doi: 10.1016/j.athoracsur.2013.01.014. doi: 10.1016/j.athoracsur.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 2.Zheng X, Chen S, Wang J, Yang T, Chen Y. Dilated cardiomyopathy with hypertension: Prevalence and response to high-dose ß1-adrenoceptor antagonist therapy. Clin Exp Pharmacol Physiol. 2009;36:945–9. doi: 10.1111/j.1440-1681.2009.05184.x. doi: 10.1111/j.1440-1681.2009.05184.x. [DOI] [PubMed] [Google Scholar]
- 3.Chan SH, Wu KL, Chang AY, Tai MH, Chan JY. Oxidative impairment of mitochondrial electron transport chain complexes in rostral ventrolateral medulla contributes to neurogenic hypertension. Hypertension. 2009;53:217–27. doi: 10.1161/HYPERTENSIONAHA.108.116905. doi: 10.1161/HYPERTENSIONAHA.108.116905. [DOI] [PubMed] [Google Scholar]
- 4.Armani C, Botto N, Andreassi MG, Centaro E. Molecular markers of cardiovascular damage in hypertension. Curr Pharm Des. 2013;19:2341–50. doi: 10.2174/1381612811319130002. [DOI] [PubMed] [Google Scholar]
- 5.Li R, Liu Y, Li Z, Yang L, Wang S, Guan MX. Failures in mitochondrial tRNAMet and tRNAGln metabolism caused by the novel 4401A>G mutation are involved in essential hypertension in a Han Chinese family. Hypertension. 2009;54:329–37. doi: 10.1161/HYPERTENSIONAHA.109.129270. doi: 10.1161/HYPERTENSIONAHA.109.129270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Y, Li R, Li Z, Wang XJ, Yang L, Wang S, et al. Mitochondrial transfer RNAMet 4435A>G mutation is associated with maternally inherited hypertension in a Chinese pedigree. Hypertension. 2009;53:1083–90. doi: 10.1161/HYPERTENSIONAHA.109.128702. doi: 10.1161/HYPERTENSIONAHA.109.128702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Z, Liu Y, Yang L, Wang S, Guan MX. Maternally inherited hypertension is associated with the mitochondrial tRNA (Ile) A4295G mutation in a Chinese family. Biochem Biophys Res Commun. 2008;367:906–11. doi: 10.1016/j.bbrc.2007.12.150. doi: 10.1016/j.bbrc.2007.12.150. [DOI] [PubMed] [Google Scholar]
- 8.Wang S, Li R, Fettermann A, Li Z, Qian Y, Liu Y, et al. Maternally inherited essential hypertension is associated with the novel 4263A>G mutation in the mitochondrial tRNAIle gene in a large Han Chinese family. Circ Res. 2011;108:862–70. doi: 10.1161/CIRCRESAHA.110.231811. doi: 10.1161/CIRCRESAHA.110.231811. [DOI] [PubMed] [Google Scholar]
- 9.McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123:19–26. doi: 10.1172/JCI62862. doi: 10.1172/JCI62862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhasin P, Kapoor S. Impact of consanguinity on cardio-metabolic health and other diseases: Findings from an Afro-Indian tribal community. J Community Genet. 2015;6:129–35. doi: 10.1007/s12687-014-0207-z. doi: 10.1007/s12687-014-0207-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abumwais JQ. Etiology of chronic renal failure in Jenin district, Palestine. Saudi J Kidney Dis Transpl. 2012;23:158–61. [PubMed] [Google Scholar]
- 12.Ross JM, Stewart JB, Hagström E, Brené S, Mourier A, Coppotelli G, et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature. 2013;501:412–5. doi: 10.1038/nature12474. doi: 10.1038/nature12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O’Connell J, et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the definition and classification of cardiomyopathies. Circulation. 1996;93:841–2. doi: 10.1161/01.cir.93.5.841. [DOI] [PubMed] [Google Scholar]
- 14.Chalmers J, MacMahon S, Mancia G, Whitworth J, Beilin L, Hansson L, et al. 1999 World Health Organization-International Society of Hypertension Guidelines for the management of hypertension. Guidelines sub-committee of the World Health Organization. Clin Exp Hypertens. 1999;21:1009–60. doi: 10.3109/10641969909061028. [DOI] [PubMed] [Google Scholar]
- 15.Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure. The sixth report of the Joint National Committee on prevention, detection, evaluation, and treatment of high blood pressure. Arch Intern Med. 1997;157:2413–46. doi: 10.1001/archinte.157.21.2413. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y, Li Y, Gao J, Zhu C, Lan Y, Yang J, et al. Molecular characterization of a Chinese family carrying a novel C4329A mutation in mitochondrial tRNAIle and tRNAGln genes. BMC Med Genet. 2014;15:84. doi: 10.1186/1471-2350-15-84. doi: 10.1186/1471-2350-15-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gómez-Carballa A, Cerezo M, Balboa E, Heredia C, Castro-Feijóo L, Rica I, et al. Evolutionary analyses of entire genomes do not support the association of mtDNA mutations with Ras/MAPK pathway syndromes. PLoS One. 2011;6:e18348. doi: 10.1371/journal.pone.0018348. doi: 10.1371/journal.pone.0018348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–65. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 19.Zhu HY, Wang SW, Martin LJ, Liu L, Li YH, Chen R, et al. The role of mitochondrial genome in essential hypertension in a Chinese Han population. Eur J Hum Genet. 2009;17:1501–6. doi: 10.1038/ejhg.2009.63. doi: 10.1038/ejhg 200963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Darr A, Small N, Ahmad WI, Atkin K, Corry P, Modell B. Addressing key issues in the consanguinity-related risk of autosomal recessive disorders in consanguineous communities: Lessons from a qualitative study of British Pakistanis. J Community Genet. 2015:12. doi: 10.1007/s12687-015-0252-2. doi: 10.1007/s12687-015-0252-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Itsara LS, Kennedy SR, Fox EJ, Yu S, Hewitt JJ, Sanchez-Contreras M, et al. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet. 2014;10:e1003974. doi: 10.1371/journal.pgen.1003974. doi: 10.1371/journal.pgen.1003974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, Salmenperä P, et al. Genetics and genotype-phenotype correlations in Finnish patients with dilated cardiomyopathy. Eur Heart J. 2015;36:2327–37. doi: 10.1093/eurheartj/ehv253. doi: 10.1093/eurheartj/ehv253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michels VV, Moll PP, Miller FA, Tajik AJ, Chu JS, Driscoll DJ, et al. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med. 1992;326:77–82. doi: 10.1056/NEJM199201093260201. [DOI] [PubMed] [Google Scholar]
- 24.Mahjoub S, Sternberg D, Boussaada R, Filaut S, Gmira F, Mechmech R, et al. A novel mitochondrial DNA tRNAIle (m.4322dupC) mutation associated with idiopathic dilated cardiomyopathy. Diagn Mol Pathol. 2007;16:238–42. doi: 10.1097/PDM.0b013e3180cc313b. doi: 10.1097/PDM.0b013e3180cc313b. [DOI] [PubMed] [Google Scholar]
- 25.Wallace DC, Brown MD, Lott MT. Mitochondrial DNA variation in human evolution and disease. Gene. 1999;238:211–30. doi: 10.1016/s0378-1119(99)00295-4. [DOI] [PubMed] [Google Scholar]
- 26.Kazuno AA, Munakata K, Nagai T, Shimozono S, Tanaka M, Yoneda M, et al. Identification of mitochondrial DNA polymorphisms that alter mitochondrial matrix pH and intracellular calcium dynamics. PLoS Genet. 2006;2:e128. doi: 10.1371/journal.pgen.0020128. doi: 10.1371/journal.pgen.0020128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Payne BA, Wilson IJ, Yu-Wai-Man P, Coxhead J, Deehan D, Horvath R, et al. Universal heteroplasmy of human mitochondrial DNA. Hum Mol Genet. 2013;22:384–90. doi: 10.1093/hmg/dds435. doi: 10.1093/hmg/dds435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wallace DC, Fan W, Procaccio V. Mitochondrial energetics and therapeutics. Annu Rev Pathol. 2010;5:297–348. doi: 10.1146/annurev.pathol.4.110807.092314. doi: 10.1146/annurev.pathol.4.110807.092314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verny C, Guegen N, Desquiret V, Chevrollier A, Prundean A, Dubas F, et al. Hereditary spastic paraplegia-like disorder due to a mitochondrial ATP6 gene point mutation. Mitochondrion. 2011;11:70–5. doi: 10.1016/j.mito.2010.07.006. doi: 10.1016/jmito.2010.07.006. [DOI] [PubMed] [Google Scholar]
- 30.Qu J, Li R, Zhou X, Tong Y, Lu F, Qian Y, et al. The novel A4435G mutation in the mitochondrial tRNAMet may modulate the phenotypic expression of the LHON-associated ND4 G11778A mutation. Invest Ophthalmol Vis Sci. 2006;47:475–83. doi: 10.1167/iovs.05-0665. doi: 10.1167/iovs.05-0665. [DOI] [PubMed] [Google Scholar]
- 31.Yang Z, Venardos K, Jones E, Morris BJ, Chin-Dusting J, Kaye DM. Identification of a novel polymorphism in the 3’UTR of the L-arginine transporter gene SLC7A1: Contribution to hypertension and endothelial dysfunction. Circulation. 2007;115:1269–74. doi: 10.1161/CIRCULATIONAHA.106.665836. doi: 01161/CIRCULATIONAHA.106.665836. [DOI] [PubMed] [Google Scholar]
- 32.Samuels DC, Li C, Li B, Song Z, Torstenson E, Boyd Clay H, et al. Recurrent tissue-specific mtDNA mutations are common in humans. PLoS Genet. 2013;9:e1003929. doi: 10.1371/journal.pgen.1003929. doi: 10.1371/journal.pgen.1003929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bai Y, Guo Z, Xu J, Zhang J, Cui L, Zhang H, et al. Single nucleotide polymorphisms in the D-loop region of mitochondrial DNA and age-at-onset of patients with chronic kidney disease. Chin Med J. 2014;127:3088–91. doi: 10.3760/cma.i.issn.0366-6999.20140708. [PubMed] [Google Scholar]
- 34.Lu Z, Chen H, Meng Y, Wang Y, Xue L, Zhi S, et al. The tRNAMet 4435A>G mutation in the mitochondrial haplogroup G2a1 is responsible for maternally inherited hypertension in a Chinese pedigree. Eur J Hum Genet. 2011;19:1181–6. doi: 10.1038/ejhg.2011.111. doi: 10.1038/ejhg.2011.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Djoussé L, Driver JA, Gaziano JM. Relation between modifiable lifestyle factors and lifetime risk of heart failure. JAMA. 2009;302:394–400. doi: 10.1001/jama.2009.1062. doi: 10.1001/jama.2009.1062. [DOI] [PMC free article] [PubMed] [Google Scholar]