

Abstract
Background
Glioblastoma (GBM) is the most frequent and aggressive primary brain tumor in adults. Recent research on cancer stroma indicates that the brain microenvironment plays a substantial role in tumor malignancy and treatment responses to current antitumor therapy. In this work, we have investigated the effect of alterations in brain tumor extracellular matrix tenascin-C (TNC) on brain tumor growth patterns including proliferation and invasion.
Methods
Since intracranial xenografts from patient-derived GBM neurospheres form highly invasive tumors that recapitulate the invasive features demonstrated in human patients diagnosed with GBM, we studied TNC gain-of-function and loss-of function in these GBM neurospheres in vitro and in vivo.
Results
TNC loss-of-function promoted GBM neurosphere cell adhesion and actin cytoskeleton organization. Yet, TNC loss-of-function or exogenous TNC had no effect on GBM neurosphere cell growth in vitro. In animal models, decreased TNC in the tumor microenvironment was accompanied by decreased tumor invasion and increased tumor proliferation, suggesting that TNC regulates the “go-or-grow” phenotypic switch of glioma in vivo. We demonstrated that decreased TNC in the tumor microenvironment modulated behaviors of stromal cells including endothelial cells and microglia, resulting in enlarged tumor blood vessels and activated microglia in tumors. We further demonstrated that tumor cells with decreased TNC expression are sensitive to anti-proliferative treatment in vitro.
Conclusion
Our findings suggest that detailed understanding of how TNC in the tumor microenvironment influences tumor behavior and the interactions between tumor cells and surrounding nontumor cells will benefit novel combinatory antitumor strategies to treat malignant brain tumors.
Keywords: glioblastoma, patient-derived GBM neurospheres, TNC, tumor microenvironment
Glioblastoma (GBM), the most common and malignant primary adult brain tumor, comprises ∼25 000 new cases annually in the United States. Despite considerable progress in modern antitumor therapy, the median survival for patients diagnosed with GBM is <2 years. The mechanisms by which GBM tumor cells exert their malignant potential and lead to poor patient prognosis originate from the tumor cells themselves, in addition to factors intrinsic to the surrounding tumor microenvironment. The tumor microenvironment includes surrounding stromal cells such as endothelial cells in blood vessels, immune cells, fibroblasts, and the extracellular matrix (ECM).1,2 Each of these stromal cells comprises a complex network of macromolecules with distinctive physical, biochemical, and biomechanical properties. Although tightly controlled during embryonic development and organ homeostasis, the ECM is commonly deregulated and disorganized in cancer.3 Abnormalities in the ECM affect cancer progression by directly promoting cellular transformation and metastasis. ECM anomalies also change the behavior of stromal cells, facilitate tumor-associated angiogenesis, and promote tumor-associated inflammation leading to the generation of tumorigenic microenvironments. In GBM, the biological impact of the tumor microenvironment (ECM) on tumor cell behavior and overall tumor progression remains poorly understood.
The extracellular matrix glycoprotein tenascin-C (TNC) is a nonfilamentous protein that mediates cell-cell and cell-matrix interactions. TNC is highly expressed during normal fetal development and plays an important role during embryogenesis and morphogenesis.4,5 In adult tissues, TNC expression is very low, yet it is upregulated under pathological conditions such as infection, inflammation, or tumor growth. TNC is highly expressed in the tumor microenvironment of solid malignancies including lung cancer, breast cancer, colon cancer, and GBM. The TNC protein consists of 6 subunits linked by bisulfide bonds; each subunit has a cysteine-rich amino terminus, 14.5 epidermal growth factor (EGF)-like repeats, 8 constitutive fibronectin type III domains, and a fibrinogen-like C-terminus.4 Due to its multidomain structure, TNC has the capacity to interact with multiple proteins influencing cell migration, adhesion, proliferation, and differentiation. For example, the 8 fibronectin-like domains interact with fibronectin and elicit both anti-adhesion and pro-adhesion functions depending on alternative splicing. In addition, TNC interacts with Toll-like receptors through its fibrinogen domains during joint inflammation.6
The biological function of TNC in development, tissue remodeling, and wound healing has been extensively studied, and TNC is implicated in tumorigenesis and metastatic tumor progression.4,5 In normal brain, TNC is expressed in the ventricular and subventricular zones where neural stem cells reside and orchestrate growth factor signaling to accelerate neural stem cell development.7 In cancers, Oskarsson et al reported that TNC creates a metastasis niche for migrating breast cancer cells.8 Whereas TNC is produced by stromal fibroblasts in the majority of solid tumors, brain tumor cells themselves are the main source of extracellular matrix TNC in glial malignancies.9,10 The intensity of TNC expression is shown to correlate with glioma grade and patient prognosis.9,11 Due to its expression in solid tumors, TNC has been used as a tumor-associated antigen to deliver antibody-conjugated radiotherapeutic agents to GBM.12
Like many other solid tumors, GBM exhibits cellular heterogeneity and differentiation hierarchy in which GBM cancer stem cells (GSCs) are at the apex.13 GSCs are characterized by a capacity for long-term self-renewal, multilineage differentiation, and tumor propagation.14–16 These GSCs are believed to contribute to GBM vascularization, therapeutic resistance, and tumor recurrence. Compared with GBM cell lines maintained in serum-containing medium (eg, U87, U251), intracranial xenografts from GSCs form highly invasive tumors that recapitulate the invasive features demonstrated in human patients diagnosed with GBM.16,17 Therefore, investigation into the interactions of GSCs and their surrounding tumor microenvironment is expected to provide novel insight into the development of therapeutic regimens against GBM. In this study, we used well-characterized, patient-derived GBM neurosphere cultures enriched in GSCs to interrogate the impact of tumor microenvironment TNC on GBM growth pattern.
Materials and Methods
Reagents and Cell Cultures
All reagents were purchased from Sigma Chemical Co. unless otherwise stated. Tenascin-C was purchased from EMD/Millipore. The GBM patient-derived neurosphere lines HSR-GBM1A (0913) and HSR-GBM1B (0627) were originally derived by Dr. A. Vescovi et al.5–17 Rat brain endothelial cells were originally cultured from microvessels isolated from the brains of Lewis rats and immortalized using the adenovirus 2 EIA gene as described in Roux et al.18
Viral Transfection
To knock down TNC expression, we used lentivirus containing a control nonsilencing (NS) sequence and 2 distinct TNC shRNAs (10 044 527 and 10 054 467) in a GIPZ viral vector (Thermoscientific) containing the green fluorescent protein (GFP) coding frame. Neurosphere cells were triturated as single cell suspension and transfected with lentivirus for 72 hour, followed by antibiotic selection with puromycin (2 µg/mL). Transfection efficiency was determined by observing GFP+ cells under fluorescent microscopy. Pooled transfectants were tested for TNC by Western blot analysis.
Neurosphere Growth Assays
Neurosphere assays were performed as previously described.15–17 Briefly, GBM neurospheres were seeded at the density of 5 × 103/cm2 in 6-well plates. Cells were incubated for 5–7 days and counted. For details, see Supplementary materials.
Cell Adhesion and Migration/Invasion Assays
Twenty-four-well plates were coated with laminin (10 μg/mL) or TNC (10 μg/mL) at 37°C overnight and rinsed twice with DMEM. After blocking with 0.5% bovine serum albumin for 1 hour, GBM neurosphere cells were plated at 4 × 105/cm2. After 4 hours, plates were washed with PBS, and the remaining cells were quantified by MTT assays. We used Boyden chambers for cell migration/invasion assays as we described in Wang et al.19 For details, see Supplementary materials.
Immunoblot Analysis
Immunoblot analysis was performed as previously described.15,16 All primary antibodies were purchased from Cell Signaling Technology except anti–TNC antibody (Millipore). For details, see Supplementary materials.
Tumor Xenografts, Immunofluorescence, and Immunohistochemistry
For intracranial xenografts, 8-week-old female SCID immunodeficient mice (NCI) received 10 000 viable control or TNC knockdown GBM neurosphere cells in 2 µL of PBS by stereotactic injection into the right caudate/putamen.20 Mice were sacrificed 6 weeks after implantation. The primary antibodies used for immunofluorescent staining are the following: monoclonal anti-Ki67 (BD Biosciences), anti-TNC (Millipore), anti-cleaved caspase 3 (Cell Signaling), anti-laminin, anti-Iba1 (Millipore), and anti-mouse MHCII (eBiosciences). All animal protocols used in this study were approved by the Johns Hopkins School of Medicine Animal Care and Use Committee.
Statistical Analysis
Data were analyzed using parametric statistics with 1-way ANOVA. Post hoc tests included the Student t test and Tukey multiple comparison test, as appropriate, using Prism (GraphPad). All experiments reported here represent at least 3 independent replications. All data are represented as mean value ± standard error of mean (SEM). Significance was set at P < .05. This study did not involve in human tissues.
Results
Expression of Tenascin-C in Patient-derived Gblioblastoma Neurosphere Cells
GBM patient-derived neurosphere lines HSR-GBM1A and HSR-GBM1B were utilized to dissect the impact of the extracellular matrix protein TNC on GBM malignancy. These cultures were enriched in GBM stem cells (GSCs) to form infiltrative intracranial xenografts, as in prior studies.15–17 TNC was highly expressed in both HSR-GBM1A and HSR-GBM1B cells (Fig. 1A). Immunoblot analysis of tumor cell extracts revealed multiple immunoreactive bands with molecular weight between 210–300 kDa in GBM neurosphere cells, consistent with TNC's alternatively spliced forms, as previously reported.21 TNC expression was also examined in the conditioned medium of HSR-GBM1A and HSR-GBM1B cells utilizing immunoblot analysis. A predominant form of TNC migrating at ∼250 kDa was detected in the conditioned medium of both GBM neurosphere lines tested (Fig. 1B).
Fig. 1.
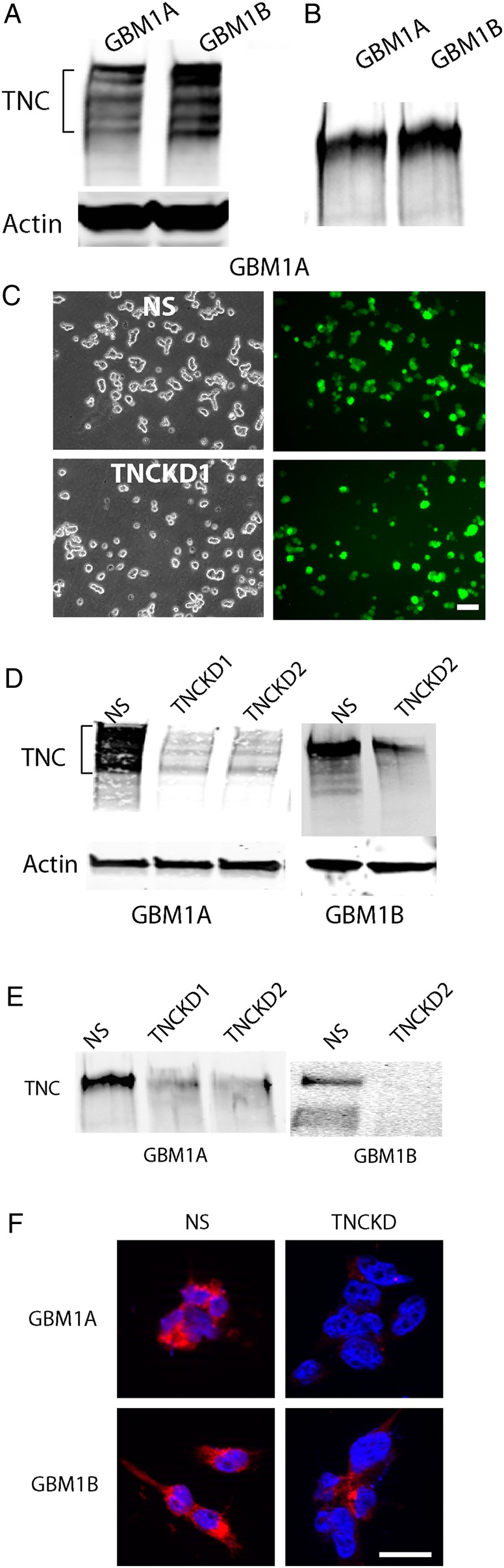
Expression of tenascin-C (TNC) in glioblastoma (GBM) neurosphere cells. (A). TNC protein was highly expressed in GBM1A and GBM 1B cells. The multiple immunoreaction bands indicate multisplicing forms of TNC. (B). TNC was detected in the conditioned medium of GBM neurosphere cells, suggesting that TNC may elicit biological function via autocrine or paracrine loop. (C). GBM neurosphere cells were transfected with lentivirals containing nonsilencing shRNA sequence (NS) or TNC shRNA together with a green fluorescent protein coding frame. After transfection, the neurosphere cells were plated as single cells and observed under fluorescence microscopy. Approximately 80%–90% of the transfected cells were GFP+. Bar = 100 µm. (D). Western blot analysis confirmed significant downregulation of TNC in cells receiving 2 distinct TNC shRNAs (TNCKD1 and TNCKD2) compared with control transfected cells (NS). (E). TNC expression in the conditioned medium of TNC knockdown GBM neurosphere cells was also decreased. (F). Immunocytostaining of TNC in GBM neurosphere cells. TNC was highly expressed in control cells, whereas the staining was much weaker in TNC knockdown cells. Bar = 20 µm.
To investigate the biological function of endogenous TNC in GBM neurosphere cells, we generated stable lines with TNC knockdown using 2 distinct TNC shRNAs. Both nonsilencing shRNA-transfected cells (designated as NS) and TNC shRNA-transfected cells (designated as TNCKD1 and TNCKD2) were labeled with GFP. Under the lentiviral vector transfection, we observed ∼80%–90% GFP+ cells in both the NS and TNC knockdown stable cell lines (Fig. 1C). Immunoblot analysis of cell extracts confirmed ∼90% and ∼70% inhibition of TNC expression in HSR-GBM1A and HSR-GBM1B cells receiving TNC shRNA1 and TNC shRNA2, respectively (Fig. 1D). TNC levels in conditioned medium were also significantly decreased (∼70%–90%) in TNC knockdown cells when compared with control cells (Fig. 1E). Immunofluorescent staining further confirmed strong TNC staining in control cells, whereas TNC expression was greatly diminished in TNC knockdown cells (Fig. 1F).
Tenascin-C Modulates Glioblastoma Neurosphere Cell Adhesion via Focal Adhesion Kinase Pathway
The extracellular matrix protein TNC is implicated in cell-matrix attachment. Using our stable TNC knockdown cell lines, we investigated the effect of TNC loss-of-function on GBM neurosphere cell adhesion. We measured GBM neurosphere cell adhesion on laminin-coated surfaces, a condition that has been previously reported to maintain tumor cell stem-like properties.22 Compared with controls, TNC knockdown resulted in increased cell adhesion on laminin (∼3–5 fold) (Fig. 2A and B). When the culture surfaces were coated with purified TNC instead of laminin, cell adhesion induced by TNC knockdown was abolished (Fig. 2A and B). Thus, TNC had an anti-adhesive role in GBM neurosphere cells, which would affect neurosphere cell migration/invasion ability. We examined neurosphere invasion on Matrigel-coated transwells and found that TNC knockdown GBM neurosphere cells were less migratory/invasive (Fig. 2C), possibly because fewer cells were adherent to transwells. To rule out the possibility that less migratory/invasive cells are due to decreased cell proliferation, we investigated the effect of TNC knockdown on GBM neurosphere cell growth in vitro. Compared with controls, there was no appreciable cell growth change in TNCKD1 and TNCKD2 GBM neurosphere cells (Fig. 2D). These results suggest that TNC decreases cell surface adhesion but does not alter the growth response of GBM neurosphere cells in vitro.
Fig. 2.

Tenascin-C (TNC) loss-of-function facilitates GBM neurosphere cell adhesion. (A). Phase contrast microphotographs of GBM neurosphere cells grown on laminin-coated (10 µg/mL) surfaces for 4 hours. Control cells loosely attached to the laminin-coated surfaces, TNC knockdown cells spread widely on laminin-coated surfaces. When the TNC knockdown cells were plated on TNC-coated surfaces, there was no difference in cell adhesion between control and TNC knockdown cells. Bar = 50 µm (B). Quantitative assays of GBM neurosphere cell adhesion on laminin- or TNC-coated surfaces. Cells were plated for 4 hours; nonadherent cells were washed away; and the remaining adherent cells were measured by MTT assays. Compared with control NS cells, more than 3-5 fold TNC knockdown cells remained on laminin-coated surfaces; whereas TNC-coated surfaces blocked cell adhesion. (C). Control and TNC knockdown cells were plated on Matrigel-coated transwells in growth factor-free GBM medium; the bottom wells were filled with 10% FCS containing DMEM medium. After 24 hours, the invading cells were quantified by counting cells in 5 selected fields per transwell. TNC knockdown cells were less invasive than control cells. (D). TNC knockdown in GBM neurosphere cells did not affect cell growth as indicated by cell number counting in cultures after 3 and 6 days. (*P < .05; ***: P < .001).
To investigate the mechanism by which TNC loss-of-function promotes GBM neurosphere cell adhesion, we studied focal adhesion and cytoskeleton structure in control and TNC knockdown cells using anti-vinculin antibody and phalloidin that detects polymerized actin (F-actin). Control cells demonstrated very weak vinculin and F-actin fluorescent staining. In contrast, strong, punctate vinculin/F-actin staining in filopodial-like cytoplasmic projections was observed in TNC knockdown cells, suggesting increased focal adhesions in these cells (Supplementary material, Fig. S1A). We further examined downstream signaling pathways that mediate the enhanced adhesion of TNC knockdown GBM neurosphere cells, including AKT, MAPK, and focal adhesion kinase (FAK) pathways.23 FAK, AKT, and MAPK activation were increased 2.0-fold, 5.1-fold, and 5.9-fold, respectively, relative to controls (Supplementary material, Fig. S1B). Given the increased expression of these second messenger molecules, we used pathway-specific pharmacological inhibitors to determine if the inhibition of FAK, AKT, and/or MAPK pathways altered TNC knockdown-mediated cell adhesion. The adhesion of TNC knockdown cells was significantly blocked by the specific FAK pathway inhibitors PF573228 (100 nmol/L, ∼75%, P < .001) and FAK inhibitor 14 (100 µmol/L, ∼80%, P < .001) (Supplementary material, Fig. S1C).19 In contrast, the Erk1/2 pathway inhibitor PD98059 (30 µmol/L) and the PI3K pathway inhibitors wortmannin (10 µmol/L) and LY294002 (10 µmol/L) had no effect on TNC knockdown-induced cell adhesion (Supplementary material, Fig. S1C), suggesting that TNC knockdown in GBM neurosphere cells leads to FAK-mediated cell adhesion.
Effect of Tenascin-C Gain-of-Function on Glioblastoma Neurosphere Cell Growth, Migration, and Invasion
We further examined the effect of exogenous TNC on GBM neurosphere cell growth, adhesion, and migration. In the presence of EGF and FGF, TNC had no obvious effect on cell growth (Supplementary material, Fig. S2A), further confirming that TNC is dispensable for GBM neurosphere cell growth. For adhesion assays, we coated cell culture surfaces with TNC (10 μg/mL) and compared cell attachment with that on laminin-coated surfaces. TNC induced a decreased cell adhesion (Supplementary material, Fig. S2B). We further examined the effect of TNC on GBM neurosphere cell migration, either as a coating agent or a chemoattractant in transwell assays. Compared with laminin-coated surfaces, cell migration was decreased on TNC-coated surfaces (Supplementary material, Fig. S2C and D), consistent with the anti-adhesive results shown in Supplementary material, Fig. S2B. However, when we examined cell migration on laminin-coated surfaces with concurrent application of TNC as a chemoattractant in bottom wells of transwell assays, TNC increased cell migration in a dose-dependent manner. Compared with control, TNC at 3 µg/mL increased cell migration by ∼2.1 fold (Supplementary material, Fig. S2E, P < .05). Similarly, TNC (10 µg/mL) promoted GBM neurosphere cell invasion through Matrigel-coated surfaces (Supplementary material, Fig. S2F, P < .05). These are consistent with an anti-attachment role of TNC in GBM neurosphere cells, which allows cells to detach from ECM-coated surface to migrate/invade.
Decreased Tenascin-C in Tumor Microenvironment Mediates Glioma “Go-or-Grow”
To study the impact of TNC expression on the tumor growth pattern, we generated intracranial xenografts using control and TNC knockdown GBM neurosphere cells. Immunofluorescent staining was used to examine TNC expression in vivo. In control tumors, TNC was highly expressed and homogenously distributed in tumor extracellular spaces; whereas TNC expression was greatly reduced in tumor extracellular spaces in TNC knockdown xenografts (Fig. 3A).
Fig. 3.

Decrease of tenascin-C (TNC) in the tumor microenvironment regulates GBM go-or-grow. (A). Immunofluorescent staining of TNC in tumor xenografts derived from control and TNCKD1 GBM neurosphere cells. Hatched areas are enlarged in the two panels on the right; “c” stands for center, “b” stands for border. TNC was highly expressed in control tumor extracellular spaces, both center and border (upper panel). In TNC knockdown xenografts, weak TNC expression was found in the extracellular space along tumor borders, and TNC expression in tumor cores was extremely low (lower panel). (B). Quantification of tumor size based on H&E staining. Xenografts from TNC knockdown cells were significantly bigger than those of control cells. (C and D). H&E staining and fluorescent images showed that TNC knockdown xenografts were less invasive than controls. In TNC knockdown xenografts, tumor cells were confined within distinct tumor borders; whereas in control tumors, cells migrated into adjacent normal brain tissues. (E). Tumor invasion was quantified by counting the number of migrating tumor cells (GFP+) beyond the tumor mass under fluorescent microscopy in 8 randomly selected fields per tumor. Control cells clearly displayed an invasive phenotype as there were 70% more migrating tumor cells beyond tumor borders (***P < .001). Bar = 200 µm.
We examined the tumor growth response and the pattern (tumor core and the tumor/normal brain interface) of xenografts derived from GBM neurosphere cells via H&E and immunofluorescent staining. Intracranial xenografts derived from TNC knockdown cells were significantly larger than those derived from control cells (27 ± 2.67 mm3 versus 6.31 ± 0.64 mm3) (Fig. 3B, P < .001). Consistent with previous reports,14,16 tumors derived from control cells grew in an invasive fashion with tumor cells infiltrating adjacent normal brain tissues, a pattern typical for GBM neurosphere cells. In contrast, tumors derived from TNC knockdown cells were less invasive, with tumor cells confined to more well-defined tumor borders (Fig. 3C and D). To quantify the effect of TNC knockdown on tumor cell invasion, we randomly selected 8 regions in the periphery of each tumor and counted the number of migrating tumor cells (GFP+) beyond the noninvasive tumor borders that were defined according to cell density. Seventy percent more tumor cells migrated beyond tumor borders in xenografts derived from control cells than those from TNC knockdown cells (Fig. 3E, P < .001).
We hypothesized that the increased tumor size in TNC knockdown xenografts could result from increased cell proliferation, decreased apoptosis, or both. We carried out double-staining in NS and TNC knockdown xenografts with anti-TNC and anti-Ki67 antibodies. NS xenografts were associated with high TNC expression and relatively low Ki67 staining. The TNC knockdown xenografts were associated with low TNC expression and higher Ki67 staining and thus were more proliferative (Fig. 4A). There were ∼32% more Ki67+ cells in TNC knockdown xenografts compared with the control xenografts (Fig. 4B, P < .001). Conversely, there was no significant difference in tumor cell death in control and TNC knockdown xenografts as revealed by cleaved caspase-3 staining (Fig. 4C). The data indicate that TNC loss-of-function increased tumor growth by promoting tumor cell proliferation in vivo, suggesting that TNC knockdown mediates the go-or-grow growth pattern of glioma in vivo, which was distinct from our in vitro findings that TNC did not affect tumor cell growth.
Fig. 4.
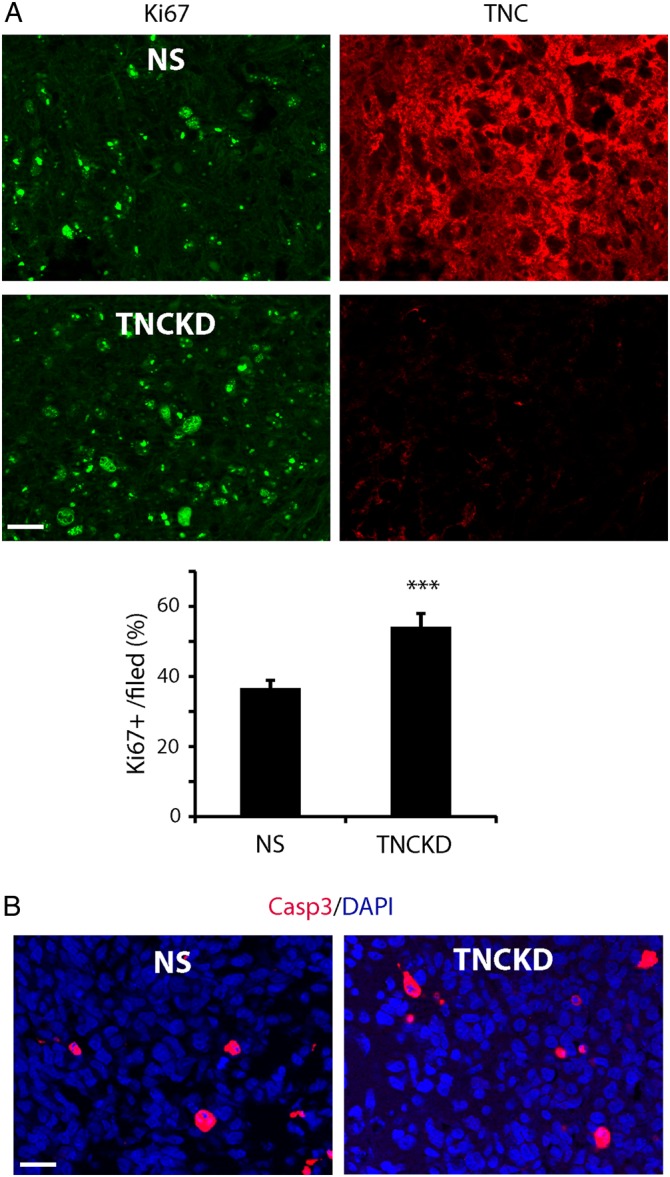
Tenascin-C (TNC) downregulation in the tumor microenvironment promotes intracranial tumor growth. (A). Co-immunostaining of Ki67 and TNC in NS and TNC knockdown xenografts. Compared with control, TNC xenografts showed lower TNC expression with relatively more Ki67+ cells, indicating that TNC knockdown increased tumor cell proliferation in vivo. (B). Quantification of Ki67 staining revealed ∼32% higher proliferation index in TNC knockdown xenografts compared with that of control xenografts (***P < .001). (C). Cleaved caspase-3 immunofluorescent staining showed no significant cell death in control and TNC knockdown xenografts. Bar = 50 µm.
Tenascin-C Changes in the Tumor Microenvironments Modulate Tumor-Stromal Interactions
Since the tumor cells are the main source of TNC in the tumor microenvironment, TNC knockdown in GBM neurosphere cells may influence tumor cell proliferation by modulating stromal cells in the tumor microenvironment including endothelial cells and microglia cells. We therefore examined tumor blood vessels by laminin staining. Substantial differences in the morphology and density of tumor blood vessels were found in xenografts derived from control and TNC knockdown cells. In control xenografts, the blood vessel density was high, and the lumen of blood vessels was small. In contrast, in TNC knockdown xenografts, the blood vessel density was low, and the lumen of the blood vessels was enlarged (Fig. 5A), suggesting distinct endothelial cell behavior in response to TNC changes in the tumor microenvironment.
Fig. 5.

Alteration of tenascin-C (TNC) in tumor extracellular spaces influences tumor cell-stromal cell interactions. (A). Laminin staining of intracranial tumors showed that TNC knockdown tumors had fewer blood vessels, but enlarged tumor blood vessel lumen, compared with control. (B). Distribution and morphology of tumor microglia stained with microglial marker Iba1. In control xenografts, the morphology of microglia in tumor center and border was similar, both with long processes, resembling the inactivated, ramified microglia in the brain. In TNC knockdown tumors, microglia in tumor border were similar to those in control tumors with long processes. However, in the center of TNC knockdown tumors, the microglia morphology resembled those activated amoeboid-like microglia with fewer processes. (C). Immunostaining and quantification of active microglia in NS and TNC knockdown xenografts. The tumors were stained with an anti-murine MHCII antibody, which recognizes active microglia in xenografts. MHCII+ cells/field were counted under fluorescent microscopy. Compared with control, TNC knockdown xenografts harbored ∼2 times more MHCII+ cells (N = 5, ***P < .001). (D). The adhesion of rat brain endothelial (RBE) cells was significantly reduced on TNC-coated surfaces compared with that of fibronectin- and laminin- coated surfaces. Phase contrast microphotograph showing RBE cell morphology after being plated on fibronectin (FN) or TNC coated surfaces for 4 hours. (E). RBE cell adhesion was quantified by MTT assays. Compared with fibronectin and laminin, RBE cells adhered poorly on TNC-coated surfaces. (F). Transwells were coated with laminin, fibronectin, or TNC (10 µg/mL). Compared with laminin- and fibronectin- coated surfaces, RBE cells migrated slowly on TNC-coated transwells (***P < .001).
We also examined the influence of TNC knockdown on microglia using Iba1 staining. The morphology of microglia cells was dramatically altered in TNC knockdown xenografts. In control tumors, the morphology of microglia throughout the entire tumors resembled inactivated microglia with long and thin processes (Fig. 5B). In TNC knockdown tumors, the morphology of microglia at tumor periphery was similar to those in control tumors with long and thin processes. Yet, at the center of TNC knockdown tumors, the morphology of microglia resembled activated microglia with fewer extensions (insets in Fig. 5B). No significant difference was found in the number of microglia cells in the control and TNC knockdown tumors (Supplementary material, Fig. S3). When stained with anti-mouse MHC Class II (I-A/I-E), the active microglia marker,24 the TNC knockdown xenografts harbored more MHCII+ microglia than control xenografts (Fig. 5C), consistent with the morphological staining findings with the Iba1 antibody.
Our findings suggested that decreased TNC levels in the tumor microenvironment significantly altered the tumor growth pattern by affecting the behavior of other stromal cells in the tumor microenvironments, especially endothelial cells. To investigate how TNC affected endothelial cell functions, we used well-characterized endothelial cultures derived from rat brains (rat brains endothelial, RBE25,26). RBE cells were normally maintained on fibronectin- or laminin-coated surfaces. Compared with fibronectin or laminin, the adhesion of RBE cells to TNC was significantly reduced (Fig. 5D and E, P < .001). We also quantified the migration of RBE cells on laminin-, fibronectin-, and TNC-coated surfaces in transwell assays. TNC significantly blocked RBE cell migration (Fig. 5F, P < .001). These results suggested TNC had an anti-adhesive role in RBE cells as well, compared with fibronectin. Thus, decreased TNC in the tumor microenvironment will impact the behavior of stroma cells, which in turn alters tumor growth pattern in xenografts.
Tenascin-C Knockdown Cells are Sensitive to Temozolomide
Our in vivo data indicate that knocking down TNC in tumor cells resulted in decreased invasion but increased tumor proliferation; which suggests that there might be a better antitumor effect if we combine TNC inhibition with anti-proliferative strategies to target both brain tumor invasion and proliferation. To test this idea, we treated control and TNC knockdown GBM neurosphere cells with the chemotherapeutic drug temozolomide. Compared with control NS cells, TNCKD GBM neurosphere cells were more sensitive to temozolomide (Fig. 6A). After a 48 hour treatment with temozolomide at 3 µM, only 18% NS GBM neurosphere cells died (P > .05), whereas more than 55% TNCKD GBM neurosphere cells died at this treatment regimen (Fig. 6B, P < .001). This suggests that down-regulation of endogenous TNC level in brain tumor cells can be combined with anti-proliferative drugs to simultaneously target brain tumors.
Fig. 6.

Simultaneous targeting of brain tumor cells by combining tenascin-C (TNC) knockdown with temozolomide. (A). Hundred thousand/well NS and TNCKD GBM neurosphere cells were plated in 6 well/plates and treated with temozolomide at 3 µM for 2 days. Phase-contrast microphotographs indicate TNC KD GBM neurosphere cells were more sensitive to temozolomide by showing small neurosphere size and dead cells. (B). NS and TNCKD GBM neurosphere cells were treated with temozolomide at indicated concentrations for 2 days; live cells were quantified by trypan blue exclusion staining and counting. Compared with NS cells, TNCKD GBM neurosphere cells were ∼10 times more sensitive to temozolomide treatment by showing significant cell death (55%) at 3 µm, whereas NS cells showed significant cell death (47%) at 30 µm temozolomide (***P < .001).
Discussion
The tumor microenvironment is an emerging source of novel therapeutic targets in cancer. Tenascin-C is the main extracellular matrix protein of malignant brain tumors and is overexpressed in invasive glioma cells both in vitro and in vivo, indicating a role for this ECM molecule in glioma pathology. Therefore, determining tumor responses to TNC changes in preclinical animal models will aid in the development of tumor microenvironment-based antitumor therapeutics. In this work, we demonstrated that TNC loss-of-function increased GBM neurosphere cell adhesion without affecting the cell proliferation rate in vitro. In intracranial xenografts, decreased TNC in the tumor microenvironment modulated the behavior of tumor stromal cells, inhibited tumor invasion, and increased tumor cell proliferation. The combination of these phenotypic phenomenon is similar to the go-or-grow glioma growth phenotype found in human neoplasms, which represent characteristic, highly proliferative tumor cores and diffuse borders with a low proliferation rate. The molecular and cellular mechanisms mediating the glioma go-or-grow phenomenon are less known.
In our GBM neurosphere models that are enriched for GSCs, TNC was highly expressed and secreted in medium. The study of Fan et al27 compared the cell surface glycoproteins of GSCs and conventional adherent GBM cells (eg, U87 and U373 cells) maintained in serum-containing medium and concluded that TNC is one of the 5 specific surface markers for GSCs.28 It has been reported that TNC is an important ECM component in the neural stem cell niche7 and the metastatic niche.8 TNC in the neural stem cell niche regulates the neural stem cell pool7 and inhibits cell differentiation.29 In our in vivo models, although TNC is dispensable for the growth of GSCs, it is conceivable that TNC plays a role in the brain tumor invasive niche and promotes tumor cell invasion. These phenotypes largely contribute to radio/chemotherapy resistance and tumor recurrence. Yet, unlike the pro-adhesive function of Id proteins30 and JAM-A,31 which have been both identified in the neural stem cell niche and GSC niche, TNC in the GBM invasive niche functions as an anti-adhesive ECM to promote migration of tumor cells away from the tumor mass. Also different from other ECMs (eg, chondroitin sulfate proteoglycans32), TNC in the tumor microenvironment favors tumor invasion, instead of inhibition.
In the literature, both anti- and pro-activities of TNC on cell adhesion have been reported; various culture conditions and cell types used in these studies may explain the discrepancy regarding TNC function in cell adhesion. Some studies used TNC as a coating material and compared cell adhesion on TNC-coated surfaces with that on noncoated surfaces, and it was concluded that TNC promotes cell adhesion.23 Some studies compared cell adhesion on TNC and fibronectin co-coated surfaces and concluded that TNC prevents cell adhesion on fibronectin-coated surfaces.33 While the majority of published studies used adherent cells such as fibroblasts, in our system, GBM neurosphere cells grow as floating neurosphere cells and require extracellular matrix such as laminin or Matrigel to assist their attachment on culture surfaces. We found that TNC loss-of-function promoted cell adhesion on laminin-coated surfaces, consistent with TNC function in neural stem cells, in which TNC modulates cell response to EGF and FGF (2 growth factors that are necessary for the formation of free-floating neurospheres).7 These findings may explain why intracranial tumors from TNC knockdown GBM neurosphere cells were less invasive, since TNC loss-of-function promoted cell adhesion and were thus less migratory. Our results were in line with a previous study in LN229 human GBM cells,34 in which endogenous tenascin-C contributed to the invasive nature of GBM. In addition, we found that FAK pathway is the pivotal mediator of the anti-adhesive effect of TNC. FAK has been previously shown downstream of TNC in the regulation of cell adhesion, migration, and epithelial-mesenchymal transition.23 Like other ECMs, TNC can regulate FAK pathway via integrins. The study of Lathia et al35 indicated that integrin α6 is enriched in GSCs. Further studies of the paracrine or autocrine loop of integrin-TNC signaling transduction will shed light on the influence of ECM on GSC biology.
In intracranial tumors, TNC knockdown not only affected tumor cell migration/invasion, but also cell proliferation. This finding differed from what we found in vitro and also differed from the study of TNC in LN229, in which TNC reduction did not affect tumor size.34 The increased GBM neurosphere cell proliferation could result from changes in surrounding stromal cells including endothelial cells. Our in vitro studies indicate that brain-derived endothelial cells adhere poorly to TNC-coated surfaces, consistent with the anti-adhesive function of TNC. It is tempting to propose that in TNC knockdown xenografts, less TNC in the tumor microenvironment may facilitate endothelial migration into tumors. In addition, reduced TNC expression in brain tumor cells may also increase tumor cell adhesion on blood vessels since one of the main components of the vascular basement membrane of blood vessels in the brain is laminin, which was shown to be expressed by nonstem tumor cells and tumor-associated endothelial cells.36 A decrease in TNC levels in the tumor microenvironment could facilitate tumor cells to better adhere to blood vessels and obtain nutritional factors for fast growth. All these may explain why TNC knockdown cells grew faster than control cells in vivo. However, since ECMs including TNC have been implicated in angiogenesis,37 there is a possibility that decreased TNC in the tumor microenvironment results in an angiogenic defect and changes vasculature structures. In the future, a detailed mechanistic study on molecular levels will shed light on how TNC decrease in the tumor microenvironment influences the interactions between brain tumor cells and their microenvironment.
TNC has been shown to modulate cellular functions through induction of pro-inflammatory cytokines.4 It has been well documented that TNC is upregulated during inflammation and plays an important role in wound healing.4,6 It is important to note that we used immunodeficient mice for our xenograft experiments, in which the adaptive immunoresponse is deficient, yet the innate immunoresponse, including microglia-mediated inflammation, is still intact.38 Therefore, it is rational to examine how TNC changes in the tumor microenvironment affect immune cell behavior in the brain since tumor growth in the brain could be considered as a non-healing wound39 and could trigger native immune responses such as inflammation.40 Indeed, we found that TNC knockdown xenografts were associated with more activated microglia. The study of Zhang et al41 showed that microglia cells in brain tumors elicit immunorepression functions and contribute to tumor progression. The biological impact of activated microglia cells on tumor growth in the TNC knockdown models require more detailed studies in the future.
Finally, our findings have several clinical implications. First, TNC-based anticancer strategies such as peptide and siRNAs have been proposed to treat brain tumors in animal models.42,43 Yet, evaluating the overall effect that targeting TNC will have on tumor growth and invasion requires a full understanding of how TNC downregulation in the tumor microenvironment influences the interactions between tumor cells and their surrounding nontumor cells, which can ultimately influence tumor responses to anticancer strategies. In fact, we examined the correlation between TNC expression levels and patient survival in patients diagnosed with gliomas. We found that TNC expression levels or gene copy numbers do not significant affect patient survival (Supplementary material, Fig. S4). These datasets independently agreed with our original conclusions that targeting TNC alone may not be sufficient to benefit patient outcomes.
Second, our in vitro data suggest that TNC knockdown cells were more sensitive to anti-proliferative strategies, which could ultimately lead to novel combinatory antitumor strategies that can target both tumor invasion and proliferation. In fact, the study of Siebzehnrubl et al44 has shown that targeting GBM invasion increases tumor sensitivity to TMZ. Thus, targeting TNC may be potentially helpful for designing more effective anti-GBM therapies when used in conjunction with anti-proliferative modalities such as chemotherapy, radiation therapy, and anti-angiogenesis strategies.
Supplementary Material
Supplementary material is available at Neuro-Oncology Journal online (http://neuro-oncology.oxfordjournals.org/).
Funding
This work is supported by NIH NS43987 (J.L.).
Conflict of interest statement. None declared.
Supplementary Material
References
- 1.Charles NA, Holland EC, Gilbertson R, et al. The brain tumor microenvironment. Glia. 2011;59(8):1169–1180. [DOI] [PubMed] [Google Scholar]
- 2.Lorger M. Tumor microenvironment in the brain. Cancers (Basel). 2012;4(1):218–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quirico-Santos T, Fonseca CO, Lagrota-Candido J. Brain sweet brain: importance of sugars for the cerebral microenvironment and tumor development. Arq Neuropsiquiatr. 2010;68(5):799–803. [DOI] [PubMed] [Google Scholar]
- 4.Jones FS, Jones PL. The tenascin family of ECM glycoproteins: structure, function, and regulation during embryonic development and tissue remodeling. Dev Dyn. 2000;218(2):235–259. [DOI] [PubMed] [Google Scholar]
- 5.Midwood KS, Hussenet T, Langlois B, et al. Advances in tenascin-C biology. Cell Mol Life Sci. 2011;68(19):3175–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Midwood K, Sacre S, Piccinini AM, et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med. 2009;15(7):774–780. [DOI] [PubMed] [Google Scholar]
- 7.Garcion E, Halilagic A, Faissner A, et al. Generation of an environmental niche for neural stem cell development by the extracellular matrix molecule tenascin C. Development. 2004;131(14):3423–3432. [DOI] [PubMed] [Google Scholar]
- 8.Oskarsson T, Acharyya S, Zhang XH, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med. 2011;17(7):867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Behrem S, Zarkovic K, Eskinja N, et al. Distribution pattern of tenascin-C in glioblastoma: correlation with angiogenesis and tumor cell proliferation. Pathol Oncol Res. 2005;11(4):229–235. [DOI] [PubMed] [Google Scholar]
- 10.Leins A, Riva P, Lindstedt R, et al. Expression of tenascin-C in various human brain tumors and its relevance for survival in patients with astrocytoma. Cancer. 2003;98(11):2430–2439. [DOI] [PubMed] [Google Scholar]
- 11.Brosicke N, van Landeghem FK, Scheffler B, et al. Tenascin-C is expressed by human glioma in vivo and shows a strong association with tumor blood vessels. Cell Tissue Res. 2013;354(2):409–430. [DOI] [PubMed] [Google Scholar]
- 12.Spenle C, Saupe F, Midwood K, et al. Tenascin-C: exploitation and collateral damage in cancer management. Cell Adh Migr. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432(7015):396–401. [DOI] [PubMed] [Google Scholar]
- 14.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64(19):7011–7021. [DOI] [PubMed] [Google Scholar]
- 15.Ying M, Sang Y, Li Y, et al. Kruppel-like family of transcription factor 9, a differentiation-associated transcription factor, suppresses Notch1 signaling and inhibits glioblastoma-initiating stem cells. Stem Cells. 2011;29(1):20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ying M, Wang S, Sang Y, et al. Regulation of glioblastoma stem cells by retinoic acid: role for Notch pathway inhibition. Oncogene. 2011;30(31):3454–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun P, Xia S, Lal B, et al. DNER, an epigenetically modulated gene, regulates glioblastoma-derived neurosphere cell differentiation and tumor propagation. Stem Cells. 2009;27(7):1473–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roux F, Couraud PO. Rat brain endothelial cell lines for the study of blood-brain barrier permeability and transport functions. Cell Mol Neurobiol. 2005;25(1):41–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang SD, Rath P, Lal B, et al. EphB2 receptor controls proliferation/migration dichotomy of glioblastoma by interacting with focal adhesion kinase. Oncogene. 2012;31(50):5132–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lal B, Goodwin CR, Sang Y, et al. EGFRvIII and c-Met pathway inhibitors synergize against PTEN-null/EGFRvIII+ glioblastoma xenografts. Mol Cancer Ther. 2009;8(7):1751–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiquet-Ehrismann R, Matsuoka Y, Hofer U, et al. Tenascin variants: differential binding to fibronectin and distinct distribution in cell cultures and tissues. Cell Regul. 1991;2(11):927–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pollard SM, Yoshikawa K, Clarke ID, et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4(6):568–580. [DOI] [PubMed] [Google Scholar]
- 23.Nagaharu K, Zhang X, Yoshida T, et al. Tenascin C induces epithelial-mesenchymal transition-like change accompanied by SRC activation and focal adhesion kinase phosphorylation in human breast cancer cells. Am J Pathol. 2011;178(2):754–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Unternaehrer JJ, Chow A, Pypaert M, et al. The tetraspanin CD9 mediates lateral association of MHC class II molecules on the dendritic cell surface. Proc Natl Acad Sci USA. 2007;104(1):234–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnston P, Nam M, Hossain MA, et al. Delivery of human fibroblast growth factor-1 gene to brain by modified rat brain endothelial cells. J Neurochem. 1996;67(4):1643–1652. [DOI] [PubMed] [Google Scholar]
- 26.Nam M, Johnston P, Lal B, et al. Endothelial cell-based cytokine gene delivery inhibits 9L glioma growth in vivo. Brain Res. 1996;731(1–2):161–170. [DOI] [PubMed] [Google Scholar]
- 27.He J, Liu Y, Xie X, et al. Identification of cell surface glycoprotein markers for glioblastoma-derived stem-like cells using a lectin microarray and LC-MS/MS approach. J Proteome Res. 2010;9(5):2565–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nie S, Gurrea M, Zhu J, et al. Tenascin-C: a novel candidate marker for cancer stem cells in glioblastoma identified by tissue microarrays. J Proteome Res. 2015;14(2):814–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Czopka T, von Holst A, ffrench-Constant C, et al. Regulatory mechanisms that mediate tenascin C-dependent inhibition of oligodendrocyte precursor differentiation. J Neurosci. 2010;30(37):12310–12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niola F, Zhao X, Singh D, et al. Id proteins synchronize stemness and anchorage to the niche of neural stem cells. Nat Cell Biol. 2012;14(5):477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lathia JD, Li M, Sinyuk M, et al. High-throughput flow cytometry screening reveals a role for junctional adhesion molecule a as a cancer stem cell maintenance factor. Cell Rep. 2014;6(1):117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silver DJ, Siebzehnrubl FA, Schildts MJ, et al. Chondroitin sulfate proteoglycans potently inhibit invasion and serve as a central organizer of the brain tumor microenvironment. J Neurosci. 2013;33(39):15603–15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Obberghen-Schilling E, Tucker RP, Saupe F, et al. Fibronectin and tenascin-C: accomplices in vascular morphogenesis during development and tumor growth. Int J Dev Biol. 2011;55(4–5):511–525. [DOI] [PubMed] [Google Scholar]
- 34.Hirata E, Arakawa Y, Shirahata M, et al. Endogenous tenascin-C enhances glioblastoma invasion with reactive change of surrounding brain tissue. Cancer Sci. 2009;100(8):1451–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lathia JD, Gallagher J, Heddleston JM, et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell. 2010;6(5):421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lathia JD, Li M, Hall PE, et al. Laminin alpha 2 enables glioblastoma stem cell growth. Ann Neurol. 2012;72(5):766–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tanaka K, Hiraiwa N, Hashimoto H, et al. Tenascin-C regulates angiogenesis in tumor through the regulation of vascular endothelial growth factor expression. Int J Cancer. 2004;108(1):31–40. [DOI] [PubMed] [Google Scholar]
- 38.Arnold L, Tyagi RK, Mejia P, et al. Analysis of innate defences against Plasmodium falciparum in immunodeficient mice. Malar J. 2010;9:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng Y, Santoriello C, Mione M, et al. Live imaging of innate immune cell sensing of transformed cells in zebrafish larvae: parallels between tumor initiation and wound inflammation. PLoS Biol. 2010;8(12):e1000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Najbauer J, Huszthy PC, Barish ME, et al. Cellular host responses to gliomas. PLoS One. 2012;7(4):e35150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang J, Sarkar S, Cua R, et al. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis. 2012;33(2):312–319. [DOI] [PubMed] [Google Scholar]
- 42.Rolle K, Nowak S, Wyszko E, et al. Promising human brain tumors therapy with interference RNA intervention (iRNAi). Cancer Biol Ther. 2010;9(5):396–406. [DOI] [PubMed] [Google Scholar]
- 43.Zukiel R, Nowak S, Wyszko E, et al. Suppression of human brain tumor with interference RNA specific for tenascin-C. Cancer Biol Ther. 2006;5(8):1002–1007. [DOI] [PubMed] [Google Scholar]
- 44.Siebzehnrubl FA, Silver DJ, Tugertimur B, et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol Med. 2013;5(8):1196–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.