

Abstract
Zebrafish represents the third vertebrate with an officially completed genome, yet it remains incomplete with additions and corrections continuing with the current release, GRCz10, having 13% of zebrafish cDNA sequences unmapped. This disparity may result from population differences, given that the genome reference was generated from clonal individuals with limited genetic diversity. This is supported by the recent analysis of a single wild zebrafish, which identified over 5.2 million SNPs and 1.6 million in/dels in the previous genome build, zv9. Re-examination of this sequence data set indicated that 13.8% of quality sequence reads failed to align to GRCz10. Using a novel bioinformatics de novo assembly pipeline on these unmappable reads, we identified 1,514,491 novel contigs covering ∼224 Mb of genomic sequence. Among these, 1083 contigs were found to contain a potential gene coding sequence. RNA-seq data comparison confirmed that 362 contigs contained a transcribed DNA sequence, suggesting that a large amount of functional genomic sequence remains unannotated in the zebrafish reference genome. By utilizing the bioinformatics pipeline developed in this study, the zebrafish genome will be bolstered as a model for human disease research. Adaptation of the pipeline described here also offers a cost-efficient and effective method to identify and map novel genetic content across any genome and will ultimately aid in the completion of additional genomes for a broad range of species.
Introduction
Zebrafish (Danio rerio) are an important model organism in biology, having provided significant advances in genetics, developmental biology, oncology, evolutionary biology, regenerative medicine, and others. These advances would not have been possible without the completion of the zebrafish reference genome.1 In the most recent release, GRCz10, more than 4000 known genome issues were resolved from the previous zv9 build and an additional 15 Mb of genome sequence was placed on chromosome 4. This included 94 previously unplaced contigs, which have now been integrated into the reference genome. Despite this improvement, large amounts of genome sequence remain unmapped, given that only 87% of known cDNA sequences can currently be placed in GRCz10.
To ensure zebrafish is a suitable model system to research human disease, it is important to examine gene syntenies between genomes. Current research suggests humans share 70% of protein-coding genes with zebrafish,1 and reciprocally, zebrafish share 69% with humans. The overlap primarily includes genes considered essential to all vertebrates. This assessment is based on genes represented within the two genome builds along with published EST data sets with the caveat that the zebrafish data set has been primarily generated from a small sampling of individuals.1,2 Selecting few, often clonal, individuals for genome development is helpful for reducing diploidy for assembly,3 but as a consequence, the resulting genome may not be representative of all genetic backgrounds. In the case of zebrafish, founder effects make it likely that the reference genome, built using common laboratory strains (Tubingen [TU] and AB), lacks genetic diversity compared to wild populations.1,4,5
In a recent study cataloging genetic differences between a single wild zebrafish and the reference genome, 5.2 million SNPs and over 1.6 million in/dels were identified.6 While the study generated a valuable sequence data set and highlighted the vast genetic differences between wild and laboratory strain zebrafish, the results only accounted for differences in published genomic regions. Resequencing projects (i.e., short read sequences mapped against the reference genome) are useful, but ignore high-quality sequences failing to map (i.e., unmappable reads). In the present study, we re-examine this raw sequence data set to catalog and map genomic regions currently absent from the zebrafish genome due to intraspecific variation. To do so, we isolated and de novo assembled unmapped reads failing to align to the current zebrafish reference genome, GRCz10. Following de novo assembly, aligned mate pair reads to unmapped contig 5′ and 3′ termini were used as anchors to determine positions within the reference genome. Novel contigs were also scanned for alignments to protein sequences from human (Homo sapiens) and Mexican tetra (Astyanax mexicanus) reference data sets to identify novel gene orthologs previously undescribed in zebrafish. Using this pipeline, our results significantly improve the zebrafish reference genome as a resource for diverse biological and biomedical research and pave the way for efficient and effective reanalysis of published data to make other genome resources more complete.
Materials and Methods
Raw zebrafish genome Illumina sequences were downloaded from the analysis of a single wild individual from Northeastern India, referred to as ASWT (ERP001723).6 The data set included paired and unpaired sequences. Before assembly, raw sequence files were checked for adaptors using Prinseq software's “tag sequence check” tool, confirming that input sequences were adaptor free. For our analysis, paired sequences were split and assembled as single reads (Fig. 1), allowing for more efficient isolation of unmappable reads compared to traditional paired-end mapping. Sequences were mapped to GRCz10 (GB: GCA_000002035.3) using default alignment parameters in Bowtie2 v2.1.0, with unmapped reads sent to a separate output file.7 These reads were filtered to remove contaminating bacterial sequences by aligning to various prokaryotic genomes with Bowtie2 and then de novo assembled using Mira v4.8 Mira was selected because of its tendency to be conservative and retain a higher portion of raw reads when compared to other assembly algorithms.9 Mira also uses several sequence quality filters before assembly, such as the SKIM filter, which ensures alignment of reads within “high confidence regions” only as determined by Phred quality scores. Therefore, poor quality reads and ends of reads tend to be ignored during assembly. Assembly was also attempted with Velvet,10 but Mira produced assemblies with higher total base-pair coverage and longer assembled contigs. Assembly parameters used were genome, de novo, accurate, and solexa as the sequencing technology.
FIG. 1.

Overview of bioinformatics pipeline utilized to generate, test, and annotate novel genomic contigs.
Prinseq v0.20.4 was used to calculate assembly statistics, including N50 contig size, GC content, and overall sequence complexity using embedded Entropy and DUST algorithms.11 Using the filter feature in Prinseq, primarily repetitive sequences with low complexity (<75 Entropy scores) were removed. In comparison with other assembly programs, Mira produces more redundant assemblies than some other algorithms.12 To account for this, our assembly was assessed for sequence redundancy using both Prinseq v0.20.411 and Simplifier v0.4.13 Both programs check for exact alignments, internal alignments, and overlapping contigs with 5′ or 3′ overhangs. For Simplifier, we ignored 5 bp on contig ends before alignment to account for possible low-quality base calls. Redundant contigs identified by either program were removed before downstream analyses.
To ensure unmapped contigs were not primarily the product of sample contamination before sequencing, we selected 1000 random contigs using Galaxy's “fasta to tabular,” “select random lines,” and “tabular to fasta” tools14–16 to identify top BLASTn hits from GenBank's nonredundant (nr) database. Bacterial, parasitic, and other nonvertebrate hits were considered to be derived from potential sample contamination, although some of these sequences may have been integrated into the zebrafish genome through horizontal gene transfer.
To map assembled contigs, raw reads were aligned to the de novo assembly using default parameters in Bowtie2. Using paired-end relationships of reads where one mate maps to the genome and the other maps to the assembly, we cross-referenced map coordinates from Bowtie2 SAM outputs to generate approximate in/del loci. This list was filtered to include only hits with strong Bowtie2 alignment scores (≥20) to both the genome and assembly. Alignment scores range from 0 to 42, with lower scores indicative of reads mapping to more possible locations and higher scores mapping to fewer locations.
The assembly was scanned for orthologs to annotated human genes to identify novel candidate zebrafish genes potentially useful for human disease research. First, we download the human “protein-coding transcript translation sequences” fasta file for the current genome build from GENCODE (gencode.v22.pc_translations.fa). All assembled contigs were queried against human protein sequences using local BLASTx analysis. The results were sorted and filtered based on shared sequence identity (≥70%) and BLAST alignment score (≥50) to include only strong alignments. Contigs with strong hits to human protein sequences were then queried against the current zebrafish protein sequence data set from Ensembl (protein.fa), and again results were filtered based on minimum sequence identity and alignment score. BLASTx results were compared between species, and the candidate gene list was revised to include genes in contigs with stronger hits to the human proteome than to zebrafish, based on sequence identity. These contigs were inspected individually using BLASTn and BLASTx search results from the broader GenBank nr database. The most frequent BLASTx hit in nr came from Mexican tetra, therefore, we downloaded its protein database from UniProt (uniprot-proteome%3AUP000018467.fasta) and repeated the annotation analysis to find additional functional content specific to teleosts. Gene names and symbols were used to search for annotations in the GRCz10 and hg38 (human) genome builds in the UCSC genome browser and NCBI gene database for additional confirmation. Genes found in the human or tetra genome build, but not the zebrafish build, are considered top candidates for novel zebrafish gene discoveries.
We downloaded zebrafish RNA sequence data (ERR202493) from the maternal to zygotic transition period.17 This data set was generated using Sanger AB-TU whole embryos. We aligned these sequences to GRCz10, our full assembly, the portion of our assembly containing candidate gene content, and 1000 randomly selected contigs as a control using default parameters in Bowtie2. Although this RNA data set does not represent a comprehensive zebrafish transcriptome, it allowed us to estimate the portions of the data set missing from the genome, but represented in our assembly. In addition, we checked for strong RNA sequence alignments to further support predicted gene discoveries from the previously described comparative species analysis.
To confirm that generated contigs were correctly assembled, we designed oligos to polymerase chain reaction (PCR) amplify sequences from total genomic DNA for laboratory strain zebrafish (Table 1). The laboratory strains used were TU, AB, WIK, and Nadia (NA). The current zebrafish genome was developed using domesticated zebrafish sequences and meiotic maps from TU and Sanger AB-TU lines,1 where WIK and NA fish are descendants of more recently caught wild fish from India.18,19 Therefore, WIK and NA fish were expected to have more unmapped genome sequences in common with the sequenced ASWT fish than the other laboratory strains. We chose three individuals from each strain, and TU, AB, and WIK DNA samples were extracted using the Dneasy Blood and Tissue Kit (Qiagen). NA DNA samples were sent to us from Dr. John Postlethwait's laboratory at the University of Oregon.
Table 1.
Oligo Sequence Pairs Utilized for Validating Contig Mapping Coordinates Amplifying in Samples
| Contig | Forward primer | Reverse primer |
|---|---|---|
| ERR163_all_c22 | GTCCGAAAACGCATACTTCC | AAGTTAGGGGCGTGACAGTG |
| ERR163_all_c455 | TGAGCAAGTGCTTGGAAATG | TTGCACACGTTACAATTACGAA |
| ERR163_all_c732 | CAAACACCACAAAAATGTTTTATCA | ACGCTTTCCAACTGATGGTC |
| ERR163_all_c757 | TCAGGGTCCAGTGTCCAAAT | TCTCAGATGTGATTTTTGATGG |
| ERR163_all_c778 | CCCCAGAAAAACAGAATGGA | CTTCCATTGGGCATCTGTC |
| ERR163_all_c782 | AAACCTATTTTCAACCAAGTATGTTGT | GGGCTCCACACAATTATTTCA |
| ERR163_all_c117602 | ATAACCCGTCCAGTGCTCAG | GGTTTGCATCTTTCGGTCTC |
| ERR163_all_c19294 | GAAGGAACTGGCCAATCAGA | TCTGTTCAGCTGTGCAGGAG |
| ERR163_all_c36029 | TTGTTGATACCCGTTTGACG | TTCCTGATAACGGAGCGTTT |
| ERR163_all_c53301 | GTCACCTTGCTGTGAGCGTA | TCTCTGGCTCTTGCAGACTTC |
| ERR163_all_c541415 | AGATCGACCCGAACACAAAG | AGGCTGATGATGCGGTAAAC |
| ERR163_all_c55651 | GGCGAATGATCTGACCTGTT | CTCACAGCAGCACAATGGAG |
| ERR163_all_c109908 | GAGGTTTGCAGCAGGAGTTC | AAACCAGCTAATGGGCCTCT |
| ERR163_all_c402004 | GGCACAAACGTAAAGCTGGT | CTTGGATTTGCTGTGTCGAA |
| ERR163_all_c67492 | TCGGTGCAGAAACTGAAGTG | GCCTGGTGATTTCTCTGAGG |
Ten contigs were selected for primer design based on BLASTn results that showed 5′ and 3′ end homology to known zebrafish sequences. Since the bulk of the sequence for these contigs showed no homology (center of contig), oligos were designed to flanking genomic sequences. An additional 10 contigs were selected with predicted orthologous gene content found in humans. Oligos for these contigs were designed surrounding or inside candidate exons. Sequences were amplified with LongAmp Taq Master Mix (NEB) according to the manufacturer's protocol with a 59°C annealing temperature and 5-min elongation step. Products were visualized and scored on a 1.2% agarose gel. Samples were exo/sap treated, labeled with the BigDye Terminator Kit (ABI), and sent to Oregon Health Sciences University DNA Services Core for Sanger sequencing to confirm correct amplification of assembled contigs. When possible, sequenced samples were preferentially amplifed from TU or AB DNA, given their use in the creation of the zebrafish reference genome allowed for more accurate comparison with the reference genome. For contigs not found in either TU or AB, the sequenced product was amplified from random WIK or NA individuals.
Results
Eighty-six percent (86.2%) of downloaded single-end and split paired-end raw read sequence reads (891,678,553) aligned to the zebrafish genome build GRCz10, while 143,012,187 (13.8%) remained unmapped. De novo assembly of unmapped reads resulted in 1,514,491 contigs covering ∼224 Mb (Supplementary Data 1; Supplementary Data are available online at www.liebertpub.com/zeb and http://archives.pdx.edu/ds/psu/16241). The assembly's N50 is 152 bp with a maximum contig length of 19,567 bp. For contigs >500 bp, an average of 493 raw sequences per contig were assembled. GC content across the de novo assembly is 34.65% ± 8.9% (Fig. 2), consistent with the reported 36.5% GC content of the zebrafish reference genome.20 Eighty-seven point nine percent (87.9%) of the assembly exhibited high complexity based on DUST (≤4) and 87.1% based on Entropy (≥75) (Fig. 3).
FIG. 2.

Distribution of GC content among ∼1.5M assembled sequences generated in this study. Mean GC content for zebrafish genome assembly is 34.65% ± 8.94%.
FIG. 3.

Sequence complexity distribution of the secondary assembly contigs as calculated by (A) DUST and (B) Entropy algorithms embedded within Prinseq v0.20.4. For DUST, lower values indicate higher complexity with ≤4 as the complexity cutoff used in analysis. For Entopy, higher values are considered more complex with ≤75 as the complexity cutoff.
For further quality control, the assembly was evaluated for possible contamination using BLASTn results from 1000 randomly selected contigs queried against the GenBank nr nucleotide database. Zebrafish sequence was the top hit for 64.9% of the contigs with an average of 91.7% identity. One third (33.4%) of the sequences showed no BLAST hits, 1.3% matched carp (Cyprinus carpio) with a mean sequence identity of 90.3%, and 0.4% had top BLAST hits to cloning vectors (mean 96.8% identity) (Fig. 4). Combined, these results suggest that our pipeline does not preferentially isolate contaminating raw sequences given only four contigs yielded nonteleost BLAST alignments as the top hit.
FIG. 4.
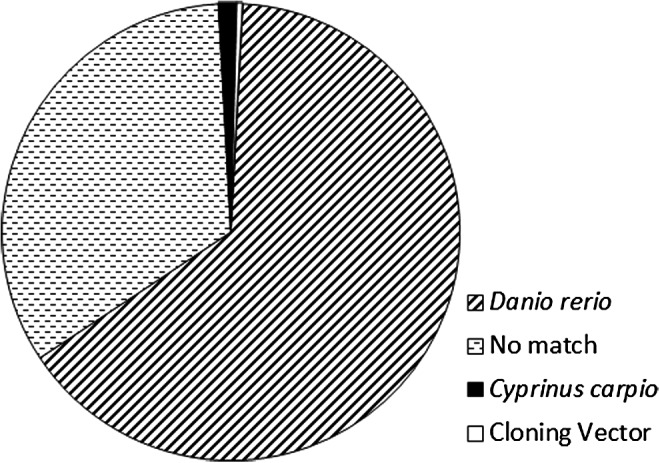
Distribution of top nucleotide BLAST hits by species from the NCBI nr database for 1000 random contigs in the assembly indicating that the majority of hits were highly related to zebrafish or other teleosts. Unknown contigs were not found in any species within the database.
Prinseq identified and removed 23 duplicates, and Simplifier identified and removed 305 additional redundant contigs from the nonrepetitive assembly. Both algorithms look for exact sequence matches and overlaps with either 5′ or 3′ overhangs. These results suggest that the pipeline generated a primarily nonredundant assembly. After filtering, our assembly represents 194.3 Mb of novel nonrepetitive zebrafish genome sequence among 1,318,296 contigs deposited at EBI Biostudies under accession S-BSMS1 and in Supplementary Data 1. Roughly 30% (447,033 contigs) yielded approximate genome loci, with at least one paired-end sequence mapping to the assembly and its mate mapping to the genome (Supplementary Table S1). Among these, 134,177 were considered high-quality mapping contigs with alignment scores for read pairs in both databases >20 (Supplementary Table S2).
We probed the unmapped zebrafish sequence assembly for novel human and Mexican tetra gene orthologs. After filtering for BLASTx hits with ≥70% sequence identity and ≥50 alignment score, the search yielded 1739 contigs with strong hits to human protein sequences and 1768 strong hits to tetra protein sequences, with 1153 contigs represented in both data sets. Comparison with the zebrafish protein sequence data set indicated that 743 contigs had no strong alignments in the zebrafish proteome. An additional 340 contigs yielded hits with higher sequence identity to non-Danio protein sequences revealing 1083 contigs with possible novel gene content (Supplementary Table S3).
Contigs were queried against the NCBI nr protein database using BLASTx with 281 contigs having ≥95% translated sequence identity to protein sequences in primarily non-Danio taxa (Supplementary Table S4). Species distributions of BLASTx hits revealed three teleosts with a higher number of protein sequence alignments than zebrafish, with the Mexican tetra at the top of the list with ∼500 (Fig. 5). Forty-eight (48) contigs contained annotated zebrafish mRNA sequences for 33 unique gene IDs, although associated genome scaffolds were not found in the current build. Two hundred three contigs (203), representing 138 unique gene IDs, contain unknown exons or novel gene duplicates of annotated zebrafish genes based on observations of BLASTx hit gene IDs that were found in the GRCz10 genome build with only weak sequence homology (≤95%) or with no matches. A number of cancer-related genes were observed in this list, including several in the ras superfamily, proto-oncogene vav-like (vav1-like), and regulator of g-protein signaling 7 (rgs7). These variants warrant follow-up research to look for potential functional differences.
FIG. 5.

Distribution of BLASTx hits by species in the NCBI nr protein database for contigs identified with stronger hits to human proteins than to zebrafish. List only uses top 10 species, and only the top 5 protein hits are reported per contig. Although not in this list, human proteins were the 22nd most frequent BLASTx hit with 38.
Twenty-two of the 281 contigs contain unknown zebrafish candidate gene content representing 10 unique gene IDs (Supplementary Table S5). Two of these genes (fbf1, lrrc16a-like) are undocumented in current zebrafish databases in both NCBI and UCSC, despite being annotated in humans, tetra, and other species. The other eight genes (dnajc13-like, cul9-like, uhrf2-like, gcp6-like, atg2b-like, myl4-like, atp11b, nek9-like) have no annotation in the UCSC genome browser, but had previous annotation information in NCBI that has been withdrawn due to lack of supporting evidence in the current genome build. While these 10 genes are the strongest candidates for 1:1 novel gene orthologs, most identified gene orthologs shared between human and zebrafish show a 1:2 ratio due to the additional genome duplication experienced within all teleosts.21 Combined with candidate unknown gene duplicates and unknown exons, we found up to 148 novel or variant genes previously unknown in zebrafish. Furthermore, the list of 1083 contigs that shares stronger sequence similarity with human or tetra genes likely contains many novel zebrafish pseudogenes. While this analysis specifically probed the assembly for genes found in humans and Mexican tetra, both well annotated species, the unmapped assembly likely contains novel zebrafish genes with no known human or tetra orthologs.
To further support the potential discovery of novel zebrafish genes identified through comparative species analysis, we aligned previously published zebrafish RNA-seq data (∼38.9 M read pairs) to the annotated portion of our assembly.17 About 3% (2.7%) of the RNA sequences aligned, at least partially, with the 1083 candidate gene contigs. Three hundred sixty-two (362) contigs had RNA sequence align with perfect Bowtie2 alignment scores of 42 (Table 2), including contigs representing six candidate zebrafish genes (fbf1, lrrc16a-like, dnajc13-like, cul9-like, uhrf2-like, atg2b-like). This contrasts with our control data set of 1000 randomly selected contigs, where only 86 had RNA sequences align with perfect scores, although these hits may indicate the presence of more functional transcribed content in the assembly not found through comparative analysis.
Table 2.
RNA-Seq Bowtie Alignment to Potential Novel Gene Sequences and Randomly Selected Sequence Contigs from Our Data set
| Contigs with alignment scores | ||||
|---|---|---|---|---|
| Contigs | Any score | ≥20 | 42a | |
| Potential novel gene contigs | 1083 | 935 | 434 | 362 |
| Randomly selected contigs | 1000 | 323 | 148 | 86 |
Contigs where RNA-seq samples and contigs mapped to single loci.
This RNA data set was also aligned to the reference genome (GRCz10) and to our entire assembly to estimate representation of this particular transcriptome in each genome resource. Eighty-one point nine percent (81.9%) of the RNA sequence mapped to GRCz10, and of the remaining unmapped reads, 33.4% aligned to our assembly (6% of the entire RNA data set). This further suggests that our assembly contains a large number of unknown zebrafish genes, and additional research to annotate the entire data set is ongoing.
Twenty contigs were chosen for PCR confirmation in three individuals from each of the four zebrafish laboratory strains; TU, AB, WIK, and NA with 15 successfully amplifying products in at least one strain. Six of 15 amplified primer sets were designed to the genome sequence template aligning to the 5′ and 3′ ends of the contigs, which appeared to represent boundaries of large in/dels. All six showed variable amplification between strains, with at least one strain not showing amplification of the target sequence in any individual (Fig. 6). One of six contigs amplified in at least one TU individual, three of six in AB, all six in WIK, and five of six in NA (Table 3). All target sequences appear as deletions in TU and/or AB strains, while they appear as insertions in WIK and/or NA strains, with the former two domesticated laboratory strains1 and the latter two recently derived from wild populations.18,19
FIG. 6.

Polymerase chain reaction (PCR) example of variable amplification in zebrafish strains. Oligos were designed to amplify contig ERR163_all_c757, and the correct product was confirmed through Sanger sequencing.
Table 3.
PCR Confirmation of Strain-Variable Structural Variants
| Contig | TU (3) | AB (3) | WIK (3) | NA (3) |
|---|---|---|---|---|
| ERR163_all_c22 | 0 | 0 | 2 | 2 |
| ERR163_all_c455 | 0 | 0 | 2 | 0 |
| ERR163_all_c732 | 0 | 2 | 3 | 3 |
| ERR163_all_c757 | 0 | 2 | 3 | 3 |
| ERR163_all_c778 | 2 | 0 | 3 | 1 |
| ERR163_all_c782 | 0 | 2 | 3 | 3 |
Numbers of columns represent the number (out of three) that successfully amplified in each contig.
PCR, polymerase chain reaction.
The remaining nine amplifying primer sets were designed to contigs with apparent protein homology lacking 5′ and 3′ alignments. These contigs are expected to be part of larger not-fully-assembled structural variants, and all designed primer sets amplified products in all strains and individuals tested. PCR products were Sanger sequenced, and all products were confirmed to amplify the correct contigs (Supplementary Data 2). Based on these sequencing results, three primer sets appeared to amplify multiple PCR products (c732, c782, c19294), however, trace files were visually inspected and base calls were made manually to confirm that the correct PCR product was prominent.
Discussion
Modern genetic research relies heavily on reference genomes as a primary resource for primer design, analysis of unknown sequence fragments, and as a launching point for large scale projects such as genome-wide association studies (GWAS) and copy number variant (CNV) analysis.22 The availability of these resources not only allows for time and cost-efficient analysis of sequence variation within species but also comparisons between species' genomes.12,23,24 To facilitate the assembly of novel reference genomes, a small number of individuals, often generated as clonal lines,1,25 are used for primary sequencing. While this reduces heterozygosity, making genome assembly easier, it also produces genomes that underestimate sequence variability among broader populations. Reduced variability is primarily due to genetic drift and founder effects, which lead to losses, or less frequently gains (i.e., loss in source population or duplication in sequenced line), of genetic diversity when compared to source populations.6,26–28 Loss of diversity is well documented for SNPs, microsatellites, small in/dels, and other genetic markers, but for large in/dels next-generation sequence reads can fall entirely within genome gaps. Thus, some sequences fail to align and are commonly ignored in downstream analyses. In this study, we demonstrate a novel method providing a fast and efficient way to discover unmapped sequences from any publicly available genome resequencing project and identify unmapped genomic content for the zebrafish reference genome containing extensive sequences with novel, functional genetic content.
Through comparative species proteome analysis, we have discovered at least 2 and possibly 10 candidate genes currently unannotated in zebrafish, and up to 138 novel duplicates or variants of previously known genes. Using a pooled embryo RNA-seq library for further confirmation, we found 362 of 1083 contigs with candidate gene content had exact RNA alignments. This finding supports the hypothesis that our assembly contains novel zebrafish genes and further shows that our pipeline uncovered real structural variants from previously analyzed data. These discoveries bolster the role of zebrafish as a model organism for human disease research, given the increased number of functional orthologous sequences identified, which are shared with humans. Considering the size of our data set, analysis of the content of the unmapped portion of the genome is still incomplete. Ongoing work will undoubtedly confirm additional functional elements (i.e., genes, ncRNAs) from other developmental time points.
This assembly was created using a large, publicly available next-generation sequence data set, which has been analyzed extensively. The previous authors used paired-end mapping coordinates to detect 354 large structural variants (≥100 bp) with an average size of 128 bp.6 By comparison, our methods targeted unmappable sequences, from the same data set, and identified 1.5 million unmapped contigs covering ∼224 Mb. As a caveat, the annotated portion of our data set sometimes exhibited duplicate gene IDs for separate contigs. This is either because these genome regions are highly copy number variable with each duplicate annotation representing a unique gene copy or the actual number of structural variants is less than the observed number due to incomplete assembly of adjacent sequences across single genes. Regardless of whether the number of independent structural variants may be reduced, the estimated ∼224 Mb of unmapped sequence would not proportionally decrease since contigs are nonredundant.
The pipeline presented here represents a way forward for the reanalysis and refinement of any publicly available sequence data set quickly and efficiently. Such reanalyses could lead to discovery and mapping of many previously unknown genes across taxa as well as reconciliation of previously discordant assays. As an example, GWAS, quantitative trait loci mapping, and CNV analyses are commonly used methods to discover genetic mechanisms underlying disease-related phenotypes in humans and in model systems, including zebrafish.29–31 Given the incompleteness of nonhuman genomes, it is possible that loci identified as associating with certain traits may not contain a “smoking-gun” candidate gene. For example, a number of studies have attempted to map the sex-determining locus (or loci) in zebrafish.19,32,33 In one particular study, Bradley et al. used SNPs to map the sex phenotype to two genomic loci, however, their results only accounted for 16% of the trait variance.33 Additional studies have mapped the sex phenotype to the telomeric q arm of chromosome 4, although no specific sex-associated genes were identified.19,32 These difficulties may result from the associated genes being deleted in the TU and/or AB zebrafish lines and therefore missing from the zebrafish reference genome. Such losses would account for the continued inability to identify specific associated genes with sex determination even though the phenotype is likely mapping correctly.19 The methods we present will serve as a fast and efficient mechanism for discovery of deleted reference genes from within these regions and could be used more broadly for any taxon or trait.
This work establishes a resource that can vastly improve the zebrafish reference genome. In addition, it serves as a proof of concept for the pipelines' ability to efficiently capture and characterize genetic diversity residing outside established genome sequences. When conducted on a large sample of individuals across multiple populations, this method should easily reveal population-specific markers, some of which may underlie important genetic traits. This pipeline offers significant advantages over both complete de novo assembly and genome resequencing methods, as the former requires tremendous computing power and specific library preparations34 and the latter is limited only to the examination of previously mapped regions. We expect, if widely adopted, our methods will lead to significant improvements in a wide variety of published reference genomes.
Supplementary Material
Acknowledgments
The authors would like to thank the researchers at the CSIR–Institute of Genomics and Integrative Biology, Wellcome Trust Sanger Institute, and the Mayo Clinic responsible for producing, analyzing, and making available sequence data for BioProject ERP001723. They would also like to thank Dr. John Postlethwait for providing Nadia DNA and the OHSU DNA Services Core for Sanger Sequencing services. All supplementary data is also available at http://archives.pdx.edu/ds/psu/16241. In addition, the nonrepetitive and nonredundant portion of this assembly is available at EBI under the accession S-BSMS1. This study was funded in part by a grant from the National Institute of Environmental Health Sciences (R00ES018892) to K.H.B.
Disclosure Statement
No competing financial interests exist.
References
- 1.Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, et al. . The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013;496:498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. . The sequence of the human genome. Science 2001;291:1304. [DOI] [PubMed] [Google Scholar]
- 3.Haridas S, Breuill C, Bohlmann J, Hsiang T. A biologist's guide to de novo genome assembly using next-generation sequence data: a test with fungal genomes. J Microbiol Methods 2011;86:368–375 [DOI] [PubMed] [Google Scholar]
- 4.Wade CM, Kulbokas EJ, Kirby AW, Zody MC, Mullikin JC, Lander ES, et al. . The mosaic structure of variation in the laboratory mouse genome. Nature 2002;420:574–578 [DOI] [PubMed] [Google Scholar]
- 5.Wright D, Nakamichi R, Krause J, Butlin RK. QTL analysis of behavioral and morphological differentiation between wild and laboratory zebrafish (Danio rerio). Behav Genet 2006;36:271–284 [DOI] [PubMed] [Google Scholar]
- 6.Patowary A, Purkanti R, Singh M, Chauhan R, Singh AR, Swarnkar M, et al. . A sequence-based variation map of zebrafish. Zebrafish 2013;10:15–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Muller WEG, Wetter T, et al. . Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res 2004;14:1147–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mundry M, Bornberg-Bauer E, Sammeth M, Feulner PGD. Evaluating characteristics of de novo assembly software on 454 transcriptome data: a simulation approach. PLoS One 2012;7:e31410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008;18:821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011;27:863–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. . A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramos RTJ, Carneiro A, Azevedo RV, Schneider MP, Barh D, Silva A. Simplifier: a web tool to eliminate redundant NGS contigs. Bioinformation 2012;8:996–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, et al. . Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol 2010;Chapter 19:Unit 19.0.1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, Shah P, et al. . Galaxy: a platform for interactive large-scale genome analysis. Genome Res 2005;15:1451–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goecks J, Nekrutenko A, Taylor J, Galaxy T. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 2010;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harvey SA, Sealy I, Kettleborough R, Fenyes F, White R, Stemple D, et al. . Identification of the zebrafish maternal and paternal transcriptomes. Development 2013;140:2703–2710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rauch G-J, Granato M, Haffter P. A polymorphic zebrafish line for genetic mapping using SSLPs on high-percentage agarose gels. Tech Tips Online 1997;2:148–150 [Google Scholar]
- 19.Wilson CA, High SK, McCluskey BM, Amores A, Yan YL, Titus TA, et al. . Wild sex in zebrafish: loss of the natural sex determinant in domesticated strains. Genetics 2014;198:1291–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han L, Zhao ZM. Comparative analysis of CpG islands in four fish genomes. Compar Funct Genomics 2008:565631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fong JH, Murphy TD, Pruitt KD. Comparison of RefSeq protein-coding regions in human and vertebrate genomes. BMC Genomics 2013;14:1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown KH, Dobrinski KP, Lee AS, Gokcumen O, Mills RE, Shi X, et al. . Extensive genetic diversity and substructuring among zebrafish strains revealed through copy number variant analysis. Proc Natl Acad Sci U S A 2012;109:529–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altshuler D, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, et al. . A map of human genome variation from population-scale sequencing. Nature 2010;467:1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K, et al. . SNP detection for massively parallel whole-genome resequencing. Genome Res 2009;19:1124–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berthelot C, Brunet F, Chalopin D, Juanchich A, Bernard M, Noel B, et al. . The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat Commun 2014;5:3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nei M, Maruyama T, Chakraborty R. Bottleneck effect and genetic-variability in populations. Evolution 1975;29:1–10 [DOI] [PubMed] [Google Scholar]
- 27.Peery MZ, Kirby R, Reid BN, Stoelting R, Doucet-Beer E, Robinson S, et al. . Reliability of genetic bottleneck tests for detecting recent population declines. Mol Ecol 2012;21:3403–3418 [DOI] [PubMed] [Google Scholar]
- 28.Tajima F. The effect of change in population-size on DNA polymorphism. Genetics 1989;123:597–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He X, Fuller CK, Song Y, Meng Q, Zhang B, Yang X, et al. . Sherlock: detecting gene-disease associations by matching patterns of expression QTL and GWAS. Am J Hum Genet 2013;92:667–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rudner LA, Brown KH, Dobrinski KP, Bradley DF, Garcia MI, Smith ACH, et al. . Shared acquired genomic changes in zebrafish and human T-ALL. Oncogene 2011;30:4289–4296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen EY, Dobrinski KP, Brown KH, Clagg R, Edelman E, Ignatius MS, et al. . Cross-species array comparative genomic hybridization identifies novel oncogenic events in zebrafish and human embryonal rhabdomyosarcoma. PLoS Genet 2013;9:e1003727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson JL, Mari AR, Braasch I, Amores A, Hohenlohe P, Batzel P, et al. . Multiple sex-associated regions and a putative sex chromosome in zebrafish revealed by RAD mapping and population genomics. PLoS One 2012;7:e40701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bradley KM, Breyer JP, Melville DB, Broman KW, Knapik EW, Smith JR. An SNP-based linkage map for zebrafish reveals sex determination loci. G3 (Bethesda) 2011;1:3–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gnerre S, MacCallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, et al. . High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A 2010;108:1513–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.